Back to Archived Journals » Journal of Receptor, Ligand and Channel Research » Volume 7
Pharmacophore modeling: advances, limitations, and current utility in drug discovery
Authors Qing X, Lee XY, De Raeymaecker J, Tame J, Zhang K, De Maeyer M, Voet A
Received 21 July 2014
Accepted for publication 14 August 2014
Published 11 November 2014 Volume 2014:7 Pages 81—92
DOI https://doi.org/10.2147/JRLCR.S46843
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Trevor W. Stone
Video abstract presented by Xiaoyu Qing and Xiaoyin Lee
Views: 10035
Xiaoyu Qing,1,* Xiao Yin Lee,2,* Joren De Raeymaeker,1 Jeremy RH Tame,3 Kam YJ Zhang,2 Marc De Maeyer,1 Arnout RD Voet1,2
1Laboratory for Biomolecular Modelling, Department of Chemistry, Katholieke Universiteit Leuven, Heverlee, Belgium; 2Structural Bioinformatics Team, Center for Life Science Technologies, RIKEN, Yokohama, Kanagawa, Japan; 3Drug Design Laboratory, Yokohama City University, Yokohama, Kanagawa, Japan
*These authors contributed equally to this work
Abstract: Pharmacophore modeling is a successful yet very diverse subfield of computer-aided drug design. The concept of the pharmacophore has been widely applied to the rational design of novel drugs. In this paper, we review the computational implementation of this concept and its common usage in the drug discovery process. Pharmacophores can be used to represent and identify molecules on a 2D or 3D level by schematically depicting the key elements of molecular recognition. The most common application of pharmacophores is virtual screening, and different strategies are possible depending on the prior knowledge. However, the pharmacophore concept is also useful for ADME-tox modeling, side effect, and off-target prediction as well as target identification. Furthermore, pharmacophores are often combined with molecular docking simulations to improve virtual screening. We conclude this review by summarizing the new areas where significant progress may be expected through the application of pharmacophore modeling; these include protein–protein interaction inhibitors and protein design.
Keywords: ADME-tox, computer-aided drug design, pharmacophore fingerprint, protein design, virtual screening
What is computer-aided drug design?
Drug design is an expensive and laborious process of developing new medicine. This process has its origin in herbal remedies dating back millennia.1 Only since the last century have drugs had a (semi)synthetic origin.2 The first hit compounds often lack both potency and safety, and must therefore be optimized. While historically this was a trial-and-error process,3,4 soon rational strategies were developed to improve potency.5,6 As with any data handling procedures, computers have become a more prominent and ubiquitous tool in drug discovery since the 1980s.7 The crossover between computational and pharmaceutical research is typically designated computer-aided drug design (CADD).8,9
CADD covers a broad range of applications spanning the drug discovery pipeline, although these are highly clustered in the early phases. The main purpose of CADD is to speed up and rationalize the drug design process while reducing costs.10 The aim of the earliest phase in drug discovery is to identify the first hit compounds, which is sometimes attempted by high-throughput screening (HTS), the testing of many thousands of compounds with a suitable activity assay. The in silico counterpart of in vitro HTS is referred to as virtual screening and aims at filtering libraries of molecules using computational methods to prioritize those most likely to be active for a given target.11 Later in the drug discovery pipeline the potency of the hit and lead compounds needs to be improved.12 New derivatives are designed with or without a different scaffold at the core of the molecule.13 The ultimate goal is to design highly potent and specific molecules which also have a suitable intellectual property position.14 This can be achieved by classical medicinal chemistry approaches, where the design can be based on the observed structure–activity relationships (SAR) or based on structural information.15 Computational methods however can also be used to create diverse derivatives based on different scaffolds,16,17 and then score them for improved potency. This prioritizes the most promising derivatives from a very wide chemical space in a relatively short time.18,19 However, the potency of the compounds is not the only consideration. Pharmacokinetic properties (absorption, distribution, metabolism, excretion) and toxicity, referred to as ADME-tox, are also of vital importance if a compound is to be clinically useful.20–22 As well as a battery of in vitro and in vivo experiments, virtual methods have also been developed to predict the ADME-tox profile of drug-like compounds early during the development process.
The basis of all CADD methods is chemo-informatics, the application of data storage, handling, and retrieval methods to chemical structures, their properties, and biological activity.23,24 Chemo-informatics also covers the calculation of molecular descriptors that describe a chemical or physical property based on the molecules’ structure, and which can be used for filtering compounds.25 In order to be able to compare and quantify (dis)similarity between molecules, molecular fingerprints are often the methods of choice.26
Another very important CADD subfield focuses on quantitative structure activity/property relationship (QSAR/QSPR), in which the physicochemical properties (as calculated by molecular descriptors) of a set of inhibitors are related to the inhibitory activity or toxicity to construct a predictive model for novel inhibitors.27–29 QSAR has become a very popular tool to profile novel inhibitors accurately in silico without going through expensive and time-consuming in vitro and in vivo assays.30
Probably the best known and most used CADD method is molecular docking simulations, whereby the 3D binding mode of a given ligand for a given biomolecular receptor (typically a protein structure) is predicted and scored for affinity. This is extremely useful for the structural analysis of protein–ligand interactions where experimental structural information is absent.31 Docking, however, has also become a very popular tool to screen for hit compounds virtually, or by reverse engineering to identify the target.32–35
The CADD methods briefly introduced earlier are some of the most widely known, but many more exist including artificial-intelligence-based methods.36,37 The topic of this review, however, is another very successful CADD method known as pharmacophore modeling.38–42 This review is aimed at medicinal chemists and others new to CADD and covers the history, progress, and current limitations of pharmacophore modeling. We do not list or compare the many different pharmacophore modeling programs or algorithms.
What is a pharmacophore?
Historical perspective
The original concept of the pharmacophore was developed by Paul Ehrlich during the late 1800s.43 At that time, the understanding was that certain “chemical groups” or functions in a molecule were responsible for a biological effect, and molecules with similar effect had similar functions in common. The word pharmacophore was coined much later, by Schueler in his 1960 book Chemobiodynamics and Drug Design, and was defined as “a molecular framework that carries (phoros) the essential features responsible for a drug’s (pharmacon) biological activity.”44 The definition of a pharmacophore was therefore no longer concerned with “chemical groups” but “patterns of abstract features.”
