")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
Pharmacology and clinical efficacy of dimethyl fumarate (BG-12) for treatment of relapsing–remitting multiple sclerosis
Authors Ruggieri S , Tortorella C, Gasperini C
Received 20 August 2013
Accepted for publication 9 December 2013
Published 27 March 2014 Volume 2014:10 Pages 229—239
DOI https://doi.org/10.2147/TCRM.S53285
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Serena Ruggieri,1 Carla Tortorella,2 Claudio Gasperini3
1Department of Neurology and Psychiatry, Sapienza University of Rome “Sapienza”, Rome, Italy; 2Department of Basic Sciences, Neurosciences and Sense Organs, University of Bari, Bari, Italy; 3Department of Neurosciences, S Camillo Forlanini Hospital, Rome, Italy
Abstract: The last two decades have seen the introduction of several therapies for multiple sclerosis (MS). These therapies are intended to work at different levels of the disease, typically targeting direct symptom management, brief corticosteroid administration for acute exacerbations, and the regular use of disease-modifying drugs. Nevertheless, in clinical practice, disease-modifying drugs or immunosuppressive treatments are frequently associated with suboptimal response in terms of efficacy and several side effects leading to poor patient adherence, so the proportion of relapsing–remitting MS patients not adequately responding to disease-modifying therapy have been reported to range from 7% to 49%. Natalizumab and fingolimod are the newest US Food and Drug Administration-approved agents that have been added to the MS treatment armamentarium, but their use is limited by a less known safety profile and recognized specific risk. Thus, there is an important need for new therapeutic strategies, especially those that may offer greater patient satisfaction and safer risk profile in order to optimize therapeutic outcomes. A number of potential therapies for MS are now in late-stage development. Effective, safe, and well-tolerated therapies may improve compliance and empower patients with a level of independence not presently possible. To meet these characteristics, most of these therapies are oral compounds. Herein, we review the pharmacology and efficacy of dimethyl fumarate (BG-12) to date and its role in the evolving marketplace.
Keywords: disease-modifying drugs, nuclear factor erythroid-derived 2 (E2)-related factor, Nrf2, oxidative stress, neuroprotection, oral treatment
Introduction
Multiple sclerosis (MS) is a chronic inflammatory immune-mediated disease of the central nervous system (CNS). Disease onset usually occurs in young adults, and it is more common in females than in males (3:1). MS affects about 2.5 million people worldwide, and incidence and prevalence are highly variable, depending on genetics and several environmental factors.1 Although having wide incidence variability, MS is still the commonest nontraumatic cause of neurological disability in young patients in Western Europe and North America.2 Most patients (80%) present with a relapsing and remitting (RR) course, which is characterized by recurring attacks of acute focal neurological deficits or exacerbations of existing deficits (relapses) followed gradually by partial or full recovery (remission).3 The combination of inflammation and neurodegeneration makes MS a progressive disease wherein the severity and specific symptoms are unpredictable and often vary from one person to another. The multifocal nature of the disease manifests clinically as a range of sensorimotor, cerebellar, visual, sphincteric, brain stem, cognitive, and neuropsychiatric symptoms. In the natural history of MS, approximately 80% of patients convert to the secondary progressive course after 20 years, in which there is acceleration of disability and accumulating irreversible neurologic deficits in the absence of clinical relapses.4 The remaining 20% with progressive clinical deterioration from the onset of the disease have primary progressive MS. According to more recent studies, the natural history of the disease in patients that have been treated with immunological agents is significantly changed, showing a more favorable prognosis.5,6
