Back to Journals » International Journal of Nanomedicine » Volume 11
Photodynamic therapy of tumors with pyropheophorbide-a-loaded polyethylene glycol–poly(lactic-co-glycolic acid) nanoparticles
Authors Liu H, Zhao M, Wang J, Pang M, Wu Z ![]() , Zhao L, Yin Z, Hong Z
, Zhao L, Yin Z, Hong Z ![]()
Received 11 May 2016
Accepted for publication 17 August 2016
Published 27 September 2016 Volume 2016:11 Pages 4905—4918
DOI https://doi.org/10.2147/IJN.S112541
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Hui Liu,1 Mei Zhao,1 Jin Wang,1 Mingpei Pang,1 Zhenzhou Wu,1 Liqing Zhao,1 Zhinan Yin,1,2 Zhangyong Hong1
1State Key Laboratory of Medicinal Chemical Biology, College of Life Sciences, Nankai University, Tianjin, 2Biomedical Translational Research Institute, International Immunology Center, Jinan University, Guangzhou, People’s Republic of China
Abstract: Photodynamic therapy (PDT) has many advantages in treating cancers, but the lack of ideal photosensitizers continues to be a major limitation restricting the clinical utility of PDT. This study aimed to overcome this obstacle by generating pyropheophorbide-a-loaded polyethylene glycol–poly(lactic-co-glycolic acid) nanoparticles (NPs) for efficient tumor-targeted PDT. The fabricated NPs were efficiently internalized in the mitochondrion by cancer cells, and they efficiently killed cancer cells in a dose-dependent manner when activated with light. Systemically delivered NPs were highly enriched in tumor sites, and completely ablated the tumors in a xenograft KB tumor mouse model when illuminated with 680 nm light (156 mW/cm2, 10 minutes). The results suggested that this tumor-specific NP-delivery system for pyropheophorbide-a has the potential to be used in tumor-targeted PDT.
Keywords: photodynamic therapy, photosensitizer, pyropheophorbide-a, nanoparticle
Introduction
Photodynamic therapy (PDT), a highly localized and tumor-specific modality, has been shown to treat cancers.1 Compared to conventional cancer-treatment modalities, including chemotherapy, radiotherapy, surgery, and more recently immunotherapy,2 PDT has the advantages of extremely low systemic toxicity, minimal invasiveness, excellent function-sparing, and tolerance of repeated therapy without cumulative toxicity.3–5 Essentially, it involves the administration of a photosensitizer (PS) followed by photoexcitation to generate cytotoxic reactive oxygen species (ROS), such as singlet oxygen (1O2), which irreversibly ablate tumor cells without damaging healthy ones.4,6 PDT can also induce immune responses and thus contribute to long-term tumor control,4 and can be used as an adjunctive therapy to reduce the residual tumor burden following surgical resection of tumors.7 Additionally, PDT agents have the potential to act as multifunctional theranostic agents to achieve simultaneous monitoring and therapeutic capabilities during cancer treatment.8,9 For these reasons, PDT has emerged as an important therapeutic option in the management of cancers, such as early lung cancer,10 Barrett’s esophagus,11 bladder cancer,12 head and neck cancers,13 skin cancer,14 and bronchial cancers.15
The essential element for effective PDT is an ideal PS, the lack of which continues to be a major roadblock restricting the clinical utility of PDT as a first-line treatment option.16 An ideal PS must be able to generate singlet oxygen efficiently upon activation with longer-wavelength light (650–900 nm) for deeper light penetration.17 Most importantly, it must preferentially accumulate in tumor tissues and be rapidly cleared from normal tissues. Over the past decade, a substantial effort has been put into the development of new types of PSs. Compared with the clinically used first-generation PS Photofrin (Aptalis, Birmingham, AL, USA; approved in 1997),18 pyropheophorbide-a (Pyro) has been shown to be much more potent, with an over 50% singlet-oxygen quantum yield and a longer wavelength absorption of 668 nm (ε=3.79×104 L·mol−1·cm−1).19 Its derivative Photochlor is now in Phase I/II clinical trials.20 However, Pyro has a relatively narrow therapeutic window with very limited tumor localization, which causes treatment-related toxicity to healthy adjacent tissues and severe cutaneous photosensitivity due to skin accumulation.17,21 Although PDT is a localized treatment,22 highly targeted accumulation of PSs to the diseased tissues is still required for accurate tumor killing without causing excessive toxicity to adjacent normal tissue. The targeted delivery of Pyro will reduce the effective dose administered and skin photosensitivity by increasing the accumulation of PSs inside tumor tissues.23,24 Maximizing tumor-tissue selectivity and reducing normal tissue or skin accumulation is very important for improving the therapeutic outcome of Pyro in PDT.
