Back to Journals » Infection and Drug Resistance » Volume 13
Phenotypic and Genomic Characterization of Virulence Heterogeneity in Multidrug-Resistant ST11 Klebsiella pneumoniae During Inter-Host Transmission and Evolution
Authors Liu C ![]() , Du P, Zhao J, Li B, Wang C, Sun L, Lu B, Wang Y, Liu Y, Cao B
, Du P, Zhao J, Li B, Wang C, Sun L, Lu B, Wang Y, Liu Y, Cao B
Received 26 December 2019
Accepted for publication 26 April 2020
Published 10 June 2020 Volume 2020:13 Pages 1713—1721
DOI https://doi.org/10.2147/IDR.S243836
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eric Nulens
Chao Liu, 1, 2 Pengcheng Du, 3 Jiankang Zhao, 2 Binbin Li, 2 Chunlei Wang, 2 Lingxiao Sun, 2, 4 Binghuai Lu, 2 Yimin Wang, 2 Yingmei Liu, 2 Bin Cao 1, 2, 4– 6
1Peking Union Medical College, Institute of Respiratory Medicine, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, Laboratory of Clinical Microbiology and Infectious Diseases, China-Japan Friendship Hospital, Beijing, People’s Republic of China; 3Institute of Infectious Diseases, Beijing Ditan Hospital, Capital Medical University and Beijing Key Laboratory of Emerging Infectious Diseases, Beijing, People’s Republic of China; 4Clinical Center for Pulmonary Infections, Capital Medical University, Beijing, People’s Republic of China; 5National Clinical Research Center of Respiratory Diseases, Beijing, People’s Republic of China; 6Tsinghua University-Peking University Joint Center for Life Sciences, Beijing, People’s Republic of China
Correspondence: Bin Cao Tel +86 10-84205288
Email [email protected]
Background: Multidrug-resistant (MDR) ST11 hypervirulent Klebsiella pneumoniae (hvKp) is emerging in China.
Purpose: The aim of this study was to track the transmission and evolution of hvKp.
Materials and Methods: A retrospective study focused on Kp infection was conducted. Clinical data were collected from electronic medical records. Whole-genome sequencing of Kp strains was performed. Single-nucleotide polymorphisms (SNPs) were analyzed and a transmission map was constructed. Sequence type, and antimicrobial and virulence-associated genes were characterized. Strains with some combination of the virulence genes, prmpA, prmpA2, iucA, iroB, and peg-344, were defined as hvKp. Kp virulence phenotypes were evaluated using the Galleria mellonella model.
Results: All 33 Kp strains were MDR-Kp and 13 (39.4%) were hvKp. Most hvKp strains (84.6%, 11/13) were hospital-acquired infections (HAIs). Two unique combinations of virulence-associated genes were detected among hvKp strains. Eleven cases were associated with prmpA2+iucA and two strains presented with peg-344+prmpA+prmpA2+iucA. Surprisingly, two community-acquired MDR-hvKp infection cases were identified. Eight hvKp strains (61.5%, 8/13) exhibited a hypervirulent phenotype in the G. mellonella model. Five MDR-hvKp strains with the hypervirulence phenotype originated from a single cluster. Additionally, nine clones were identified among the two clades, six of which were hvKp. Moreover, the hvKp in clade 1 carried the IncHI1B plasmid replicon, whereas none of the hvKp strains in clade 2 harbored IncHI1B. These data, showing that different hvKp clones distributed into separate clades, indicate that transmission and evolution occurred within the hospital.
Conclusion: During inter-host evolution and transmission, various virulence clusters of the epidemic clone, MDR-ST11, converged, conferring phenotypic virulence heterogeneity and spread within the hospital and possibly the community. Mobile/conjugative genetic elements associated with virulence-encoding gene clusters might emerge and have been transmitted within the hospital, suggesting that enhanced ongoing surveillance is essential.
Keywords: hypervirulent Klebsiella pneumoniae, multidrug resistance, whole-genome sequencing, hospital-acquired infection, community-acquired infection
Introduction
Klebsiella pneumoniae (Kp) is a major emerging Gram-negative bacteria involved in hospital-acquired infection, particularly various fatal infections.1 There are two distinct pathotypes of Kp: hypervirulent (hvKp) and classical (cKp),2–5 where cKp is notorious for acquiring antibiotic-resistant genes, representing a threat to public health. Further, cKp is rapidly becoming resistant to the majority of available antibiotics, primarily driven by worldwide dissemination of a specific multidrug-resistant clone, defined as clone group (CG) 258, particularly the ST11 in China.6,7 Kp ST11 is typically encountered in isolates producing Kp carbapenemase (KPC).6,8 Further, the hvKp pathotype is frequently associated with aggressive, invasive community infections, such as bacteremia and pyogenic liver abscesses, in immunocompetent ambulatory younger adults with no underlying disease.9–11 Previously, hvKp was defined by its hypermucoviscosity phenotype (string test, >5 mm);9 however, recent evidence from in vitro and in vivo models shows that the hypermucoviscosity phenotype is not actually closely associated with virulence.4,12-15. Further, compared with genetic traits, hypermucoviscosity is an inferior marker for differentiating hvKp and cKp,16 A combination of five virulence-associated genes, peg-344, iroB, iucA, prmpA, and prmpA2, showed higher diagnostic accuracy for hvKp than phenotype or other virulence-associated genes alone.16 Moreover, cKp strains are at considerably higher risk of acquiring virulence genes to become hypervirulent and drug resistant, relative to hvKp clones.17 Importantly, a previous study reported that the epidemic clone in China (ST11) is becoming hypervirulent due to acquisition of various virulence genes or a pLVKP-like plasmid.18
Most previous studies have primarily focused on outbreaks of cKp. Although the multidrug resistance (MDR) and extended-spectrum-β-lactamase (ESBL) producing hvKp, particularly those resistant to colistin and carbapenems, are emerging in China,19–21 few reports have demonstrated that hvKp can be the source of hospital infection outbreaks.18 Most importantly, reports of the dynamic genomic evolution of hvKp strains are rare.