The exact cause of MS is still unknown, but breakdown of the blood–brain barrier (BBB) is a requirement for immune system access to the CNS, infiltration of the inflammatory cells into the CNS, and subsequent autoimmune demyelination. Pathological studies have shown that inflammatory cells not only lead to demyelination, but may also be involved in axon injury.7,8 MS and its animal model experimental autoimmune encephalomyelitis (EAE) have long been regarded as primarily T-helper (Th) cell type 1-mediated diseases; however, recent evidence suggests that Th17 cells, a mostly unexplored subset of Th cells, may be even more pathogenic than Th1 cells. In the EAE model, this cell type is crucial for the recruitment of leukocytes into the CNS and for triggering parenchymal inflammation. In humans, Th17 cells are found in acutely active and on the borders of chronically active lesions. Overall, CD4+ T-cells only recognize antigens presented on major histocompatibility complex (MHC) class II complexes, and these are seldom found in the CNS. MHC class I, in contrast, can be induced on neurons and myelin. This also makes CD8+ T-cells promising candidates as effector cell types. Indeed, CD8+ T-cells outnumber CD4+ T-cells in the lesions of MS patients and can induce axonal pathology.7 MS lesions contain many inflammatory components, including T-cells (CD4, CD8, and gamma delta), plasma cells, macrophages, antibodies, and complement factors. Activated T-cells cross the BBB, a process that is mediated by interaction of very late antigen-4 on the T-cell surface and vascular cell adhesion molecule-1 (VCAM-1) on the brain vascular endothelium; infiltrating T-cells increase the permeability of the BBB by matrix metalloproteinases, which degrade the extracellular matrix.8 Activated immune cells in the brain interact with their responsive antigen presented by macrophages or microglia and secrete cytokines and chemokines that further permeabalize the BBB and further reinforce the immune response, thus resulting in tissue damage. Even macrophages, which are the predominant cell type along the periphery of lesions, secrete tumor necrosis factor alpha (TNF-α) and oxygen-free radicals, leading to tissue damage. B-cells can both provide signaling to T-cells and differentiate into plasma cells, secreting antibodies that can bind to their antigen and activate complement, leading to myelin breakdown. The oligodendrocytes are damaged by a combination of cytokines released during the inflammatory reaction and possibly by direct cellular contact. Axons may be damaged and transected by direct attack by inflammatory cells and their cytokines, or by loss of trophic support or protection provided by the oligodendrocytes and myelin membranes, respectively.9 Noninflammatory mechanisms, such as mitochondrial dysfunction, may also contribute to neurodegeneration in MS.10 Excessive release of free radicals may play an important role in MS pathogenesis and promote transendothelial leukocyte migration, as well as contribute to oligodendrocyte damage and axonal degeneration.11,12 Thus, oxidative stress, stemming from different cell types and targeting several cellular components of the CNS to a variable extent, is involved in this detrimental concert.
On the basis of this pathologic pathway and immune cascade, most research is addressed at blocking a particular step of the immunological cascade.
Background
The arsenal for MS therapy includes treatment of acute relapses with corticosteroids, symptomatic treatment with appropriate agents, and disease modification with immunomodulatory agents. Relapses of MS are commonly treated with high-dose intravenous methylprednisolone given over a period of 3–5 days. This shortens relapse duration, but long-term effects have been poorly investigated.13,14 The past 20 years have seen the introduction of mainly immunomodulatory agents for MS, such as interferon (IFN)-β and glatiramer acetate (GA). The main therapeutic goals of these treatments consist of clinical relapse rate reduction, extension of time to next relapse, reduction of new lesions detectable by magnetic resonance imaging (MRI), and decreasing the long-term accumulation of disability. The US Food and Drug Administration (FDA) approved the first disease-modifying drug (DMD) for MS, subcutaneous IFN-β-1b (marketed as Betaseron® [Bayer HealthCare Pharmaceuticals, Montville, NJ, USA] in USA and as Betaferon [Bayer Schering Pharma AG, Berlin-Wedding, Germany] in the rest of the world), for RRMS in 1993. IFN-β acts as an anti-inflammatory drug and has several mechanisms of action, including a reduction in the production of IFN-γ and TNF-α, inhibition of T-cell activation and clonal expansion, modulation of cytokine and matrix metalloproteinase production, and inhibition of T-cell migration and entry into the CNS.15 Currently approved immunomodulator treatments for RRMS include GA and recombinant interferons (IFN-β-1a; AVONEX®; Biogen Idec Inc, Cambridge, MA, USA, and Rebif®; Merk Serono, Merck KGaA, Darmstadt, Germany [EMD Serono in the United States and Canada], and IFN-β-1b [Betaseron®/Extavia, Bayer HealthCare Pharmaceuticals Inc., Montville, NJ USA]) that represent first-line therapies for MS. Mitoxantrone (Novantrone®; Merk Serono, Merck