Nanoparticles (NPs) are ideal carriers to retain therapeutic agents within tumor tissues through the enhanced permeability-and-retention effect,25,26 and currently several NP-based drugs have been either approved or are being tested in clinical trials with very encouraging results.27 The liposomal NP formulation of the PS verteporfin, named Visudyne, is used in clinics for the treatment of patients with age-related macular degeneration or subfoveal choroidal neovascularization,28,29 indicating the promising clinical potential of the NP-based strategy for PS delivery. Among various NP-based delivery systems, one based on the biodegradable copolymer poly(lactic-co-glycolic acid) (PLGA), a US Food and Drug Administration-approved material for human use, has received much attention for its high stability and high drug loading.21,30 Using this strategy, taxol-loaded polyethylene glycol (PEG)ylated PLGA NPs entered a Phase I clinical trial.31,32 Modifying the surface of NPs with PEG can greatly increase their blood-circulation time and subsequently increases their accumulation in tumors.33,34 Published reports have also demonstrated that the encapsulation of PSs inside PLGA NPs improves the photocytotoxic efficiency, reduces the administered dose, and minimizes undesirable effects.35–37 Here, PEG-PLGA NPs were used as the delivery carriers for tumor-targeted delivery of Pyro to solve the limitations associated with Pyro for clinical applications, while leaving the Pyro structure intact without chemical modification. Additionally, due to the high tumor targeting, this system may act as a theranostic for simultaneous fluorescence imaging and PDT in a “see and treat” manner.
Here, the preparation and evaluation of Pyro-loaded PEG-PLGA NPs both in vitro and in vivo was reported. The objectives were to develop a sterilizable polymeric drug-delivery system for Pyro, which was intended to be intravenously injected, to improve the PD activity of this PS. The resulting NP formulation was characterized, and the size, drug loading, entrapment efficiency, and in vitro release profile were examined. In vitro cellular uptake, intracellular localization, and phototoxicity of these NPs were evaluated in a variety of cancer cells. Finally, in vivo distribution and PDT activity were also investigated in KB tumor-bearing xenograft mice using this promising NP formulation.
Materials and methods
Materials
PEG5,000-PLGA (LA:GA ratio 50:50; molecular weight: 50 kDa) was purchased from Jinan Daigang Biomaterial Co Ltd (Jinan, People’s Republic of China [PRC]). Polyvinyl alcohol (PVA) was purchased from Sigma-Aldrich (St Louis, MO, USA). DAPI (4′,6-diamidino-2-phenylindole) was purchased from Hoffman-La Roche Ltd (Basel, Switzerland), and MTT was purchased from Solarbio (Beijing, PRC). MitoTracker green FM and LysoTracker green FM were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Paraformaldehyde and phosphate-buffered saline (PBS) were purchased from Sangon (Shanghai, PRC). Penicillin–streptomycin, fetal bovine serum, trypsin–ethylenediaminetetraacetic acid, Modified Eagle’s Medium (MEM), and Dulbecco’s MEM (DMEM) for cell culture were purchased from HyClone (South Logan, UT, USA). Deionized water was obtained using the Barnstead Nanopure water-purification system from Thermo Fisher Scientific. All other chemicals and reagents used were of analytical grade and obtained commercially unless stated otherwise.
Cells and animals
KB cells (human nasopharyngeal epidermoid carcinoma cells), HeLa cells (human cervical carcinoma cells), and A549 cells (adenocarcinomatous human alveolar basal epithelial cells) were purchased from Saierbio (Tianjin, People’s Republic of China). Both A549 cells and HeLa cells were cultured in DMEM, and KB cells were cultured in Roswell Park Memorial Institute 1640 medium. All cells were incubated in medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in humidified air supplemented with 5% CO2.
Female BALB/c nude mice (6–8 weeks of age, 15–20 g) were purchased from the Academy of Military Science (Beijing, PRC). The mice were maintained in a germ-free environment with free access to food and water. All animals were treated in accordance with the guidelines of the Committee on Animals of Nankai University (Tianjin, PRC). All animal procedures were approved by the Nankai University Experimental Animal Ethics Committee.