Since 1997, a pharmacophore has been defined by the International Union of Pure and Applied Chemistry as:
A pharmacophore is the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response.45
The pharmacophore should be considered as the largest common denominator of the molecular interaction features shared by a set of active molecules. Thus a pharmacophore does not represent a real molecule or a set of chemical groups, but is an abstract concept. Despite this clear definition, the term pharmacophore is often misused by many in medicinal chemistry to describe simple yet essential chemical functionalities in a molecule (such as guanidine or sulfonamides), or common chemical scaffolds (such as flavones or prostaglandins). Often the long definition is simplified to “A pharmacophore is the pattern of features of a molecule that is responsible for a biological effect,” which captures the essential notion that a pharmacophore is built from features rather than defined chemical groups.
Pharmacophore concepts in CADD
While the pharmacophore concept predates any form of electronic computer, it has nevertheless become an important tool in CADD. Every type of atom or group in a molecule that exhibits certain properties related to molecular recognition can be reduced to a pharmacophore feature. These molecular patterns can be labeled as hydrogen bond donors or acceptors, cationic, anionic, aromatic, or hydrophobic, and any possible combinations.46 Different molecules can be compared at the pharmacophore level; this usage is often described as “pharmacophore fingerprints.” When only a few pharmacophore features are considered in a 3D model the pharmacophore is sometimes described as a “query.”
Pharmacophore fingerprint
While molecules are 3D entities, the pharmacophore representation reduces a molecule to a collection of features at the 2D or 3D level.47,48 A pharmacophore fingerprint is an extension of this concept, and typically annotates a molecule as a unique data string. All possible three-point or four-point sets of pharmacophore features (points) are enumerated for each ligand.49 The distance between the feature points is counted in bonds (for topological fingerprints), or by distance-binning when using 3D fingerprints (Figure 1). The resulting fingerprint is a string describing the frequency of every possible combination at predefined positions within the string. Several variants of pharmacophore fingerprints have been designed and are frequently used.
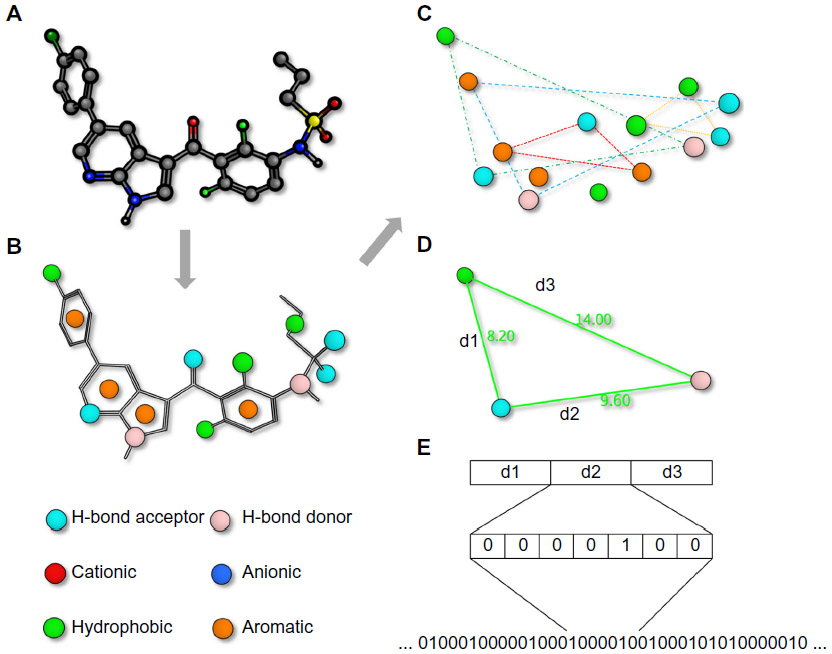 | Figure 1 Pharmacophore fingerprints. |
Such a fingerprint can be used to analyze the similarity between molecules or among a library of molecules. Alternatively, a fingerprint model can be used to analyze the common elements of active ligands to identify the key contributing features to the biological function.
Pharmacophore model or query
A pharmacophore model consists of a few features organized in a specific 3D pattern.50 Each feature is typically represented as a sphere (although variants exist) with a radius determining the tolerance on the deviation from the exact position (Figure 2). The features can be labeled as a single feature or any logic combination consisting of “AND,” “OR,” and “NOT” to combine different interaction patterns within one label. Additional features can describe forbidden volume interactions (typically to represent the receptor boundary).
 | Figure 2 Pharmacophore query. |
Such pharmacophore features are typically used as queries to screen small molecule libraries of compounds.51 In these libraries all the compounds are present in their low-energy biorelevant conformations. Each of these conformations is fitted to the pharmacophore query by aligning the pharmacophore features of the molecule and the query is composed. If a molecule can be fitted inside the spheres representing the query features it is considered a hit molecule. Often the pharmacophore query can be too complex to find hit molecules from a given library, and partial matching may be allowed. In such cases only certain features considered essential for activity are matched. Additional uses of such models are to align molecules or facilitate molecular docking simulations.52–54
Depending on the situation and the type of experiment, multiple strategies are available to construct pharmacophore models, either manually or using automated algorithms, and this is discussed in the next chapter.
Pharmacophore modeling in virtual screening
Pharmacophore modeling is most often applied to virtual screening in order to identify molecules triggering the desired biological effect. For this purpose, researchers create a pharmacophore model (query) that most likely encodes the correct 3D organization of the required interaction pattern. Depending on how much is known about the particular protein target, different options are available to construct such a query (Figure 3).
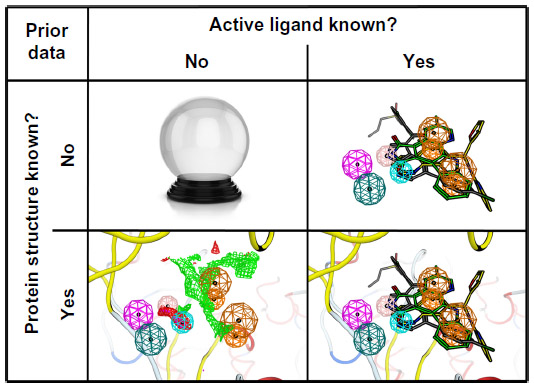 | Figure 3 Four different situations for the pharmacophore search. |
In general, it is good practice to divide the ligand data into two sets, a training and an evaluation set to validate the generated pharmacophore query, when multiple active ligands (and inactive derivatives) are known.55
While in all these cases pharmacophore queries are considered positive filters to identify compounds, they may in fact also be used as negative filters to avoid side effects as well.56,57
No protein structure and no ligand structure is known
If the target structure and all its ligands are unknown, pharmacophore modeling is impossible. The only option to employ the pharmacophore principle would be to design a diverse library employing a diversity metric based on pharmacophore fingerprints to ensure optimal diversity of the library, containing a wide variety of molecules with different pharmacophore feature composition. Indeed, considering the large number of available and potential compounds, the trend is to design libraries very carefully in order to cover chemical space efficiently in any search process.47,48,58,59
No protein structure, but active ligand structures are known
The other scenario is that the structure of the receptor (and any complex with the ligand) is unknown. This is frequently the case in drug discovery. If only a single active molecule is known, then it is impossible to map the key contributing pharmacophore features onto the molecule, and the only option may be to use similarity searches (such as using pharmacophore fingerprints) to retrieve similar molecules.60 Once these have been tested, a set of multiple active and inactive compounds may be known and more advanced pharmacophore modeling can be utilized.