Here, during surveillance, we discovered an increasing number of ST11 hypermucoviscosity and MDR Kp strains highly clustered in the hospital for approximately 1 year and associated with poor prognosis. Therefore, we conducted this study to characterize the genomic differences and phylogenetic relationships of Kp, to reconstruct its transmission route and study the genomic basis underlying the poor outcomes of patients infected with this pathogen. Surprisingly, during transmission and evolution, ST11 MDR-Kp evolved into two different pathotypes, resulting in multiple clones transmitted via different routes.
Materials and Methods
Patients Enrolled
A retrospective study was conducted from March 2017 to June 2018 at a hospital, comprising three sites: a headquarters, and west and north campuses. The respiratory department spanned two sites, the headquarters and the north campus, and the medical staff worked shifts at the different sites. Patient clinical characteristics were obtained from medical records, including basic demographic features, underlying disease, antibiotic agent exposure, mechanical ventilation, use of invasive devices, and outcome. Poor prognosis (death or withholding life-sustaining therapy) within 30 days was defined as the primary endpoint. Sequential organ failure assessment (SOFA) score and acute physiology and chronic health evaluation II (APACHE II) were conducted for patients positive for Kp by culture. The main inclusion criteria were: 1) age > 18 years; 2) Kp cultured twice or more and simultaneous clinical infection symptoms. Exclusion criteria were: 1) insufficient clinical data or bacterial strain samples; and 2) duplicate isolates from the same patient within two weeks. Hospital-acquired (HA) and community-acquired (CA) Kp infections were defined as previously described.9 CA and HA infections were differentiated for all patients.
The protocol for this study was approved by the China–Japan Friendship Hospital Ethics Committee (2018-GZR-199), and the Guidelines for Human Experimentation (PRC) were followed throughout. Informed consent was not needed, due to the retrospective nature of the study. Additionally, data for all patients enrolled in this study were anonymized.
Antimicrobial Resistance and Virulence-Associated Phenotype
All isolates were stored at −80°C and analyzed by MALDI-TOF mass spectrometry. HvKp was defined as peg-344, iroB, iucA, prmpA, or prmpA2 positive.16 The primers used are described in Supplementary Table 1.
Antimicrobial susceptibility testing was performed as previously described, and the results were interpreted according to the 2017 Clinical and Laboratory Standards Institute (CLSI) guidelines. The antibiotics used for testing included amikacin, gentamicin, tobramycin, ampicillin/sulbactam, aztreonam, cefazolin, cefepime, ceftriaxone, ceftazidime, ciprofloxacin, levofloxacin, piperacillin/tazobactam, and trimethoprim/sulfamethoxazole. An MDR strain was defined as a strain resistant to three or more different antimicrobial categories, as described previously.5
The hypermucoviscous phenotype was detected by string test, as described previously.9 The virulence phenotype was also evaluated using the wax moth, Galleria mellonella, model (insects of approximately 300 mg). Overnight cultures of Kp strains were adjusted using physiological saline to 1 × 106 CFU/mL. Then, Kp isolates were injected into the G. mellonella, as described previously18 and their survival rate recorded at 12, 24, 36, and 48 h, respectively. The survival rate of G. mellonella infected with the K1 hypervirulent Kp isolate was used as reference to define the hypervirulent phenotype.
Serum resistance assays of all isolates were conducted as previously described.22 Briefly, bacterial suspensions were collected and then mixed with healthy human serum at 1:3. The mixture was agitated at 37°C and clone counts recorded at time 0 and after 30, 60, 120, and 180 min of culture for 24 h. Survival percentages at different time points were used to determine serum susceptibility/resistance. Each strain was tested three times.
Whole-Genome Sequencing to Identify Genomic Features
All strains were sequenced using the Illumina HiSeq 2500 sequencing platform by constructing paired-end libraries to obtain 150 bp reads. Raw data were filtered to remove low-quality reads, then assembled using SPAdes v3.13. Sequencing type (ST) was identified using MLST 2.0 (Center for Genomic Epidemiology). Draft genome sequences were annotated using Prokka. Capsular types were analyzed using Kaptive. Antimicrobial resistance genes, virulence genes, and plasmid replicon types were annotated by comparison with relevant databases (ResFinder, Virulence Factor Database, plasmidFinder), using BLAST software. Antimicrobial resistance and virulence genes were identified using thresholds of 90% identity and minimum length coverage of 80%. Additionally, PCR was applied for MLST and peg-344, iroB, iucA, prmpA, and prmpA2, to validate our sequencing findings.