Preparation of Pyro-loaded PEG-PLGA NPs
PEG-PLGA polymer particles were prepared using a water–oil emulsion solvent-evaporation method. Briefly, Pyro (2 mg/mL dissolved in 125 μL trichloromethane) was mixed with PEG-PLGA (40 mg/mL dissolved in 125 μL trichloromethane). The mixture was added to PVA (2% dissolved in 2 mL deionized water) and vortexed for 1 minute. Then, the primary emulsion was sonicated (20 W, 40% duty cycle, 20 cycles) using a microtip-probe sonicator over an ice bath, and the organic solvents were evaporated with an air pump. Large particles were removed by centrifugation (3,000 rpm, 10 minutes), and the resulting particles were collected using high-speed centrifugation (12,000 rpm, 30 minutes) and washed twice with PBS.
Characterization of particle size
The size distribution of the prepared particles was characterized with a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK), and the morphology of the NPs was assessed by transmission electron microscopy (Tecnai G2 F20; Hillsboro, OR, USA).
Pyro-loading/encapsulation efficiency and in vitro release profile
For determination of the loading efficiency (LE) and encapsulation efficiency (EE) of the prepared NPs, the NPs were dissolved in acetonitrile and PBS (1:1), and the Pyro concentration inside was analyzed using high-performance liquid chromatography (HPLC; LC-20AT; Shimadzu, Kyoto, Japan). HPLC was performed using a 150×4.6 mm C18 column with a 0.6 mL/min flow rate at λ=680 nm. The column was initially held at 10% CH3CN (0.14% TFA), 90% H2O (0.14% TFA). The concentration of CH3CN was increased to 50% in 10 minutes and then to 90% in 35 minutes; this was maintained for 5 minutes. The column was washed with 100% CH3CN for 15 minutes and allowed to equilibrate at the initial mobile phase conditions for 15 minutes before the next injection. LE and EE were calculated according to the following formulae:
|
|
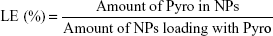
|
|
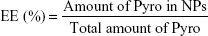
To determine Pyro-release behavior in vitro, the Pyro-loaded NPs were resuspended in 500 μL fresh PBS solution (pH 7.4) and gently shaken using a shaker set at 37°C and 110 rpm. At each projected interval, the buffer was replaced with the same volume of PBS, followed by centrifugation at 12,000 rpm for 10 minutes. The obtained supernatant was analyzed with HPLC. Release profiles are reported in terms of cumulative Pyro released versus time, and values are expressed as mean ± standard error of mean from five samples.
Cytotoxicity assays
KB, A549, and HeLa cells were grown for 12 hours in 96-well plates at a concentration of 5×103 cells/well. Next, the cells were incubated with various concentrations (0–250 μg/mL) of Pyro-loaded PEG-PLGA NPs. After 16 hours’ incubation at 37°C with 5% CO2, the cells were washed twice with ice-cold PBS buffer, and 100 μL of cell-growth medium was added. In vitro PDT treatment was performed by putting the 96-well plate under a 660 nm continuous-wavelength laser (40 mW/cm2) and illuminated for different times (0–5 minutes). The cells were then grown for 24 hours, at which time MTT was added to the medium at 0.5 mg/mL. The plates were incubated for an additional 4 hours at 37°C. After the supernatant was removed, 100 μL dimethyl sulfoxide was added to dissolve the crystals fully. Absorbance at 570 nm was measured on a microplate reader (EXL-800; Bio-Tek, Winooski, VT, USA). Radiant dose was calculated using the following formula:
Radiant dose (J/cm2) = Irradiance (W/cm2) × T (s) | (3) |
Confocal microscopy
The cellular uptake assays and subcellular colocalization assays were performed on A549 cells. For the cellular uptake assays, A549 cells were seeded in a 24-well cell-culture plate on sterile coverslips at 104 cells/well. After 12 hours, the Pyro-loaded PEG-PLGA NPs were added to the culture medium at a variety of particle concentrations (3.75–60 μg/mL) and incubated at 37°C for 4 hours, with free Pyro as control. The cells were washed three to five times with PBS, fixed with 100 μL 10% formalin for 15 minutes, and then stained with DAPI (blue fluorescence) for the nuclei. For the subcellular colocalization study, A549 cells were seeded in a 24-well plate on sterile coverslips at 104 cells/well. Twelve hours later, Pyro-loaded PEG-PLGA NPs were added to the culture medium at a concentration of 60 μg/mL and incubated for 4 hours. The cells were incubated with 100 nM MitoTracker green FM and LysoTracker green FM for 30 minutes at 37°C, then washed three to five times with PBS and fixed with 100 μL of 10% formalin for 15 minutes and the nuclei stained with DAPI. After being washed with PBS, the cells were mounted and visualized on a confocal microscope (TCS SP8; Leica Microsystems, Wetzlar, Germany) using 405 nm excitation and 410–480 nm emission for blue (DAPI), 488 nm excitation and 513–592 nm emission for green, and 633 nm excitation and 638–759 nm emission for red (Pyro) settings. Each stain was performed on three individual groups, and the photographs in the figures are representative of each group.