When a set of active ligands of known structure, with similar or different scaffolds, is available, then it is possible to use ligand-based pharmacophore modeling.52,61 The elucidation of the putative pharmacophore involves two steps. First, the conformational space of the flexible molecules needs to be covered extensively since the bioactive conformations are unknown. Second, the molecules need to be aligned by common pharmacophore features, which can be retained in a 3D model. Using inactive derivatives, the essence of the features as well as the permitted steric arrangement of the ligands can be mapped as well. The Catalyst-HypoGen algorithm in particular stands out from the variety of tools available for this purpose.62 This is a combination of QSAR and the pharmacophore method. It attempts to correlate structure and activity values (Ki or half maximal inhibitory concentration [IC50]) by constructing a pharmacophore model. Thus, HypoGen not only identifies a query compound as “active” or “inactive” in the traditional function of a pharmacophore model, but also predicts activity value based on regression of the training dataset.
Protein and ligand structures are known
In the third case, structural information is present for both ligands and the receptor protein. Usually a pharmacophore model represents the key features of a small molecule that allow it to bind to some receptor molecule, but this idea can be reversed and pharmacophore queries built from features of a protein active site.63 These features describe the principle interactions between the protein and its ligands, and can be mapped onto the bioactive conformation of the ligand. Ideally the structural model is derived from crystallographic or nuclear magnetic resonance data, but homology models or other structural data can be used as well. Although a structure for one ligand may be enough, it is beneficial to have 3D information for multiple ligands to identify the common interactions. While this approach is compatible with the majority of pharmacophore modeling methods, LigandScout is notable as the first software package able to construct automatically a query from one or more Protein Data Bank (PDB) files based on protein–ligand interactions.64
Such structure-based pharmacophore queries have multiple applications. They can be used for virtual screening, ligand binding pose prediction, and comparison of binding sites.65
Only the protein structure is known
In the last case, structural information for the protein receptor, but no active ligands, is known. In this case, a putative pharmacophore model can be constructed by analyzing the chemical properties of the binding site of interest. There are several different computational approaches that can directly convert 3D atomic structures of protein binding sites into queries. The interaction maps of the de novo drug design tool LUDI can be used to create a pharmacophore query.66 HS-Pharm is a knowledge-based method that uses machine-learning algorithms to prioritize the most interesting interacting atoms and to generate an interaction map within the binding site.67 Subsequently, the interaction map is converted into pharmacophore features. The GRID package is another approach to analyze the pocket in order to identify the key interactions.68 Using molecular interaction fields, the most favorable positions of atomic probes in the binding site can be identified and converted into pharmacophore features.69 Although many successes have been reported, the absence of any ligand structural information is a distinct disadvantage to drug design, since in the absence of a molecular scaffold it is hard to map the features in 3D space which can still be covered by atoms that are restrained by bond lengths and angles in the ligands.
In all of these examples, pharmacophore queries are utilized to identify active molecules that fulfill certain geometric and chemical restraints. Because of the simple yet versatile character of a pharmacophore query, it can be used not only to identify active molecules, as suggested from the IUPAC (International Union of Pure and Applied Chemistry) definition of a pharmacophore, it can in fact also be used as a negative query, in order to identify molecules with undesirable properties.
In recent work by Voet et al, for example, a double pharmacophore query was utilized to identify strict human androgen receptor (hAR) antagonists.70 Prostate cancer therapy often relies on anti-androgens that antagonize the hAR function.71 However, resistant mutations tend to appear in the hAR so that the antagonists become agonists. In their work, the available structural information of the hAR with compounds in the agonist and the antagonist conformations were used. A pharmacophore query was generated based on the known antagonists in agonistic conformation, and remapped in 3D onto a second query in the antagonistic conformation. Following a combined pharmacophore screening method, compounds were identified that only fulfilled the antagonist query, but not the agonistic query. Experimental evaluation of the compounds confirmed the strictly antagonistic activity of the compounds toward both wild-type hAR and drug-resistant mutants.
Pharmacophore methods in docking simulations
As indicated in the previous section, pharmacophore models are very suitable as queries for virtual screening of databases. Nevertheless, one of the more common approaches in virtual screening is a so-called hierarchical approach in which different methods are combined consecutively. This is also known as the funnel principle, where at each consecutive step the compounds most unlikely to be active are removed, leaving the most promising compounds for virtual screening.72 Typically, every step of the hierarchical approach consists of a more complex, computationally demanding step than the previous one. As such, pharmacophore models are often utilized as a filter to identify compounds that fulfill simple geometric and chemical functionality requirements of the query, prior to more complicated and computationally demanding approaches such as molecular docking.