KGaA, Darmstadt, Germany [EMD Serono in the United States and Canada]), natalizumab (Tysabri®; Biogen Idec Inc), and fingolimod (Gilenya®; Novartis, Basel, Switzerland) are also available for treatment of MS in many countries. The IFN products are all thought to have similar mechanisms of action, although they differ in the route of administration, time of onset of action, and risk of induction of neutralizing antibodies.16 In contrast, GA – a synthetic copolymer of glutamic acid, lysine, alanine, and tyrosine – is believed to activate Th2 regulatory cells in the periphery. These activated Th2 cells cross the BBB and enter the CNS, where they shift the immune response from proinflammatory to anti-inflammatory by secreting cytokines that downregulate the inflammatory response and inhibit proinflammatory Th1 cells.17 Mitoxantrone is an antineoplastic agent that inhibits DNA and RNA synthesis of B- and T-cells. While approved for the treatment of RRMS and secondary progressive MS,18 it has only shown a clear benefit for patients still experiencing relapses and developing new lesions that are detectable using MRI. Increasing recognition of short- and long-term risks of cardiotoxicity, acute leukemia, and bone marrow suppression has limited its use.19 Natalizumab is the only monoclonal antibody approved for the treatment of MS. It works by blocking leukocyte integrin α-4 and thus limits the migration of lymphocytes and monocytes through the BBB into the CNS. Large Phase III studies have been conducted in RRMS, where natalizumab as monotherapy was found to reduce MS relapses by 68% compared to placebo,20 but its use is restricted by its association with the development of progressive multifocal leukoencephalopathy (PML). As a result of this circumstance, health authorities granted approval of natalizumab only as a treatment for patients not responding to first-line drugs or as a first choice in patients with aggressive RRMS. The risk of PML (ranging from 1/10,000 to 1/100) increases according to the presence or absence of three risk factors: positive status with respect to anti-JC virus antibodies; prior use of immunosuppressant; and increasing duration of natalizumab treatment.21 Fingolimod is the first oral drug approved for MS. The results of two clinical trials led to the approval of fingolimod by the FDA as first-line treatment, and by the European Medicines Agency for highly active RRMS; however, safety concerns include the known pharmacodynamics effect of the drug on heart rate and conduction, reports of macular edema in the fingolimod transplant program and in treated MS patients, and seven cases of melanoma and squamous cell and basal cell carcinomas.22,23 Moreover, there were increased risks of infection, including two deaths from herpes virus infections in the original Phase III trial, with one case of herpes virus encephalitis and one case of primary disseminated varicella zoster.23 All these mentioned warnings occurring with fingolimod treatment require monitoring.
Nevertheless, in clinical practice, DMDs are frequently associated with suboptimal response in terms of efficacy and several side effects leading to poor patient adherence, so the proportion of RRMS patients not adequately responding to disease-modifying therapy has been reported to range from 7%–49%, depending on the criteria used.24 Despite the positive therapeutic effects observed in clinical trials, many patients with RRMS continue to exhibit disease activity in daily clinical practice: 20%–50% of patients who receive DMD agents experience a marked increase in disability or a high number of relapses within a short period of time (<6 years) after the onset of treatment.24 Moreover, there is a lack of an effective treatment option for the progressive phase of the disease.
On the other hand, second-line treatment use is often limited by safety concerns, as already mentioned. Therefore, it is clear that there are some issues related to the treatment of MS patients that must be addressed. MS treatment priorities should be based on a better understanding of MS pathogenesis and heterogeneity to guide development of better therapies and monitoring methods; additional treatment options for RRMS that are more effective, more convenient, and/or better tolerated, as well as some that include neuroprotective and repair properties, are emerging.
Several oral therapies have been evaluated in clinical trials, and results presented following completion of Phase III trials. Fingolimod, teriflunomide, laquinimod, and dimethyl fumarate (DMF) results are briefly summarized in Table 1. The latest BG-12, will be thoroughly reviewed here, starting from its mechanism of action to its possible role in MS treatment strategies.