In vivo imaging
The nude mice were subcutaneously inoculated with 3×106 KB cells in the right flank. When tumor volumes were approximately 50 mm3, Pyro-loaded NPs resuspended in PBS (200 μL) were injected via the tail vein at a dose of 75 mg/kg (equal to 3 mg/kg Pyro), and the mice in the control group were injected with the same volume of PBS or same amount of free Pyro. In vivo fluorescence analysis was performed before (prescan images) and after the injection at various time points using a Xenogen in vivo imaging system with a Cy5.5 filter (excitation: 615–665 nm, emission: 695–770 nm). The mice were euthanized 48 hours postinjection (for free Pyro group, the mice were euthanized 24 hours postinjection), and major organs, including the heart, liver, spleen, lungs, and kidneys, as well as tumors, were collected. Fluorescence images were then acquired.
PDT treatment of tumors in vivo
For PDT treatment, the mice were injected with KB cells (3×106 cells) in the right flank. When tumor volumes were approximately 50 mm3, the mice were randomly assigned to eight groups with at least five mice per group for the following treatments: 1) 75 mg/kg Pyro-loaded NPs (equal to 3 mg/kg Pyro) in 200 μL PBS were injected intravenously every other day for a week and irradiation performed at 94 J/cm2 (156 mW/cm2 for 10 minutes) 24 hours after the injection; 2) the same volume of PBS injected intravenously and no irradiation performed; 3) 75 mg/kg Pyro-loaded NPs in 200 μL PBS injected intravenously every other day for a week, and no irradiation performed; 4) the same volume of PBS injected intravenously and irradiation performed; 5) 3 mg/kg free Pyro injected intravenously and irradiation performed at 94 J/cm2 (156 mW/cm2 for 10 minutes) 24 hours after the injection; 6) same dose of Pyro injected intravenously and no irradiation performed; 7) 75 mg/kg empty PEG-PLGA NPs injected intravenously and irradiation performed; 8) 75 mg/kg of empty PEG-PLGA NPs injected intravenously and no irradiation performed. All mice were monitored daily, and their tumor volume and body weight were measured every day. The tumor volume was calculated using the following formula:
Volume = Length × Width × Width ×0.5 | (4) |
Statistical analysis
The results from the MTT assays and tumor treatments are shown as mean ± standard error of mean. Tumor-growth curves were compared and analyzed using two-way analysis of variance, performed by GraphPad Prism 5.0 for Windows.
Results and discussion
Synthesis and characterization of Pyro-loaded PEG-PLGA
Pyro-loaded PEG-PLGA NPs were prepared using a double-emulsion method to generate Pyro with the PEG-PLGA polymer in the presence of 2% PVA. The absorbance spectra of the Pyro-loaded PEG-PLGA NPs and free Pyro are shown in Figure 1A and B. The size distribution and zeta-potential of Pyro-loaded NPs and empty NPs were measured with dynamic light scattering. Figure 1C and D show that the size of the empty PEG-PLGA NPs was 108±18 nm and of Pyro-loaded NPs was 96±14 nm, which showed no significant difference in the size distribution between the empty PEG-PLGA NPs and Pyro-loaded NPs. Figure 1E shows that Pyro-loaded NPs had slightly negative zeta-potential, while empty PEG-PLGA NPs were almost neutral. Surface morphology was revealed to be a uniform spherical structure, as determined by TEM (Figure 1F). To confirm further whether the Pyro-loaded NPs were stable in physiological conditions, they were incubated in PBS in a water-bath shaker at 37°C for 24 hours. The size distribution (Figure S1A) and zeta-potential (Figure S1B) of these NPs showed negligible alteration after the incubation. This result suggests that the NPs are stable in physiological condition. The high solubility under physiological conditions and the appropriate size suggest that Pyro-loaded PEG-PLGA NPs can passively target tumor tissues.