Molecular docking simulations are computational methods that aim to predict the binding mode of a compound for a given receptor as well as the quality of the interaction, often by attempting to predict the affinity (free energy of binding) using a scoring function.31 Often molecular docking simulations are used to screen large datasets of compounds for a given target, and compounds are ranked according to their predicted affinity. Due to the high number and diversity of the screening compounds, as well as the knowledge that most of the screened compounds are in fact probably inactive, the top scoring compounds are most likely inactive and better compounds are ranked below them. Although this ranking can still be better than random, typically only a few compounds are selected from those scoring best, and many of them often turn out to be inactive.73,74
Several options are available for combining docking-based virtual screening with pharmacophore-based virtual screening:
- The database of ligands can be pre-filtered using a pharmacophore query, prior to evaluation using docking simulations.72
- The docking simulations can be post-filtered using a pharmacophore query to remove any compounds that fail to bind according to the pharmacophore query. The method can also discard compounds that would have scored well in a pure pharmacophore search, but that fail to bind according to some hypothesis taking more information into account, such as incompatibility of the overall ligand structure with the receptor site. In such a case, the ligands are evaluated in absolute conformation and should not be allowed to align with the pharmacophore features.75
- Another alternative is to use the pharmacophore alignment to guide the placement during the docking simulations.76,77 The pharmacophore model can in this case be used for the placement of the ligand, similarly to the fitting of a molecule into the pharmacophore query; or to guide the placement by using a constraint while scoring the different docking poses. The pharmacophore query could originate from a user-defined query or an automatically generated receptor-based pharmacophore query.53
Pharmacophore models are very useful for enriching the top scoring docking results with active compounds. This was demonstrated in the recent SAMPL4 virtual screening challenge where competitors were asked to rank a set of compounds for a given target, HIV-1 Integrase, without any knowledge of activity of the compounds in the library.78 The top results were obtained for the group using a hierarchical method consisting of pharmacophore pre-filtering as well as pharmacophore post-filtering of the docking results.60
Applications of pharmacophores in ADME-tox
Poor ADME-tox is a major contributing factor to failures during drug development and clinical trials.79,80 It is, therefore, widely accepted that the ADME-tox properties should be profiled early during the drug discovery process, and pharmacophore modeling approaches are often used for such ADME-tox predictions.81 The pharmacophore models can be used to identify possible interactions of drugs with drug-metabolizing enzymes by matching the equivalent chemical groups of test molecules to those of drug molecules with a well-known ADME-tox profile.82
The enzymes of major importance for observed ADME-tox profile are the cytochrome P450s (CYP) that initiate drug breakdown. It has been estimated that only six CYP isoenzymes (1A2, 2C9, 2C19, 2D6, 2E1, and 3A4) are responsible for over 90% of drug metabolism.83 Based on the observed interactions of known drugs with the CYP enzymes, receptor-based pharmacophore models have been generated that are able to predict the binding of a drug-like compound to a certain CYP and assess the possibility of degradation by this enzyme.84–87
Similarly, ADME-tox pharmacophore models have been generated for the uridine 5′-diphospho-glucuronosyltransferases, which are enzymes related to drug clearance, and transporters such as P-glycoprotein and organic cation transporter.88,89
Pharmacophore-guided drug target identification
While typically the aim of CADD is to identify and optimize drug-like molecules for a given target, the opposite situation also exists. Often drug molecules are known, but the mechanism of action is unclear. These compounds are often derived from herbal medicine, or phenotypically developed drugs. In such cases, CADD may help identify the target. Chemoinformatical fingerprint-based similarity tools are employed to identify close analogue compounds with a known mechanism of action.90,91 Nevertheless, pharmacophore modeling may also be an option, rather than screening compounds with a pharmacophore query. The molecule itself may become the query and the aim is to identify the most likely pharmacophore model that fits the molecule. Such collections of pharmacophore models may be constructed manually or automatically generated from the PDB database.64 Similarly, this approach may also be used to fish for a target for a given compound with a yet unknown activity.
One example of such an approach was reported by Rollinger et al.92 Using LigandScout, several plant metabolites were investigated and multiple potential drug targets were identified for these compounds. Experimental testing of the compounds for the given targets validated the applicability of this method. It may be expected that pharmacophore models will play a significant role in the future, as polypharmacology or drug repositioning become more widespread.56,91,93 Alternatively, this approach may also help to predict possible side effects or off-targets that can be taken into account to design more specific compounds.57
Limitations of pharmacophore methods
Despite the abundance of successful cases of drug design relying on pharmacophore modeling, as with any method, it is not failsafe and one should be cautious about the limitations of this technique.94
The major limitation in virtual screening by pharmacophore is the absence of good scoring metrics. Whereas docking simulations are based on scoring functions trying to predict the affinity, and similarity searches utilize similarity metrics such as the Tanimoto score, pharmacophore queries do not have a reliable, general scoring metric. Most commonly, the quality of fitting the ligand into a pharmacophore query is expressed by the root mean square deviation between the features of the query and atoms of the molecule.55 This metric, however, is unable to take any similarity into account with known inhibitors, and also is unable to predict the overall compatibility with the receptor protein, and thus molecules that hit a pharmacophore query may be very different from other inhibitors and have functional groups which are not complementary with the receptor binding site, rendering them inactive despite being a perfect match.
A second limitation is the dependency of a pharmacophore-based virtual screen on a pre-computed conformation database. These databases only contain a limited number of low-energy conformations per molecule.95,96 It may be possible that an active molecule cannot be identified as the conformation is missing. This is especially the case for the many different conformations of rotatable bonds of small molecular functionalities such as hydroxyl groups. Different rotations would be very hard to be distinguished during the conformation generation in terms of root mean square deviation differences, and thus may not be thoroughly sampled. Often pharmacophore search tools are able to rotate such bonds during the fitting process to obtain conformations with correct directions on the small flexible polar groups.
Finally, a major limitation is that there is no one clear way to construct a pharmacophore query. In many cases, pharmacophore models are able to retrieve molecules, but different models may have worked. One example is the case of Christ et al versus De Luca et al, where for a similar target, a similar yet slightly different pharmacophore was created.97,98 While screens were performed on a similar dataset, very different molecules were identified. Although this is just one example, it is very likely that there are many more. This is also clear from the analysis of a wide variety of kinase inhibitors. In many cases, kinase inhibitors are very similar to each other and yet have very different activity profiles for the kinome. Pharmacophore approaches aimed at identifying kinase inhibitors would without any doubt identify kinase-inhibitor-like molecules; nevertheless, there would not be a clear guarantee that these molecules would be active for the targeted kinase.99
In conclusion, plenty of experience and a certain dose of serendipity may be required for successful results. The influence of expert knowledge for in silico screening, also known as the in cerebro step, has been demonstrated during the virtual screening SAMPL4 challenge.60
While target identification, prediction of side effects, and ADME-tox profiling appear to be promising applications for pharmacophore modeling, success is limited for new molecule classes as information is lacking for such compounds or targets.100
Future perspectives on pharmacophore modeling
Pharmacophore modeling has been around since the beginning of CADD and has evolved from a basic concept into a well-established CADD method with applications including similarity metrics, virtual screening, ligand optimization, scaffold hopping, target identification, and so on. Given the simplicity and versatility of the pharmacophore concept, it can be anticipated that further developments will be made in the future for different applications.
Fragment-based drug design
Over the last two decades, fragment-based drug design has become a well-established method for the rational development of novel drugs.101 Rather than screening drug-like molecules (with molecular weights of around 500 Da), smaller molecules with a molecular weight up to 350 Da (referred to as fragments) are being screened for affinity with a receptor using highly sensitive biophysical methods. Fragments showing some affinity for the target are grown into bigger and more potent compounds, and fragments binding to adjacent areas can be linked as well.