For phylogenetic analysis, we identified single-nucleotide polymorphisms (SNPs) by mapping sequencing reads to the genome sequence of Kp strain HS11286 (GenBank accession: NC_016845.1) using bowtie2 software, followed by filtering the results using Samtools. High-quality SNPs (hqSNPs), supported by more than 5 reads, were retained and adjacent mutations within 5-bp filtered, to avoid recombination. Finally, the concatenated sequences were used to perform phylogenetic analysis using FastTree v2.1.10 with the maximum likelihood method. We deposited the read data in the Sequencing Read Archive database (SRP186665 and SRP229572).
Results
Clinical Characteristics
Thirty-three cases were enrolled in the study. Thirteen strains (13/33, 39.4%) were defined as hvKp. The mean age of patients was 65.09 ± 20.02 years and 17 (51.5%) were female. Thirty-one cases (93.9%) were identified as hospital-acquired infection (HAI) and two (6.1%) with community-acquired infection, triggered by hvKp. The main infection type was pneumonia (n = 26, 78.8%), followed by urinary tract infection (n = 6, 18.2%) and bacteremia (n = 1, 3.0%). Twenty-four patients (24/33, 72.7%) presented with severe infection (sepsis or septic shock) and seventeen patients (17/33, 51.5%) were immunosuppressed. All patients had been exposed to antibiotics and three (9.1%) were not intubated. For 22 (66.7%) patients, APACHE II scores were ≥16, while 23 (69.7%) had SOFA scores ≥6. Twenty-five (75.8%) patients had Charlson Comorbidity Index scores ≥ 3. Twelve patients (36.4%) died within 30 days and life-sustaining therapy was withheld for 3 patients (9.1%) (Table 1).
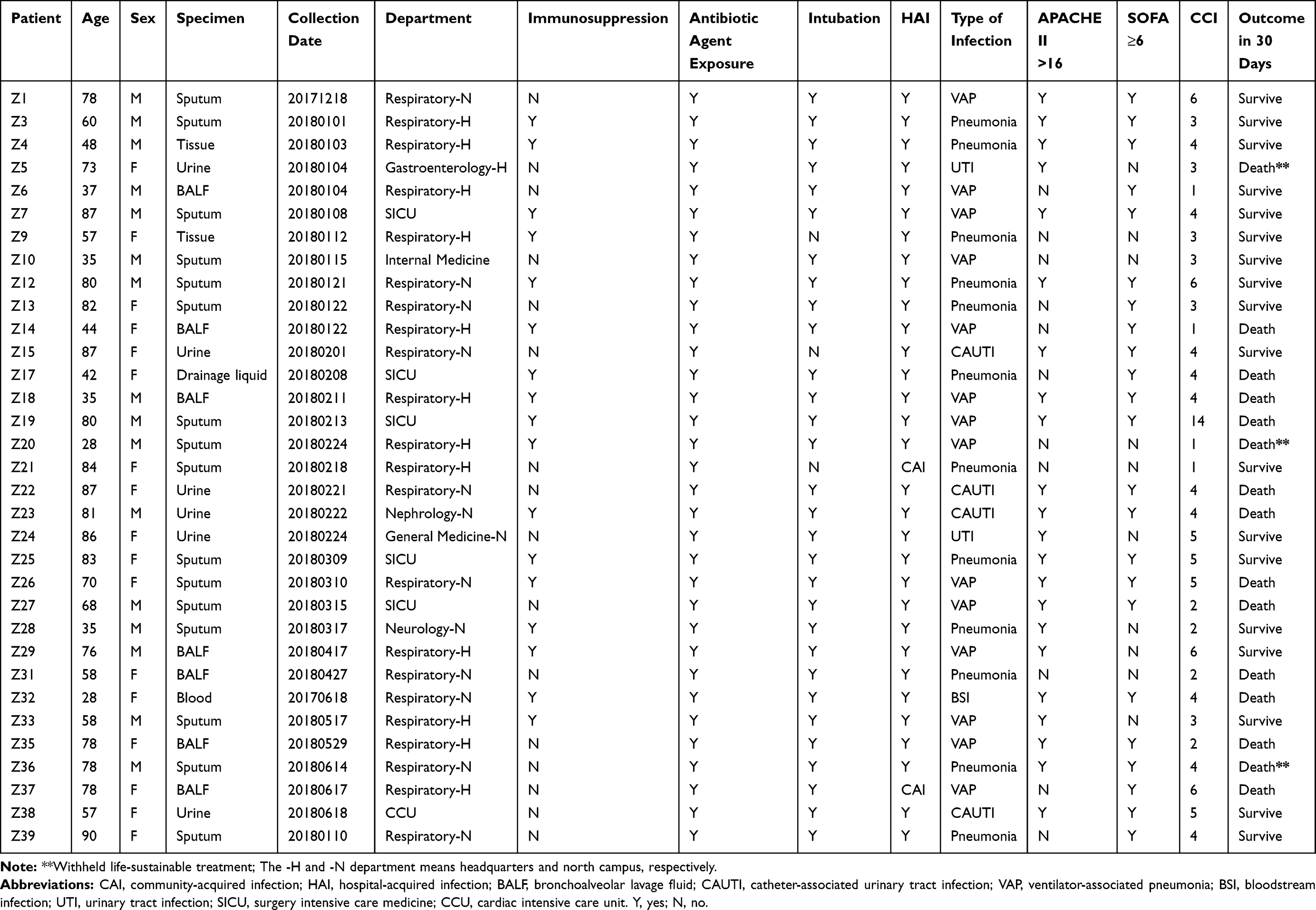 |
Table 1 Clinical Characteristics of Patients with Klebsiella pneumonia Infection |
Phylogenetic Relationships and Distribution of the Isolates
In total, 1313 hqSNPs were identified and used to perform phylogenetic analysis (Supplementary Table 2). Although all strains were sequence type 11 (ST11), they clustered into two separate clades (Figures 1 and 2). The separation into two clades was supported by 749 hqSNPs, including 372 unique to clade 1 and 377 unique to clade 2. Clade 1 comprised six isolates distributed among the International Medical Service (n = 1), Respiratory (n = 4), and Neurology (n = 1) departments. Clade 2 contained two clusters: clade 2a and clade 2b, comprising 11 and 16 isolates, respectively. In clade 2, the majority of strains were from the Respiratory department (Respiratory-H and Respiratory-N) (16/27) and SICU (5/27).