 | Figure 1 Characterization of Pyro-loaded PEG-PLGA NPs. |
LE and EE of Pyro (for HPLC analysis of Pyro, see Figure S2) were 4.17%±0.23% and 49.81%±1.23%, respectively. Figure 1G shows the in vitro release profile of Pyro from the particles after incubating the particles in a water-bath shaker at 37°C. It was found that 20% and 80% of the entrapped Pyro were released from the particles during the first 3 and 24 hours, respectively. After 72 hours, 97% of the entrapped Pyro was released. This rapid and sustained drug release in the first 1 or 2 days is ideal for PDT therapy, as PDT can then be carried out with laser illumination 1 or 2 days after injection of the PSs.
In vitro cytotoxicity studies
To explore the PDT activity of the Pyro-loaded PEG-PLGA NPs in vitro, a series of standard MTT viability assays was perfomed with A549, HeLa, and KB cells after PDT. As shown in Figure 2, there was no significant cytotoxicity associated with exposure to Pyro-loaded NPs alone in the absence of excitation light or with light exposure alone in the absence of NPs in any of the three cell lines, while cell viabilities were significantly reduced by the increase in excitation light and NPs when they were administered simultaneously. For example, as shown in Figure 2A, the viability of the KB cells was 45% following exposure to light (40 mW/cm2 for 2.5 minutes) in the presence of 1.8 μg/mL Pyro-loaded PEG-PLGA NPs, but viability was reduced to nearly zero in the presence of 3.75 μg/mL NPs, indicating that the survival percentage was significantly reduced as the concentration of Pyro-loaded PEG-PLGA NPs increased. The cell viability of KB cells was also significantly influenced by the dose of excitation light in the presence of NPs: it was 45% when light exposure was 23.5 J/cm2 (40 mW/cm2 for 2.5 minutes), but viability was reduced to 24% following exposure to 47 J/cm2 light (40 mW/cm2 for 5 minutes) in the presence of 1.8 μg/mL NPs. Similar results were obtained for HeLa and A549 cells treated with Pyro-loaded PEG-PLGA NPs (Figure 2B and C). Free Pyro had a similar PDT effect on KB (Figure 2D), HeLa (Figure 2E), and A549 (Figure 2F) cells. These data show that the phototoxicity of Pyro-loaded PEG-PLGA or free Pyro was dependent on the dose of light exposure and NPs as the two essential elements of PDT.
 | Figure 2 In vitro PDT activity against tumor cells. |
Cell-morphology changes during PDT with Pyro-loaded NPs
To verify further the effect of PDT using Pyro-loaded NPs on tumor cells, morphological changes were observed in the target cells after PDT using microscopy (Figure 3). As shown in Figure 3A, cellular swelling, bleb formation, and rupture of vesicles were observed in the A549 tumor cells after exposure to Pyro-loaded NP-based PDT; however, they showed no significant morphological changes following exposure to Pyro-loaded NPs alone in the absence of excitation light. HeLa (Figure 3B) and KB (Figure 3C) cells also showed similar morphological changes after PDT. These results suggest that Pyro-loaded PEG-PLGA NPs and light exposure were essential for PDT. As the control, the free Pyro had a similar PDT effect against A549 (Figure 3D), HeLa (Figure 3E), and KB cells (Figure 3F).
 | Figure 3 Cell morphological changes observed by microscopy during Pyro-loaded PEG-PLGA NP-based and free Pyro-based PDT. |
Cellular uptake studies
To analyze further the internalization efficacy of Pyro-loaded PEG-PLGA NPs, different concentrations of NP solutions were incubated with A549 cells and KB cells for 4 hours in vitro, with free Pyro as control, and the cells were then observed with confocal laser-scanning microscopy. The imaging showed that Pyro signals were dispersed throughout the cells, and the signal intensity increased gradually with the increase in NP (Figure 4) and free Pyro concentration (Figure 5A). The internalization of sufficient amounts of PSs is very important for efficient PDT, as the intracellular PS is responsible for inducing phototoxicity. The high internalization efficiency of these NPs by cancer cells indicated that they can potentially be used for efficient PDT. To determine the subcellular localization of the Pyro-loaded PEG-PLGA NPs and free Pyro, MitoTracker green FM and LysoTracker green FM were used to stain the mitochondria and lysosome, respectively, and DAPI to stain the nuclei. Confocal microscopy analysis showed that Pyro fluorescence in the PLGA NP group had partially colocalized with LysoTracker green fluorescence (Figure 4B) and a high degree of colocalization with MitoTracker green fluorescence (Figure 4C), but was absent from the nucleus, demonstrating that the Pyro-loaded NPs were predominantly internalized in the mitochondria, which are known to be very effective targets for phototoxicity damage.38 However, the Pyro fluorescence in the free Pyro group was mainly colocalized with LysoTracker green fluorescence (Figure 5B) and partially colocalized with MitoTracker green fluorescence (Figure 5C).