Since the diversity of small molecule fragments can easily be sampled with a few hundred compounds, in silico screening methods are highly suitable for fragment-based design. CADD methods such as docking and pharmacophore modeling have therefore also been used to identify fragment-like compounds in silico prior to testing in vitro; subsequent fragment recombination can be used for the de novo design of inhibitors.66,102,103
In a first approach, the starting point is a single pharmacophore query that spans two (or more) sub-pockets in the receptor binding site. An additional pharmacophore feature is added that does not represent a molecular recognition feature, but represents an atom in the fragments, where the two fragments of the different pockets may overlap and will be linked to each other.
Then fragments are identified that fulfill the features present in a sub-pocket of the pharmacophore query, as well as on the linking feature. Then the compatibility of the fragment hits for the respective sub-pockets is evaluated in terms of possibility to maintain the correct conformation after linking the two fragments. Subsequently the de novo designed compounds can be synthesized and evaluated.
In the following example, using a different yet similar strategy, Cavalluzzo et al designed a novel small molecule inhibitor binding to the LEDGF/p75 protein, based on an inhibitory peptide.104 They used predefined amino acid side chain fragments taken from the inhibitory peptide, and constructed a pharmacophore query to link the two predefined fragments with a third scaffold fragment that mimicked similar interactions as the peptide. All possible compounds were enumerated virtually, and for the compounds that were able to adopt a conformation similar to the pharmacophore query after linking all fragments, the chemical synthesizability was assessed. Following synthesis, the inhibitory potency of the compound was found to be 30 μM IC50 compared to 7.4 μM IC50 for the most potent inhibitory peptide.105
Even when active fragments have been identified using the classical in vitro methods, computational pharmacophore methods can be applied to identify novel derivatives. For example, pharmacophore fingerprint-based similarity searching and the generation of 3D pharmacophore queries are suitable means to identify bigger and more potent molecules from small molecule libraries.
Protein–protein interaction (PPI) inhibition
Although once thought to be undruggable, “high-hanging fruits on the drug discovery tree,” PPIs have drawn a great deal of attention in recent years.106 The undruggable image has disappeared and an increasing number of small molecule inhibitors of PPIs (SMPPII) have been reported. Most of the early inhibitors originate from HTS.107 Structural analysis of proteins in PPI complexes and inhibitor complexes show that the interactions at the PPI interface are being mimicked by the ligand.108 SMPPII are found to copy the natural interaction not only in terms of shape and chemistry, but even at the electrostatic potential level.109 This mimicry suggests that the pharmacophore queries created from PPI complex structures can be used to identify SMPPII via virtual screening.110 Different methods can be employed to map the pharmacophore features onto the amino acids present at the PPI interface.111 Several SMPPII discoveries have been achieved, thanks to pharmacophore searches using manually created search features,112,113 or a consensus of interactions at the PPI interface,114,115 or using automated methods,116 or by identification of the key interactions using molecular interaction field analysis.69
PPIs are especially promising targets for controlling inappropriate signaling, as found in diseases such as cancer. The usefulness of pharmacophore modeling to create queries encoding the key interactions at the PPI interface will probably strongly stimulate the discovery of novel SMPPII using pharmacophores, both as a stand-alone virtual screening tool and incorporated into pipelines with other methods.
A potential role in protein design?
Although pharmacophore modeling originated as a drug design concept and, as indicated earlier, is nowadays a key element of CADD, pharmacophore modeling shows promise in the currently burgeoning field of computational protein design.117 Rather than designing drugs for a given protein target, the aim in computational protein design is to derive an amino acid sequence that will fold into a given structure with a desired function. In many cases, this may involve protein–small molecule ligand interactions,118 and for these it can easily be imagined that pharmacophores may be used simply by reversing the process of small molecule drug design for a known protein structure.
First of all, suitable protein templates (enzymes or otherwise) should be identified for the protein redesign process. The ligand of interest could serve as a query to try to identify possible binding proteins, which can then later be redesigned to give optimum complementarity to the ligand. Second, during the virtual protein design process, often multiple rotamers of different amino acids are sampled to identify the most desirable ones.119 Similar to ligand fitting with a pharmacophore query, the protein side chains can be fitted to features describing the complementary interactions required at the protein–ligand interface.
Conclusion
The pharmacophore concept was first put forward as a useful picture of drug interactions almost a century ago, and with the rise in computational power over the last few decades, has become a well-established CADD method with numerous different applications in drug discovery. Depending on the prior knowledge of the system, pharmacophores can be used to identify derivatives of compounds, change the scaffold to new compounds with a similar target, virtual screen for novel inhibitors, profile compounds for ADME-tox, investigate possible off-targets, or just complement other molecular methods. While there are limitations to the pharmacophore concept, multiple remedies are available at any time to counter them. Given this versatility, it is expected that pharmacophore modeling will maintain a dominant role in CADD for the foreseeable future, and any medicinal chemist should be aware of its benefits and possibilities.
Acknowledgments
Xiaoyu Qing acknowledges Kuleuven IDO/11/008 for funding, Xiao Yin Lee acknowledges Ajinomoto scholarship for ASEAN international students for funding, Joren De Raeymaeker acknowledges IWT 120050 project NEMOA for funding, and Arnout RD Voet acknowledges RIKEN for the FPR fellowship.
Disclosure
The authors report no conflicts of interest in this work.