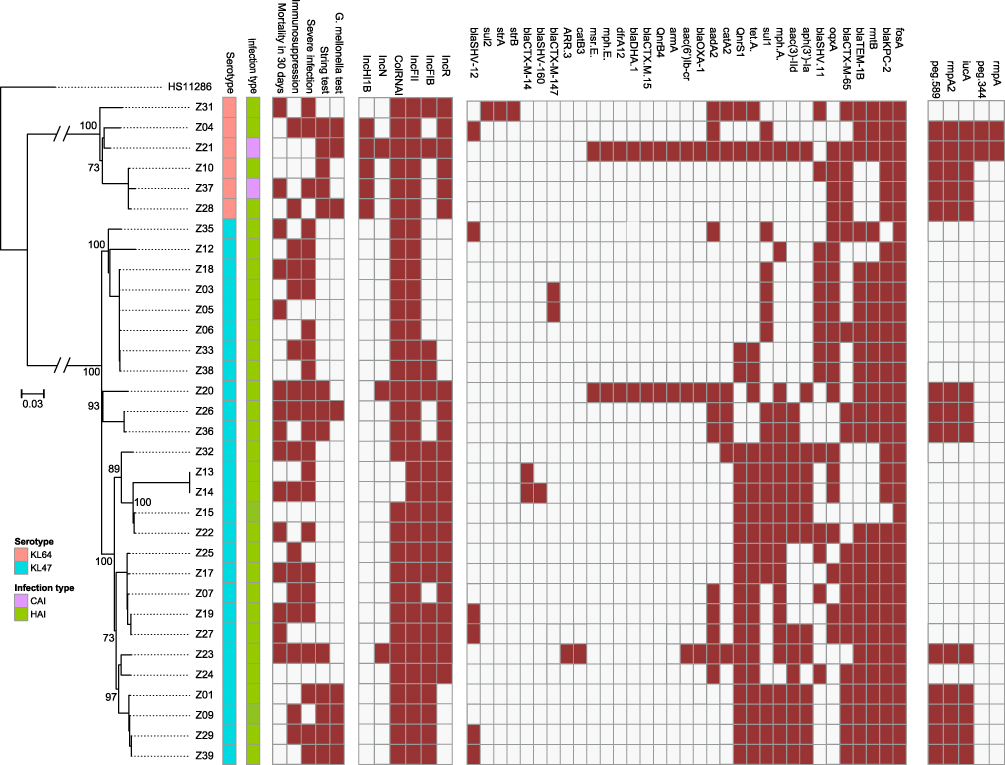 |
Figure 1 Phylogenetic tree of MDR-Kp strains. Brown: positive. Green: hospital-acquired infection. Purple: community-acquired infection. Blue: KL47. Pink: KL64. Hypervirulent phenotype: G mellonella mortality is not lower than the reference strain (K1-hvKp) at 48 h. |
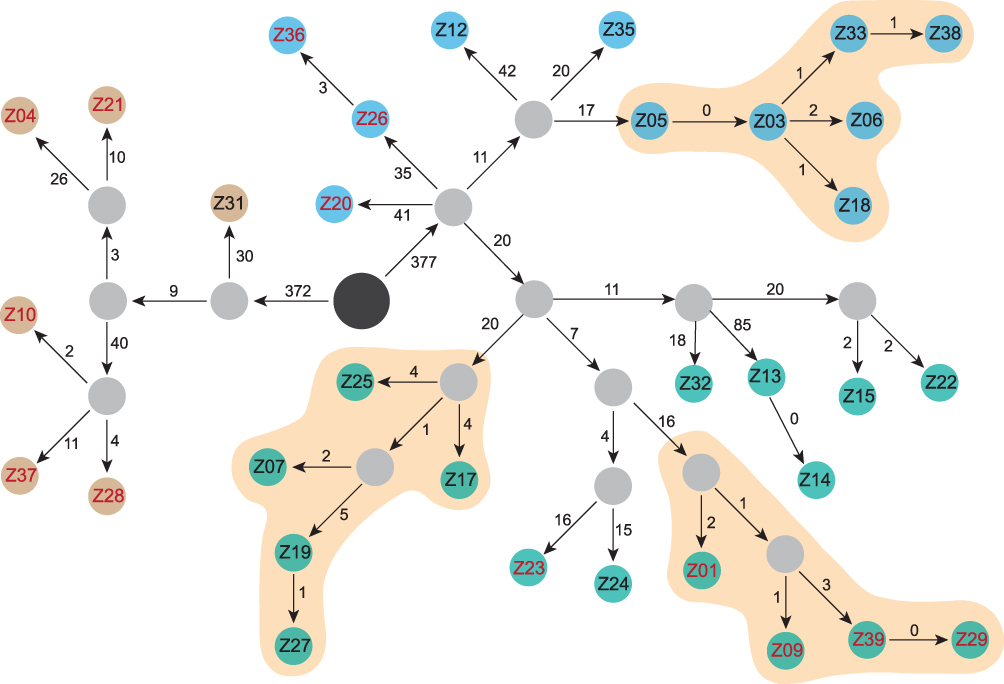 |
Figure 2 Evolutionary and transmission map of isolated Kp strains. Brown: clade 1 strains. Green: clade 2a strains. Blue: clade 2b strains. Black circle, ancestral strain determined by SNP analysis. Gray, strains during transmission not captured in this study. Red numbers, hvKp defined by the combination of five key virulence genes. The numbers on the arrows are SNPs. |
Antimicrobial Resistance and Virulence Profiles
All strains exhibited multidrug resistant phenotypes (Supplementary Table 3), with diverse drug resistance mechanisms detected (Figure 1). Of 33 strains, 31 carried blaKPC-2, as well as resistance to carbapenem. RmtB and blaTEM were co-detected in 25 strains, while 21 strains carried tet(A). Surprisingly, blaOXA-1 and blaDHA-1 were co-identified in two strains and diverse blaCTX-M genotypes were present among these strains. Notably, although there was no evidence of pre-exposure to fosfomycin, all strains carried fosA.
Further, 13 strains were hypermucoviscous and all strains carried the yersiniabactin gene (Supplementary Table 4). Thirteen strains possessed iucA, and prmpA2 was also detected in 13 strains, two of which were also positive for prmpA and peg-344, whereas none harbored iroB. Peg-589, a factor strongly associated with poor prognosis,16 was also detected in the 13 prmpA2-positive strains; therefore, these strains were defined as hvKp, due to the presence of five virulence genes, and two types of virulence profile were identified. Eleven cases were associated with prmpA2, iucA, and peg-589, while the other two strains carried peg-344+peg-589+prmpA+prmpA2+iucA. No isolate possessed iroB+peg-344+iucA+prmpA+prmpA2; however, surprisingly, 61.5% (8/13) hvKp strains exhibited a hypervirulent phenotype in the G. mellonella model (Supplementary Figure 1). Common serotypes (ie, K1, K2, K5, K20, and K54) were not detected among these strains; clade 1 strains were all serotype KL64 and those in clade 2 were all KL47. Additionally, no strain exhibited serum resistance phenotype.