 | Figure 4 In vitro cellular uptake and intracellular localization of Pyro-loaded PEG-PLGA NPs. |
 | Figure 5 In vitro cellular uptake and intracellular localization of free Pyro. |
In vivo distribution of Pyro-loaded PEG-PLGA NPs in tumor-bearing mice
The tumor-accumulating ability of Pyro-loaded PEG-PLGA NPs was investigated by a whole-body-imaging method in a xenograft tumor model by subcutaneously inoculating KB cells (3×106 cells per mouse) in nude mice. When tumors were approximately 50 mm3 in volume, 75 mg/kg Pyro-loaded PEG-PLGA NPs (or 3 mg/kg free Pyro as control) were intravenously injected into the mice via the tail vein, and the distribution of Pyro-loaded NPs was determined by the presence of Pyro fluorescence as a function of time. As shown in Figure 6A, at 6 hours the fluorescence of Pyro was distributed throughout the mouse body, with intense fluorescence signals in the liver and kidney. Over time, the fluorescence signals in normal organs decreased, while the tumor-to-background ratio increased. At 24 hours postinjection, only very weak fluorescence signals were detected in normal organs, while the fluorescence intensity in the tumors was still very high; this fluorescence could last up to 48 hours. Figure 6B shows the fluorescence distribution of Pyro in the mice injected with free Pyro. The signals were already very weak at 6 hours postinjection, with most of them accumulated in the kidney, and then almost disappeared after 9 hours postinjection. It was hard to detect the accumulation of Pyro fluorescence in the tumor site.
 | Figure 6 In vivo distribution of Pyro-loaded PEG-PLGA NPs in xenograft KB tumor mouse model. |
The mice injected with Pyro-loaded NPs were then euthanized at 48 hours postinjection, and fluorescence intensity in the organs and tumors was examined. As shown in Figure 6C and D, the fluorescence signals of mice injected with Pyro-loaded NPs in the tumor were very strong, with some signals detectable in the liver, while the fluorescence in the kidney, heart, thymus, and spleen was significantly lower. Accordingly, the mice injected with free Pyro were euthanized at 24 hours postinjection, and fluorescence signals of Pyro were not detected in either organs or tumors (Figure 6E).
The specific accumulation of the prepared Pyro-loaded NPs in tumor sites could be attributed to the highly permeable vascular structure of the neoplasm, which always leads to a passive accumulation of nanosize materials. The long duration of fluorescence of the NPs in tumor sites may be attributed to the effect of PEGylation of the NPs, which is generally a very efficient strategy to improve the systemic circulation time of NP-based drugs.
Pyro-loaded PEG-PLGA NP-based PDT-induced tumor shrinkage
Efficacy of the fabricated Pyro-loaded PEG-PLGA NPs was further evaluated as PDT agents for the treatment of KB tumors in BALB/c nude mice in vivo. KB tumors were inoculated under the skin of the nude mice, and PDT was carried out when the tumor volume was approximately 50 mm3. The mice were intravenously injected with 75 mg/kg Pyro-loaded PEG-PLGA NPs (3 mg/kg Pyro) and then irradiated with a 680 nm laser (156 mW/cm2, 10 minutes) 24 hours after the injection of NPs (group 1). As controls, group 2 received no Pyro-loaded PEG-PLGA injection or laser exposure, group 3 received the same dose of Pyro-loaded PEG-PLGA NPs but no laser exposure, group 4 received laser exposure (156 mW/cm2, 10 minutes) but no Pyro-loaded PEG-PLGA NPs, group 5 received 3 mg/kg free Pyro and laser exposure (156 mW/cm2, 10 minutes) 24 hours after the injection of free Pyro, group 6 received the same dose of Pyro but no laser exposure, group 7 received 75 mg/kg empty PEG-PLGA NPs and laser exposure (156 mW/cm2, 10 minutes) 24 hours after the injection of empty NPs, and group 8 received 75 mg/kg empty PEG-PLGA NPs but no laser exposure.