References
Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70(3):461–477. | |
Lourenço AM, Ferreira LM, Branco PS. Molecules of natural origin, semi-synthesis and synthesis with anti-inflammatory and anticancer utilities. Curr Pharm Des. 2012;18(26):3979–4046. | |
Wikberg JES, Spjuth O, Eklund M, Lapins M. Chemoinformatics Taking Biology into Account: Proteochemometrics. In: Guha R, Bender A, editors. Computational Approaches in Cheminformatics and Bioinformatics. Hoboken: John Wiley & Sons; 2011:57–92. | |
Reardon S. Project ranks billions of drug interactions. Nature. 2013; 503(7477):449–450. | |
Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162(6):1239–1249. | |
Krasavin M, Karapetian R, Konstantinov I, et al. Discovery and potency optimization of 2-amino-5-arylmethyl-1,3-thiazole derivatives as potential therapeutic agents for prostate cancer. Arch Pharm (Weinheim). 2009;342(7):420–427. | |
Kaul P. Drug discovery: Past, present and future. In: Jucker E, editor. Progress in Drug Research, Volume 50. Berlin: Springer Science and Business Media; 1998:9–105. | |
Veselovsky AV, Zharkova MS, Poroikov VV, Nicklaus MC. Computer-aided design and discovery of protein-protein interaction inhibitors as agents for anti-HIV therapy. SAR QSAR Environ Res. 2014;25(6):457–471. | |
Song CM, Lim SJ, Tong JC. Recent advances in computer-aided drug design. Brief Bioinform. 2009;10(5):579–591. | |
Taft CA, Da Silva VB, Da Silva CH. Current topics in computer-aided drug design. J Pharm Sci. 2008;97(3):1089–1098. | |
Bajorath J. Integration of virtual and high-throughput screening. Nat Rev Drug Discov. 2002;1(11):882–894. | |
Hopkins AL, Keserü GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2):105–121. | |
Ballester PJ, Mangold M, Howard NI, et al. Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J R Soc Interface. 2012;9(77):3196–3207. | |
Boyd MR. The position of intellectual property rights in drug discovery and development from natural products. J Ethnopharmacol. 1996;51(1–3):17–25; discussion 25–27. | |
Thiel KA. Structure-aided drug design’s next generation. Nat Biotechnol. 2004;22(5):513–519. | |
Schuffenhauer A. Computational methods for scaffold hopping. Wiley Interdiscip Rev Comput Mol Sci. 2012;2(6):842–867. | |
Sun H, Tawa G, Wallqvist A. Classification of scaffold-hopping approaches. Drug Discov Today. 2012;17(7–8):310–324. | |
Schneider G, Schneider P, Renner S. Scaffold-Hopping: How Far Can You Jump? QSAR Comb Sci. 2006;25(12):1162–1171. | |
Langdon SR, Westwood IM, van Montfort RL, Brown N, Blagg J. Scaffold-focused virtual screening: prospective application to the discovery of TTK inhibitors. J Chem Inf Model. 2013;53(5):1100–1112. | |
Li AP. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov Today. 2001;6(7):357–366. | |
Yu H, Adedoyin A. ADME–Tox in drug discovery: integration of experimental and computational technologies. Drug Discov Today. 2003;8(18):852–861. | |
Ekins S, Boulanger B, Swaan PW, Hupcey MA. Towards a new age of virtual ADME/TOX and multidimensional drug discovery. Mol Divers. 2000;5(4):255–275. | |
Agrafiotis DK, Bandyopadhyay D, Wegner JK, Vlijmen Hv. Recent advances in chemoinformatics. J Chem Inf Model. 2007;47(4):1279–1293. | |
Valerio LG Jr, Choudhuri S. Chemoinformatics and chemical genomics: potential utility of in silico methods. J Appl Toxicol. 2012;32(11):880–889. | |
Hong H, Xie Q, Ge W, et al. Mold(2), molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. J Chem Inf Model. 2008;48(7):1337–1344. | |
Vogt M, Bajorath J. Chemoinformatics: a view of the field and current trends in method development. Bioorg Med Chem. 2012;20(18):5317–5323. | |
Kapetanovic IM. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem Biol Interact. 2008;171(2):165–176. | |
Karelson M, Lobanov VS, Katritzky AR. Quantum-Chemical Descriptors in QSAR/QSPR Studies. Chem Rev. 1996;96(3):1027–1044. | |
Gozalbes R, Doucet JP, Derouin F. Application of topological descriptors in QSAR and drug design: history and new trends. Curr Drug Targets Infect Disord. 2002;2(1):93–102. | |
Perkins R, Fang H, Tong W, Welsh WJ. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ Toxicol Chem. 2003;22(8):1666–1679. | |
Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. | |
Paul N, Kellenberger E, Bret G, Müller P, Rognan D. Recovering the true targets of specific ligands by virtual screening of the protein data bank. Proteins. 2004;54(4):671–680. | |
Grinter SZ, Liang Y, Huang SY, Hyder SM, Zou X. An inverse docking approach for identifying new potential anti-cancer targets. J Mol Graph Model. 2011;29(6):795–799. | |
Kharkar PS, Warrier S, Gaud RS. Reverse docking: a powerful tool for drug repositioning and drug rescue. Future Med Chem. 2014;6(3):333–342. | |
Lee M, Kim D. Large-scale reverse docking profiles and their applications. BMC Bioinformatics. 2012;13:S6. | |
Ivanciuc O. Drug Design with Artificial Intelligence Methods. In: Meyers RA, editor. Encyclopedia of Complexity and Systems Science. Berlin: Springer; 2009:2113–2139. | |
Duch W, Swaminathan K, Meller J. Artificial intelligence approaches for rational drug design and discovery. Curr Pharm Des. 2007;13(14):1497–1508. | |
Shin WJ, Seong BL. Recent advances in pharmacophore modeling and its application to anti-influenza drug discovery. Expert Opin Drug Discov. 2013;8(4):411–426. | |
Braga RC, Andrade CH. Assessing the performance of 3D pharmacophore models in virtual screening: how good are they? Curr Top Med Chem. 2013;13(9):1127–1138. | |
Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today. 2010;15(11–12):444–450. | |
Güner O, Clement O, Kurogi Y. Pharmacophore modeling and three dimensional database searching for drug design using catalyst: recent advances. Curr Med Chem. 2004;11(22):2991–3005. | |
Krautscheid Y, Senning CJÅ, Sartori SB, et al. Pharmacophore modeling, virtual screening, and in vitro testing reveal haloperidol, eprazinone, and fenbutrazate as neurokinin receptors ligands. J Chem Inf Model. 2014;54(6):1747–1757. | |
Ehrlich P. Über den jetzigen Stand der Chemotherapie. Ber Dtsch Chem Ges. 1909;42(1):17–47. | |
Schueler FW. Chemobiodynamics and Drug Design. New York; McGraw-Hill: 1960. | |
Wermuth CG, Ganellin CR, Lindberg P, Mitscher LA. Glossary of terms used in medicinal chemistry (IUPAC recommendations 1998). Pure Appl Chem. 1998;70:1129–1143. | |