All strains contained the IncFII plasmid replicon and 93.9% (31/33) carried the ColRNAI plasmid replicon. Further, 60.6% (20/33) and 63.6% (21/33) were positive for the IncFIB and IncR plasmid replicons, respectively. The aforementioned were the common replicon types in our strains. Five (15.2%,5/33) strains harbored IncHI1B and three isolates contained IncN (Figure 1). More than three types of replicon were present in 27/33 (81.8%) isolates, indicating that they carried multiple plasmids. Interestingly, five of six strains in clade 1 carried the IncHI1B plasmid replicon. In contrast, all hvKp strains in clade 2 harbored IncFIB, but not the IncHI1B plasmid replicon, suggesting that a type of plasmid, or mobile genetic element, harboring virulence gene clusters may be present.
Virulence Shift During Evolution and Transmission
Based on the combined SNP and epidemiological information, we determined potential transmission routes (Figure 2). For clade 1, based on SNP variants, we estimate that the hvKp ancestor may have separated from a common ancestor with Z31, and then evolved into two clones, which further evolved into Z04 and Z21. The other clone evolved into Z10, Z28, and Z37. Notably, hvKp strains in clade 1 acquired different virulence gene clusters; however, interestingly, only the Z04, Z21, and Z28 strains exhibited hypervirulence in the G. mellonella model. Moreover, patients infected with the other clade 1 strains (Z31 and Z37) died within 30 days. Notably, phenotypic virulence heterogeneity was detected in the G. mellonella model, suggesting that it may be possible to omit the use of G. mellonella for identification of the virulence phenotype. Fortunately, the two clones were present in different wards and did not result in an outbreak.