The effect of PDT on the tumor growth was then assessed by monitoring the tumor volumes over a period of 2 weeks (Figure 7). The tumor growth in group 1 was significantly inhibited, with almost no detected tumor mass at day 14 (Figure 7A[a] and B), while the tumors in groups 2 (Figure 7A[b]), 3 (Figure 7A[c]), 4 (Figure 7A[d]), 5 (Figure 7A[e]), 6 (Figure 7A[f]), 7 (Figure 7A[g]), and 8 (Figure 7A[h]) grew very rapidly. The weight of the mice (Figure 7C) showed no obvious differences among the groups, indicating that not only irradiation but also Pyro-loaded PEG-PLGA NPs had no effect on mouse weight. The serum level of TNFα of these mice was determined, and shown to be a little higher than control mice, but was within normal range (Figure 7D). The serum level of IL-6 (Figure 7E), AST (Figure 7F), and ALT (Figure 7G) of the mice injected with Pyro-loaded NPs showed no difference from mice injected with PBS. The results suggest that Pyro-loaded NPs do not cause systemic toxicity or inflammatory response. The extremely high efficacy of the fabricated Pyro-loaded PEG-PLGA NPs in KB tumors demonstrate the high clinical potential of this strategy.
 | Figure 7 In vivo PDT in a xenograft KB tumor mouse model. |
Conclusion
In conclusion, PEG-PLGA-encapsulated Pyro-loaded NPs were fabricated for tumor-targeted PDT treatment, with the aim of solving the limitations associated with the clinical application of Pyro. The NPs can be activated in the near-infrared spectroscopy range (670 nm), and were efficiently internalized by cancer cells, where they were predominantly located in the mitochondrion. Following in vitro PDT with these NPs, the cancer cells, including KB, A549, and HeLa cells, were efficiently killed in a dose-dependent manner when illuminated with light. After administration via systemic delivery, the NPs were selectively enriched in tumor sites, and completely ablated the tumors in the KB xenograft model following activation with 680 nm light (156 mW/cm2, 10 minutes). Studies suggest that this tumor-specific NP-delivery system for Pyro has the potential to be used for tumor-targeted PDT.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (31270926), National High Technology Research and Development Program of China (SS2014AA021601), and the Major Program of the National Natural Science Foundation of China (31527801).
Disclosure
The authors report no conflicts of interest in this work.
References
Wang D, Fei B, Halig LV, et al. Targeted iron-oxide nanoparticle for photodynamic therapy and imaging of head and neck cancer. ACS Nano. 2014;8(7):6620–6632. | ||
Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. 2012;9(3):193–199. | ||
Juarranz A, Jaén P, Sanz-Rodríguez F, Cuevas J, González S. Photodynamic therapy of cancer: basic principles and applications. Clin Transl Oncol. 2008;10(3):148–154. | ||
Dolmans DE, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat Rev Cancer. 2003;3(5):380–387. | ||
Brown SB, Brown EA, Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5(8):497–508. | ||
Xiao L, Gu L, Howell SB, Sailor MJ. Porous silicon nanoparticle photosensitizers for singlet oxygen and their phototoxicity against cancer cells. ACS Nano. 2011;5(5):3651–3659. | ||
Dougherty TJ, Marcus SL. Photodynamic therapy. Eur J Cancer. 1998;28(10):1734–1742. | ||
Yu MK, Park JH, Jon SY. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2(1):3–44. | ||
Cheng Y, Samia AC, Meyers JD, Panagopoulos I, Fei B, Burda C. Highly efficient drug delivery with gold nanoparticle vectors for in vivo photodynamic therapy of cancer. J Am Chem Soc. 2008;130(32):10643–10647. | ||
Furuse K, Fukuoka M, Kato H, et al. A prospective phase II study on photodynamic therapy with photofrin II for centrally located early-stage lung cancer. J Clin Oncol. 1993;11(10):1852–1857. | ||
Overholt BF, Lightdale CJ, Wang KK, et al. Photodynamic therapy with porfimer sodium for ablation of high-grade dysplasia in Barrett’s esophagus: international, partially blinded, randomized phase III trial. Gastrointest Endosc. 2005;62(4):488–498. | ||