Gund P. Evolution of the pharmacophore Concept in Pharmaceutical Research. In: Güner OF, editor. Pharmacophore Perception, Development, and Use in Drug Design. La Jolla: Internat’l University Line. | |
McGregor MJ, Muskal SM. Pharmacophore fingerprinting. 1. Application to QSAR and focused library design. J Chem Inf Comput Sci. 1999;39(3):569–574. | |
McGregor MJ, Muskal SM. Pharmacophore fingerprinting. 2. Application to primary library design. J Chem Inf Comput Sci. 2000;40(1):117–125. | |
Mason JS, Morize I, Menard PR, Cheney DL, Hulme C, Labaudiniere RF. New 4-point pharmacophore method for molecular similarity and diversity applications: overview of the method and applications, including a novel approach to the design of combinatorial libraries containing privileged substructures. J Med Chem. 1999;42(17):3251–3264. | |
Geppert TD, Lipsky PE. Antigen presentation at the inflammatory site. Crit Rev Immunol. 1989;9(4):313–362. | |
Sheridan RP, Rusinko A 3rd, Nilakantan R, Venkataraghavan R. Searching for pharmacophores in large coordinate data bases and its use in drug design. Proc Natl Acad Sci U S A. 1989;86(20):8165–8169. | |
Jones G, Willett P, Glen RC. A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J Comput Aided Mol Des. 1995;9(6):532–549. | |
Goto J, Kataoka R, Hirayama N. Ph4Dock: pharmacophore-based protein-ligand docking. J Med Chem. 2004;47(27):6804–6811. | |
Wolber G, Seidel T, Bendix F, Langer T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov Today. 2008;13(1–2):23–29. | |
Langer T, Hoffman RD. Pharmacophores and Pharmacophore Searches. Mannhold R, Kubinyi H, Folkers G, editors. Hoboken: John Wiley & Sons; 2006:395. | |
Liu X, Zhu F, Ma XH, et al. Predicting targeted polypharmacology for drug repositioning and multi- target drug discovery. Curr Med Chem. 2013;20(13):1646–1661. | |
Thai KM, Ngo TD, Tran TD, Le MT. Pharmacophore modeling for antitargets. Curr Top Med Chem. 2013;13(9):1002–1014. | |
Luu TT, Malcolm N, Nadassy K. Pharmacophore modeling methods in focused library selection – applications in the context of a new classification scheme. Comb Chem High Throughput Screen. 2011;14(6):488–499. | |
Jose RA, Voet A, Broos K, et al. An integrated fragment based screening approach for the discovery of small molecule modulators of the VWF-GPIbalpha interaction. Chem Commun (Camb). 2012;48(92):11349–11351. | |
Voet AR, Kumar A, Berenger F, Zhang KY. Combining in silico and in cerebro approaches for virtual screening and pose prediction in SAMPL4. J Comput Aided Mol Des. 2014;28(4):363–373. | |
Hähnke V, Schneider G. Pharmacophore alignment search tool: influence of scoring systems on text-based similarity searching. J Comput Chem. 2011;32(8):1635–1647. | |
Catalyst (r). Vol San Diego: Accelrys, Inc.; 2014. Available from: http://accelrys.com/products/discovery-studio/pharmacophore-ligand-based-design.html. Accessed September 5, 2014. | |
Sanders MPA, McGuire R, Roumen L, et al. From the protein’s perspective: the benefits and challenges of protein structure-based pharmacophore modeling. Med Chem Commun. 2012;3:28–38. | |
Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45(1):160–169. | |
Desaphy J, Azdimousa K, Kellenberger E, Rognan D. Comparison and druggability prediction of protein-ligand binding sites from pharmacophore-annotated cavity shapes. J Chem Inf Model. 2012;52(8):2287–2299. | |
Böhm HJ. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des. 1992;6(1):61–78. | |
Barillari C, Marcou G, Rognan D. Hot-spots-guided receptor-based pharmacophores (HS-Pharm): a knowledge-based approach to identify ligand-anchoring atoms in protein cavities and prioritize structure-based pharmacophores. J Chem Inf Model. 2008;48(7):1396–1410. | |
Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28(7):849–857. | |
Tintori C, Corradi V, Magnani M, Manetti F, Botta M. Targets looking for drugs: a multistep computational protocol for the development of structure-based pharmacophores and their applications for hit discovery. J Chem Inf Model. 2008;48(11):2166–2179. | |
Voet A, Helsen C, Zhang KY, Claessens F. The discovery of novel human androgen receptor antagonist chemotypes using a combined pharmacophore screening procedure. ChemMedChem. 2013;8(4):644–651. | |
Helsen C, Van den Broeck T, Voet A, et al. Androgen receptor antagonists for prostate cancer therapy. Endocr Relat Cancer. 2014;21(4): T105–T118. | |
Kumar A, Zhang KYJ. Hierarchical virtual screening approaches in small molecule drug discovery. Methods. Epub July 27, 2014. | |
Dunbar JB Jr, Smith RD, Yang CY, et al. CSAR benchmark exercise of 2010: selection of the protein-ligand complexes. J Chem Inf Model. 2011;51(9):2036–2046. | |
Damm-Ganamet KL, Smith RD, Dunbar JB Jr, Stuckey JA, Carlson HA. CSAR benchmark exercise 2011-2012: evaluation of results from docking and relative ranking of blinded congeneric series. J Chem Inf Model. 2013;53(8):1853–1870. | |
Hindle SA, Rarey M, Buning C, Lengaue T. Flexible docking under pharmacophore type constraints. J Comput Aided Mol Des. 2002;16(2):129–149. | |
Hu B, Lill MA. Protein pharmacophore selection using hydration-site analysis. J Chem Inf Model. 2012;52(4):1046–1060. | |
Hu B, Lill MA. PharmDock: a pharmacophore-based docking program. J Cheminform. 2014;6:14. | |
Mobley DL, Liu S, Lim NM, et al. Blind prediction of HIV integrase binding from the SAMPL4 challenge. J Comput Aided Mol Des. 2014;28(4):327–345. | |
Lin JH, Lu AY. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev. 1997;49(4):403–449. | |
Alavijeh MS, Palmer AM. The pivotal role of drug metabolism and pharmacokinetics in the discovery and development of new medicines. IDrugs. 2004;7(8):755–763. | |
Guner OF, Bowen JP. Pharmacophore modeling for ADME. Curr Top Med Chem. 2013;13(11):1327–1342. | |
Yamashita F, Hashida M. In silico approaches for predicting ADME properties of drugs. Drug Metab Pharmacokinet. 2004;19(5):327–338. | |
Tanaka E. Clinically important pharmacokinetic drug-drug interactions: role of cytochrome P450 enzymes. J Clin Pharm Ther. 1998; 23(6):403–416. | |
de Groot MJ, Ekins S. Pharmacophore modeling of cytochromes P450. Adv Drug Deliv Rev. 2002;54(3):367–383. | |