The common ancestor of clade 2a evolved into three different clones, two of which did not acquire virulence genes or present a virulence phenotype. Combined with clinical information, these data indicate that the two clones were spread in the SICU and Respiratory-N departments, respectively. Interestingly, two SICU doctors were responsible for medical care of five patients. Overall, our data confirm that a genuine hypervirulent clone is emerging and has spread in the Respiratory department of our hospital. Among the hypervirulent clone cluster, five of six strains acquired iucA+rmpA2+peg-589 and most of them exhibited hypervirulence in G. mellonella. Of patients infected with the hypervirulent clone, 80% (4/5) suffered from septic shock. Our data, which indicate that Kp strains combining hypervirulent and MDR profiles are emerging, are alarming. Worse, patients infected with the Z21 and Z37 strains were defined as community-acquired infection, due to the presence of MDR-hvKp, representing a warning that the nosocomial MDR-hvKp strain could have spread into the community.
Discussion
In this study, we mapped inter-host evolution and transmission of ST11, which has acquired unique hvKp virulence genes, including iucA and rmpA2 (but not iroB, rmpA, or peg-344), conferring a heterogeneous hypervirulent phenotype. Specifically, these clones have spread within the hospital for years and may have transmitted to the community. In addition, we detected the interesting phenomenon that key virulence genes (iucA and rmpA2) that can frequently be located in the unique plasmid, pVir-CR-hvkp4 or pLVPK, emerged in different clones from clade 1 and clade 2a clusters, as well as a rare, sporadic strain in clade 2b. Moreover, strains in clade 2 carried the IncFIB, but not the IncHI1B, plasmid replicon, suggesting that the conjugative plasmid, or other mobile elements associated with virulence-encoding genes, may emerge within the hospital.
Multidrug-resistant Kp strains have become prominent in the last decade, particularly in China. In a Chinese survey of carbapenem-resistant Enterobacteriaceae, conducted from 2012 to 2016 at multiple centers, 66.7% were CR-Kp.6 The rapid rise in the rates of MDR strains has been attributed to the dominant emergence and expansion of a specific clone of Kp, referred to as ST11.6,8 In China, ST11 Kp is the predominant strain of cKp, and exhibits reduced susceptibility to most available antibiotics, mainly due to the insertion of KPC-2.6,23 Recently, increasing numbers of outbreaks caused by ST11-MDR-Kp have been reported.24–26 Importantly, the emergence of ST11 Kp has coincided with its increasing carriage of virulence-associated genes, which are crucial for hospital infections and outbreaks.18 HvKp is most commonly isolated from patients with community-acquired infection, sensitive to various antibiotics. Importantly, hvKp transmitted within the hospital, where it has been exposed to various antibiotics, could emerge as MDR-hvKp. Our study confirms that MDR-hvKp, the so-called “super bug”, could be transmitted from hospital into the community, warning that ongoing surveillance should focus more closely on the community.
The emergence of MDR-hvKp threatens the viability of the current therapeutic approach,18,27 and clear understanding of its evolution and transmission characteristics is essential. In our study, 53.8% (7/13) of patients infected with hvKp were immunodeficient. Within the immunodeficient patient who hospitalized long term, diverse phenotype and genotype of Kp might emerge and cause persistent infection.22 This phenomenon has been observed in many pathogens. For example, a previous study demonstrated that mutation of the mutS gene in Salmonella resulted in a hypermutator phenotype and diversification during evolution in an immunosuppressive patient.28 Additionally, Pseudomonas aeruginosa during long-term infections evolves their iron acquisition metabolism pathways from the promoter mutations of the phu system.29 The SNP mutations in hvKp strains distributed in different Clades and sporadic strains were detected (Supplementary Figures 2–4); however, no significant mutations were detected in any of the hvKp strains. Enhanced surveillance of immunosuppressed patients may be warranted.
Previous studies of outbreaks have suggested that they are primarily associated with cKp, with few reports demonstrating that outbreaks were triggered by hvKp.18 To date, only ST11 CR-hvKp has been reported as associated with HAI outbreaks. One reason may be the original capsule,17 while another possible explanation is a strong association with plasmid compatibility.30 Interestingly, a fatal outbreak induced by hvKp reported by Gu et al also originated from the same clone of ST11;18 however, in this study, various ST11-MDR-hvKp clones were involved inter-host evolution and transmission and distributed among different clades. It is alarming that hvKp, as a source of hospital infection, is tending toward polymorphism, indicating that an increase in ongoing surveillance is urgently required. Yang et al30 reported a conjugative virulence plasmid that can rapidly enhance dissemination of virulence-encoding elements among Gram-negative bacterial pathogens, particularly Kp. In our study, all hvKp strains in clade 2 carried the IncFIB, but not the IncHI1B, plasmid replicon and harbored the same virulence gene cluster (iucA+prmpA), similar to the pVir-CR-hvkp4 plasmid, suggesting that conjugative virulence-encoding plasmid, or other mobile elements, may be present.