Nseyo UO, DeHaven J, Dougherty TJ, et al. Photodynamic therapy (PDT) in the treatment of patients with resistant superficial bladder cancer: a long-term experience. J Clin Laser Med Surg. 1998;16(1):61–68. | ||
Biel M. Advances in photodynamic therapy for the treatment of head and neck cancers. Lasers Surg Med. 2006;38(5):349–355. | ||
Cairnduff F, Stringer MR, Hudson EJ, Ash DV, Brown SB. Superficial photodynamic therapy with topical 5-aminolaevulinic acid for superficial primary and secondary skin cancer. Br J Cancer. 1994;69(3):605–608. | ||
Monnier P, Savary M, Fontolliet C, et al. Photodetection and photodynamic therapy of ‘early’ squamous cell carcinomas of the pharynx, oesophagus and tracheo-bronchial tree. Lasers Med Sci. 1990;5(2):149–169. | ||
Lovell JF, Liu TW, Chen J, Zheng G. Activatable photosensitizers for imaging and therapy. Chem Rev. 2010;110(5):2839–2857. | ||
Ethirajan M, Chen Y, Joshi P, Pandey RK. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem Soc Rev. 2011;40(1):340–362. | ||
Bonnett R. Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chem Soc Rev. 1995;24(1):19–33. | ||
Zhang M, Zhang Z, Blessington D, et al. Pyropheophorbide 2-deoxyglucosamide: a new photosensitizer targeting glucose transporters. Bioconjug Chem. 2003;14(4):709–714. | ||
Huang Z. A review of progress in clinical photodynamic therapy. Technol Cancer Res Treat. 2005;4(3):283–293. | ||
Chatterjee DK, Li SF, Yong Z. Nanoparticles in photodynamic therapy: an emerging paradigm. Adv Drug Deliv Rev. 2008;60(15):1627–1637. | ||
Reddy GR, Bhojani MS, Mcconville P, et al. Vascular targeted nanoparticles for imaging and treatment of brain tumors. Clin Cancer Res. 2006;12(22):6677–6686. | ||
Yoon I, Li JZ, Shim YK. Advance in photosensitizers and light delivery for photodynamic therapy. Clin Endosc. 2013;46(1):7–23. | ||
Celli JP, Spring BQ, Rizvi I, et al. Imaging and photodynamic therapy: mechanisms, monitoring, and optimization. Chem Rev. 2010;110(5):2795–2838. | ||
Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent SMANcs. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Dan P, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Zarbin MA, Arlow T, Ritch R. Regenerative nanomedicine for vision restoration. Mayo Clin Proc. 2013;88(12):1480–1490. | ||
Gupta A, Avci P, Sadasivam M, et al. Shining light on nanotechnology to help repair and regeneration. Biotechnol Adv. 2013;31(5):607–631. | ||
Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. | ||
Anand P, Nair HB, Sung B, et al. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol. 2009;79(3):330–338. | ||
Kim TY, Kim DW, Chung JY, et al. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10(11):3708–3716. | ||
Kohler N, Fryxell GE, Zhang M. A bifunctional poly(ethylene glycol) silane immobilized on metallic oxide-based nanoparticles for conjugation with cell targeting agents. J Am Chem Soc. 2004;126(23):7206–7211. | ||
Yoo JW, Chambers E, Mitragotri S. Factors that control the circulation time of nanoparticles in blood: challenges, solutions and future prospects. Curr Pharm Des. 2010;16(21):2298–2307. | ||
Bechet D, Couleaud P, Frochot C, Viriot ML, Guillemin F, Barberi-Heyob M. Nanoparticles as vehicles for delivery of photodynamic therapy agents. Trends Biotechnol. 2008;26(11):612–621. | ||
Gomes AJ, Lunardi LO, Marchetti JM, Lunardi CN, Tedesco AC. Photobiological and ultrastructural studies of nanoparticles of poly(lactic-co-glycolic acid)-containing bacteriochlorophyll-a as a photosensitizer useful for PDT treatment. Drug Deliv. 2005;12(3):159–164. | ||
Jia X, Jia L. Nanoparticles improve biological functions of phthalocyanine photosensitizers used for photodynamic therapy. Curr Drug Metab. 2012;13(8):1119–1122. | ||
Kessel D, Luguya R, Vicente MG. Localization and photodynamic efficacy of two cationic porphyrins varying in charge distribution. Photochem Photobiol. 2003;78(5):431–435. |
Supplementary materials
 | Figure S1 Characterization of Pyro-loaded PEG-PLGA NPs after incubation in PBS for 24 hours. |
 | Figure S2 HPLC of Pyro. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.