Ekins S, de Groot MJ, Jones JP. Pharmacophore and three-dimensional quantitative structure activity relationship methods for modeling cytochrome p450 active sites. Drug Metab Dispos. 2001;29(7):936–944. | |
Masimirembwa CM, Ridderström M, Zamora I, Andersson TB. Combining pharmacophore and protein modeling to predict CYP450 inhibitors and substrates. Methods Enzymol. 2002;357:133–144. | |
Schuster D, Laggner C, Steindl TM, Langer T. Development and validation of an in silico P450 profiler based on pharmacophore models. Curr Drug Discov Technol. 2006;3(1):1–48. | |
Sorich MJ, Miners JO, McKinnon RA, Smith PA. Multiple pharmacophores for the investigation of human UDP-glucuronosyltransferase isoform substrate selectivity. Mol Pharmacol. 2004;65(2):301–308. | |
Sorich MJ, Smith PA, McKinnon RA, Miners JO. Pharmacophore and quantitative structure activity relationship modelling of UDP-glucuronosyltransferase 1A1 (UGT1A1) substrates. Pharmacogenetics. 2002;12(8):635–645. | |
Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25(2):197–206. | |
Koutsoukas A, Simms B, Kirchmair J, et al. From in silico target prediction to multi-target drug design: current databases, methods and applications. J Proteomics. 2011;74(12):2554–2574. | |
Rollinger JM, Schuster D, Danzl B, et al. In silico target fishing for rationalized ligand discovery exemplified on constituents of Ruta graveolens. Planta Med. 2009;75(3):195–204. | |
Hu Y, Bajorath J. Polypharmacology directed compound data mining: identification of promiscuous chemotypes with different activity profiles and comparison to approved drugs. J Chem Inf Model. 2010;50(12):2112–2118. | |
Scior T, Bender A, Tresadern G, et al. Recognizing pitfalls in virtual screening: a critical review. J Chem Inf Model. 2012;52(4):867–881. | |
Kirchmair J, Wolber G, Laggner C, Langer T. Comparative performance assessment of the conformational model generators omega and catalyst: a large-scale survey on the retrieval of protein-bound ligand conformations. J Chem Inf Model. 2006;46(4):1848–1861. | |
Kirchmair J, Laggner C, Wolber G, Langer T. Comparative analysis of protein-bound ligand conformations with respect to catalyst’s conformational space subsampling algorithms. J Chem Inf Model. 2005;45(2):422–430. | |
De Luca L, Barreca ML, Ferro S, et al. Pharmacophore-based discovery of small-molecule inhibitors of protein-protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. ChemMedChem. 2009;4(8):1311–1316. | |
Christ F, Voet A, Marchand A, et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol. 2010;6(6):442–448. | |
Vancraenenbroeck R, De Raeymaecker J, Lobbestael E, et al. In silico, in vitro and cellular analysis with a kinome-wide inhibitor panel correlates cellular LRRK2 dephosphorylation to inhibitor activity on LRRK2. Front Mol Neurosci. 2014;7:51. | |
Schomburg KT, Bietz S, Briem H, Henzler AM, Urbaczek S, Rarey M. Facing the challenges of structure-based target prediction by inverse virtual screening. J Chem Inf Model. 2014;54(6):1676–1686. | |
Kumar A, Voet A, Zhang KY. Fragment based drug design: from experimental to computational approaches. Curr Med Chem. 2012;19(30):5128–5147. | |
Böhm HJ. A novel computational tool for automated structure-based drug design. J Mol Recognit. 1993;6(3):131–137. | |
Lippert T, Schulz-Gasch T, Roche O, Guba W, Rarey M. De novo design by pharmacophore-based searches in fragment spaces. J Comput Aided Mol Des. 2011;25(10):931–945. | |
Cavalluzzo C, Voet A, Christ F, et al. De novo design of small molecule inhibitors targeting the LEDGF/p75-HIV integrase interaction. RSC Adv. 2012;2:974. | |
Cavalluzzo C, Christ F, Voet A, et al. Identification of small peptides inhibiting the integrase-LEDGF/p75 interaction through targeting the cellular co-factor. J Pept Sci. 2013;19(10):651–658. | |
Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450(7172):1001–1009. | |
Wilson AJ. Inhibition of protein-protein interactions using designed molecules. Chem Soc Rev. 2009;38:3289–3300. | |
Fry DC. Drug-like inhibitors of protein-protein interactions: a structural examination of effective protein mimicry. Curr Protein Pept Sci. 2008;9(3):240–247. | |
Voet A, Berenger F, Zhang KY. Electrostatic similarities between protein and small molecule ligands facilitate the design of protein-protein interaction inhibitors. PLoS One. 2013;8(10):e75762. | |
Voet A, Zhang KY. Pharmacophore modelling as a virtual screening tool for the discovery of small molecule protein-protein interaction inhibitors. Curr Pharm Des. 2012;18(30):4586–4598. | |
Voet A, Banwell EF, Sahu KK, Heddle JG, Zhang KY. Protein interface pharmacophore mapping tools for small molecule protein: protein interaction inhibitor discovery. Curr Top Med Chem. 2013;13(9):989–1001. | |
Reddy TR, Li C, Fischer PM, Dekker LV. Three-dimensional pharmacophore design and biochemical screening identifies substituted 1,2,4-triazoles as inhibitors of the annexin A2-S100A10 protein interaction. ChemMedChem. 2012;7(8):1435–1446. | |
Voet A, Callewaert L, Ulens T, et al. Structure based discovery of small molecule suppressors targeting bacterial lysozyme inhibitors. Biochem Biophys Res Commun. 2011;405(4):527–532. | |
Mustata G, Li M, Zevola N, et al. Development of small-molecule PUMA inhibitors for mitigating radiation-induced cell death. Curr Top Med Chem. 2011;11(3):281–290. | |
Voet ARD, Akihiro I, Hirohama M, et al. Discovery of small molecule inhibitors targeting the SUMO–SIM interaction using a protein interface consensus approach. Med Chem Commun. 2014;5: 783–786. | |
Corradi V, Mancini M, Manetti F, Petta S, Santucci MA, Botta M. Identification of the first non-peptidic small molecule inhibitor of the c-Abl/14-3-3 protein-protein interactions able to drive sensitive and Imatinib-resistant leukemia cells to apoptosis. Bioorg Med Chem Lett. 2010;20(20):6133–6137. | |
Baker D. Centenary Award and Sir Frederick Gowland Hopkins Memorial Lecture. Protein folding, structure prediction and design. Biochem Soc Trans. 2014;42(2):225–229. | |
Tinberg CE, Khare SD, Dou J, et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501(7466):212–216. | |
Nivón LG, Moretti R, Baker D. A Pareto-optimal refinement method for protein design scaffolds. PLoS One. 2013;8:e59004. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.