During their evolution and transmission, some ST11 strains have acquired virulence-associated genes, but do not exhibit a genuine virulence phenotype in the G. mellonella model. Previous study reported that carriage of virulence plasmids is not always associated with hypermucoviscosity and hypervirulence phenotypes in Kp.31 Indeed, the hypermucoviscosity phenotype was not lost in this study. On one hand, it may be necessary to reassess the definition of hvKp and the animal model for identifying hvKp in further studies, particularly for strains isolated from China. On the other hand, there might exist a new virulence-associated gene that could confer hypervirulence phenotype, which should be confirmed by further study.2
Outbreaks of various pathogens have been reported, where major reservoirs included water, healthcare workers, endoscopes, and other medical equipment.32–34 Infection control measures are essential for routine clinical practice. A previous study suggested that clinical departments previously infected with MDR-hvKp should be disinfected and left unoccupied for more than 2 weeks before new patients are admitted.18 This may be a good strategy to prevent large outbreaks, particularly in critically ill, mechanically ventilated patients. Regarding the potential emerging conjugative virulence-associated plasmid, our hospital has improved awareness among healthcare staff using online and offline education, strengthened active hand hygiene, encouraged use of disposable gloves, and ensured sterilization of breathing apparatus, among other measures. Although screening of fecal carriage status was implemented previously, the frequency of this procedure should be increased in the ICU department.
The main limitations of our study were that it had a retrospective design and was conducted at a single center. Further, the number of strains detected was small. We strongly believe that further prospective studies involving international collaboration are essential to address these limitations. Additionally, the use of long-read sequencing techniques may assist deep exploration of the plasmid and genomic content of Kp.
Conclusions
In summary, in this study, we observed that various virulence clusters of the epidemic clone, MDR-ST11, converged and subsequently spread as they evolved and were transmitted within the hospital, and possibly to the community. Heterogeneity of phenotypic virulence during inter-host transmission and evolution was also detected using the G. mellonella model. The detection of new types of plasmid, including virulence genes, may indicate that conjugative/mobile genetic elements have emerged and triggered spread of the organism within the hospital. Therefore, ongoing surveillance should be enhanced to determine whether this trend is also occurring elsewhere.
Acknowledgments
We thank the teams of curators at the Institute Pasteur MLST and whole-genome MLST databases for curating the data and making them publicly available at http://bigsdb.pasteur.fr.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Magill SS, O’Leary E, Janelle SJ, et al. Changes in prevalence of health care-associated infections in U.S. hospitals. N Engl J Med. 2018;379(18):1732–1744. doi:10.1056/NEJMoa1801550
2. Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32(3):e00001–19.
3. Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 2013;4(2):107–118. doi:10.4161/viru.22718
4. Catalan-Najera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: two different but complementary Klebsiella spp. phenotypes? Virulence. 2017;8(7):1111–1123. doi:10.1080/21505594.2017.1317412
5. Liu C, Shi J, Guo J. High prevalence of hypervirulent Klebsiella pneumoniae infection in the genetic background of elderly patients in two teaching hospitals in China. Infect Drug Resist. 2018;11:1031–1041. doi:10.2147/IDR.S161075
6. Wang Q, Wang X, Wang J, et al. Phenotypic and genotypic characterization of carbapenem-resistant enterobacteriaceae: data from a longitudinal large-scale CRE study in China (2012–2016). Clin Infect Dis. 2018;67(suppl_2):S196–S205. doi:10.1093/cid/ciy660
7. Toth A, Damjanova I, Puskas E, et al. Emergence of a colistin-resistant KPC-2-producing Klebsiella pneumoniae ST258 clone in Hungary. Eur j Clin Microbiol Infect Dis. 2010;29(7):765–769. doi:10.1007/s10096-010-0921-3
8. Qi Y, Wei Z, Ji S, Du X, Shen P, Yu Y. ST11, the dominant clone of KPC-producing Klebsiella pneumoniae in China. J Antimicrob Chemother. 2011;66(2):307–312. doi:10.1093/jac/dkq431
9. Li W, Sun G, Yu Y, et al. Increasing occurrence of antimicrobial-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae isolates in China. Clin Infect Dis. 2014;58(2):225–232. doi:10.1093/cid/cit675
10. Siu LK, Yeh KM, Lin JC, Fung CP, Chang FY. Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect Dis. 2012;12(11):881–887. doi:10.1016/S1473-3099(12)70205-0
11. Pomakova DK, Hsiao CB, Beanan JM, et al. Clinical and phenotypic differences between classic and hypervirulent Klebsiella pneumonia: an emerging and under-recognized pathogenic variant. Eur j Clin Microbiol Infect Dis. 2012;31(6):981–989. doi:10.1007/s10096-011-1396-6
12. Liu C, Guo J. Characteristics of ventilator-associated pneumonia due to hypervirulent Klebsiella pneumoniae genotype in genetic background for the elderly in two tertiary hospitals in China. Antimicrob Resist Infect Control. 2018;7:95. doi:10.1186/s13756-018-0371-8
13. Zhang Y, Zhao C, Wang Q, et al. High prevalence of hypervirulent Klebsiella pneumoniae infection in China: geographic distribution, clinical characteristics, and antimicrobial resistance. Antimicrob Agents Chemother. 2016;60(10):6115–6120. doi:10.1128/AAC.01127-16
14. Lin YC, Lu MC, Tang HL, et al. Assessment of hypermucoviscosity as a virulence factor for experimental Klebsiella pneumoniae infections: comparative virulence analysis with hypermucoviscosity-negative strain. BMC Microbiol. 2011;11:50. doi:10.1186/1471-2180-11-50
15. Russo TA, Olson R, MacDonald U, Beanan J, Davidson BA. Aerobactin, but not yersiniabactin, salmochelin, or enterobactin, enables the growth/survival of hypervirulent (hypermucoviscous) Klebsiella pneumoniae ex vivo and in vivo. Infect Immun. 2015;83(8):3325–3333. doi:10.1128/IAI.00430-15
16. Russo TA, Olson R, Fang CT, et al. Identification of biomarkers for differentiation of hypervirulent Klebsiella pneumoniae from classical K. pneumoniae. J Clin Microbiol. 2018;56(9). doi:10.1128/JCM.00776-18.