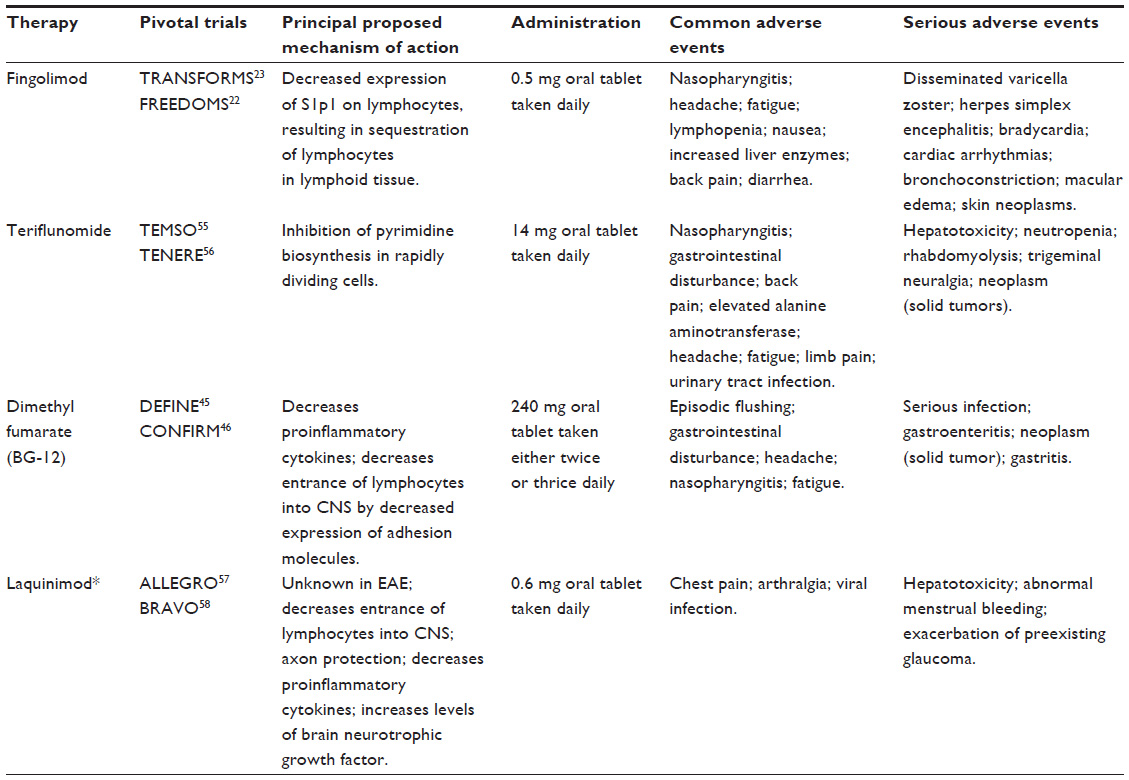 | Table 1 Summary of oral drugs for the treatment of relapsing-remitting multiple sclerosis |
Introduction to the compound
Fumaric acid esters (FAEs) are a group of similarly structured compounds that have been used in the treatment of psoriasis since 1958, originally proposed by the German biochemist Schweckendiek,25 who was afflicted by the disease himself. FAEs have been used off-label for treatment of psoriasis in Europe for some time before a mixture of compounds consisting of DMF and three salts of ethyl hydrogen fumarate were licensed in Germany in 1994 as oral therapy for severe psoriasis under the brand name Fumaderm® (Biogen-Idec, Weston, MA, USA) (Figure 1).
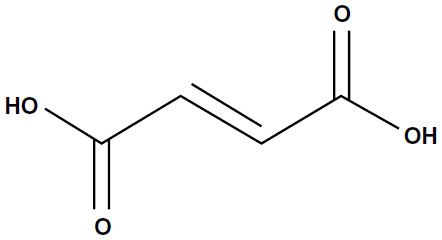 | Figure 1 Chemical structure of dimethyl fumarate (BG-12). |
Fumaderm® is approved as second-line therapy for severe systemic psoriasis in Germany but not in the USA.26 Today, the overall treatment experience comprises more than 180,000 patient years and suggests a beneficial side effect profile. Over the past 15 years, there have been many clinical trials that demonstrated the immunomodulatory efficacy and safety profile of oral FAEs in systemic psoriasis.27
Based on similarities in the inflammatory cascade of psoriasis and MS, researchers hypothesized that FAEs may also have beneficial effects in CNS autoimmune disease, where MS represents the prototypic inflammatory autoimmune disorder of this system. This hypothesis became stronger with the accidental observation of MS symptom relief and disease stabilization in patients treated with oral fumarate for concomitant psoriasis.
Pharmacological properties and mechanism of action of FAEs
After oral intake, DMF, the main component of Fumaderm®, is rapidly hydrolyzed by esterases to its metabolite monomethyl fumarate (MMF). Ingestion of DMF is associated with lower gastrointestinal side effects compared with MMF. After complete absorption in the small intestine,28 it can interact with immune cells in the blood circulation.29 Absorption of MMF is decreased by concurrent ingestion of food, though it remains highly bioavailable.29 MMF is the most bioactive metabolite,30 and typically reaches serum peak concentrations around 20 μM. MMF is eliminated mainly through breathing, while only small amounts of intact MMF are excreted through urine or feces. There is no evidence for a cytochrome P450-dependent metabolism in the liver,29 thus few drug interactions would be expected. MMF’s half-life in vivo is around 12 hours.
The exact mechanism of action by which DMF and its primary metabolite MMF exerts its effects is still unclear and under research. It is now accepted that it could work at different levels, acting at different stages and modulating different cells involved in the immune cascade (Figure 2).
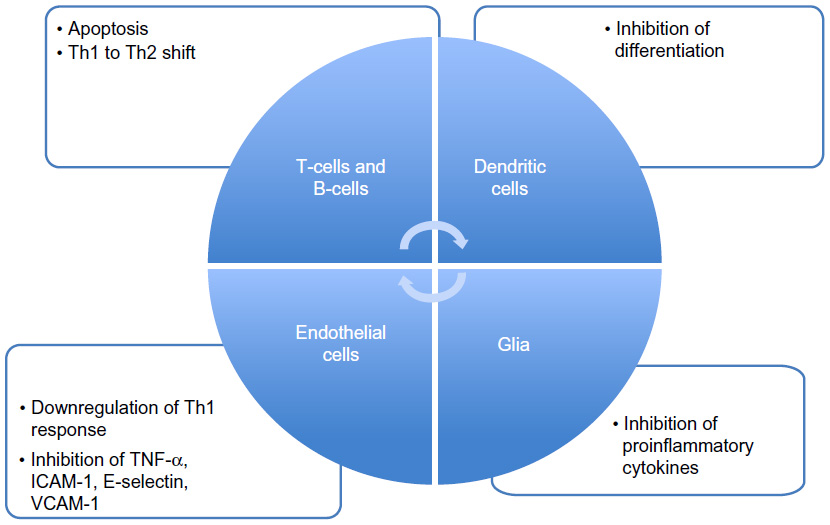 | Figure 2 Effects of fumaric acid esters on immune and accessory cells. |
DMF seems to interfere with cellular redox system by modulating intracellular thiols and thereby increasing the level of reduced glutathione. The major involved transcription factor in this process is nuclear factor erythroid-derived 2 (E2)-related factor (Nrf2), which is released from binding to Keap1 via the activity of fumarates. BG-12 induces the cleavage of Kelch-like erythroid cell-derived protein with cap “n” collar homology-associated protein 1 (Keap1) from Nrf2 in the cell cytoplasm. By cleaving this Keap1–Nrf2 complex, Nrf2 is free to cross the nuclear membrane and interact with other nuclear transcription factors to upregulate the antioxidant response element.31 Moreover, studies in Nrf2 knockout mice showed that most of the therapeutic ability of FAEs was abolished in the absence of Nrf2. Further support for these mechanisms appeared in a recently published study32 that better characterizes the potential neuroprotective, as well as cytoprotective effects, of DMF and MMF on cellular resistance to oxidative damage in primary culture of CNS cells. DMF and MMF treatment increase the cellular redox potential, glutathione as well as adenosine triphosphate (ATP) levels, and the mitochondrial membrane potential in a concentration-dependent manner. Similarly, both DMF and MMF improve cell viability after reactive oxygen species challenge. This effect is lost in cells that have eliminated or reduced Nrf2 levels. These data suggest that DMF and MMF are cytoprotective for neurons and astrocytes against oxidative stress-induced cellular injury and loss via upregulation of an Nrf2-dependent antioxidant response.32 Nrf2 also contributes to the long-term effect of DMF in neuronal cells, which may also involve other reported mechanisms such as the inhibition of the nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB); this means an inhibition of the translocation of NF-κB into the nucleus and a decreased expression of NF-κB-dependent genes that regulate the expression of a cascade of inflammatory cytokines, chemokines, and adhesion molecules.33