17. Wyres KL, Wick RR, Judd LM, et al. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019;15(4):e1008114. doi:10.1371/journal.pgen.1008114
18. Gu D, Dong N, Zheng Z, et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis. 2018;18(1):37–46. doi:10.1016/S1473-3099(17)30489-9
19. Zhang R, Lin D, Chan EW, Gu D, Chen GX, Chen S. Emergence of carbapenem-resistant serotype K1 hypervirulent Klebsiella pneumoniae strains in China. Antimicrob Agents Chemother. 2016;60(1):709–711. doi:10.1128/AAC.02173-15
20. Zhang Y, Zeng J, Liu W, et al. Emergence of a hypervirulent carbapenem-resistant Klebsiella pneumoniae isolate from clinical infections in China. J Infect. 2015;71(5):553–560. doi:10.1016/j.jinf.2015.07.010
21. Gu DX, Huang YL, Ma JH, et al. Detection of colistin resistance gene mcr-1 in hypervirulent Klebsiella pneumoniae and escherichia coli isolates from an infant with diarrhea in China. Antimicrob Agents Chemother. 2016;60(8):5099–5100. doi:10.1128/AAC.00476-16
22. Jousset AB, Bonnin RA, Rosinski-Chupin I, et al. A 4.5-year within-patient evolution of a colistin-resistant Klebsiella pneumoniae carbapenemase-producing K. pneumoniae sequence type 258. Clin Infect Dis. 2018;67(9):1388–1394. doi:10.1093/cid/ciy293
23. Li H, Zhang J, Liu Y, et al. Molecular characteristics of carbapenemase-producing Enterobacteriaceae in China from 2008 to 2011: predominance of KPC-2 enzyme. Diagn Microbiol Infect Dis. 2014;78(1):63–65. doi:10.1016/j.diagmicrobio.2013.10.002
24. Baraniak A, Izdebski R, Fiett J, et al. NDM-producing Enterobacteriaceae in Poland, 2012–14: inter-regional outbreak of Klebsiella pneumoniae ST11 and sporadic cases. J Antimicrob Chemother. 2016;71(1):85–91. doi:10.1093/jac/dkv282
25. Voulgari E, Zarkotou O, Ranellou K, et al. Outbreak of OXA-48 carbapenemase-producing Klebsiella pneumoniae in Greece involving an ST11 clone. J Antimicrob Chemother. 2013;68(1):84–88. doi:10.1093/jac/dks356
26. Yang J, Ye L, Guo L, et al. A nosocomial outbreak of KPC-2-producing Klebsiella pneumoniae in a Chinese hospital: dissemination of ST11 and emergence of ST37, ST392 and ST395. Clinical Microbiol Infect. 2013;19(11):E509–515. doi:10.1111/1469-0691.12275
27. Guo Y, Wang S, Zhan L, et al. Microbiological and clinical characteristics of hypermucoviscous Klebsiella pneumoniae isolates associated with invasive infections in China. Front Cell Infect Microbiol. 2017;7:24. doi:10.3389/fcimb.2017.00024
28. Tanner JR, Kingsley RA. Evolution of salmonella within hosts. Trends Microbiol. 2018;26(12):986–998. doi:10.1016/j.tim.2018.06.001
29. Marvig RL, Damkiaer S, Khademi SM, Markussen TM, Molin S, Jelsbak L. Within-host evolution of Pseudomonas aeruginosa reveals adaptation toward iron acquisition from hemoglobin. mBio. 2014;5(3):e00966–00914. doi:10.1128/mBio.00966-14
30. Yang X, Wai-Chi Chan E, Zhang R, Chen S. A conjugative plasmid that augments virulence in Klebsiella pneumoniae. Nature Microbiol. 2019;4(12):2039–2043. doi:10.1038/s41564-019-0566-7
31. Shu L, Dong N, Lu J, et al. Emergence of OXA-232 carbapenemase-producing Klebsiella pneumoniae that carries a pLVPK-like virulence plasmid among elderly patients in China. Antimicrob Agents Chemother. 2018;63. doi:10.1128/AAC.02246-18.
32. Eyre DW, Sheppard AE, Madder H, et al. A Candida auris outbreak and its control in an intensive care setting. N Engl J Med. 2018;379(14):1322–1331. doi:10.1056/NEJMoa1714373
33. Epstein L, Hunter JC, Arwady MA, et al. New Delhi metallo-beta-lactamase-producing carbapenem-resistant Escherichia coli associated with exposure to duodenoscopes. JAMA. 2014;312(14):1447–1455. doi:10.1001/jama.2014.12720
34. Kizny Gordon AE, Mathers AJ, Cheong EYL, et al. The hospital water environment as a reservoir for carbapenem-resistant organisms causing hospital-acquired infections-a systematic review of the literature. Clin Infect Dis. 2017;64(10):1435–1444. doi:10.1093/cid/cix132
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.