Evidence shows that DMF can modulate different type of cells in the immune system. In almost all patients treated with Fumaderm®, a decrease of T-cells was observed.34 A subsequent in vitro study detected how fumarate can induce apoptosis in human T-cells.35 MMF can induce an immune deviation, with the skewing of IFN-γ inducing antigen-specific Th1 cells to switch to an interleukin (IL)-4-dominated Th2 phenotype. Th2 cells are still reactive to antigen, but provide a different cytokine pattern.36 MMF was shown to increase the production of the Th2 cytokines IL-4 and IL-5 without having an effect on the production of the Th1 cytokines IFN-γ and IL-2, or the proliferation of T-cells.36 In another study, MMF-treated monocyte-derived dendritic cells downregulated Th1 cell responses by significantly decreasing the production of IFN-γ after stimulation by lipopolysaccharide.37
In an experiment with umbilical vein endothelial cell cultures, DMF inhibited the TNF-α-induced expression of intercellular adhesion molecule-1, E-selectin, and VCAM-1, impairing the migration of immune cells inside tissues.38,39 This may correlate with the described activity of FAEs on NF-κB. Another consequence of the decreased expression of NF-κB can be speculated as an effect on B-cells. Although there have not been any studies on direct FAE effects on B-cells, there is evidence that the downregulation of NF-κB in turn inhibits the antiapoptotic protein Bcl-2, thus leading to apoptosis in B-cells.40
Efficacy of oral fumarate and clinical trials
Given the immunomodulatory properties of FAEs, it was suggested that they could be also a beneficial effect in MS. Thus, the effects of FAEs in myelin oligodendrocyte glycoprotein-induced EAE was investigated.41 Both esters had a significant therapeutic effect on the disease course, and histology showed a strongly reduced macrophage inflammation in the spinal cord. Cytokine analysis detected an increase of IL-10 in the treated animals.
Phase II studies
An open-label, exploratory, prospective study in ten subjects with RRMS demonstrated that FAEs produced significant reductions from baseline in number (P<0.05) and volume (P<0.01) of gadolinium-enhancing (Gd+) lesions after 18 weeks of treatment at a target dose of 720 mg/day administered orally; this effect persisted during the second 48-week treatment phase at half the target dose, after a 4-week washout period. Expanded Disability Status Scale (EDSS) score, ambulation index, and nine-hole peg test remained stable or slightly improved from baseline in all patients.42
A Phase II, randomized, placebo-controlled, double-blind study of DMF in 257 RRMS patients has been completed.43 All 257 patients were randomized to DMF 120, 360, or 720 mg/day for 6 months. There was a subsequent 6-month safety extension study, in which the placebo group received the highest dose of 720 mg/day. When treated with 240 mg DMF three times daily, a 69% reduction in the mean cumulative number of new Gd+ lesions was observed when compared with placebo (P=0.002). Treated subjects also demonstrated advantages versus placebo in other MRI parameters: a 48% reduction in the mean number of new or newly enlarging T2-hyperintense lesions (P<0.001) and a 53% reduction in the mean number of new T1-hypointense lesions (P=0.014). Although the study was not powered to evaluate the effects of DMF on clinical measures, clinical efficacy end points of the intent-to-treat population were evaluated as exploratory measures. There was a 32% reduction in annualized relapse rate (ARR) and a 24% reduction in the proportion of relapsing subjects in the group treated with a high dose (240 mg three times daily), which was not statistically significant when compared with placebo. A retrospective analysis44 of the BG-12 Phase II trial44 was performed to evaluate the evolution of the new Gd+ lesions, developed between weeks 4 and 12, into T1 black holes. The odds ratio (OR) for this MRI parameter in the 720 mg/day BG-12 group compared with placebo was 0.51 (P<0.0001). The treatment effect was greater for smaller lesions (OR 0.30) than for large lesions (OR 0.62).43 Analysis of the lower dose was not reported since it did not show a significant reduction in enhancing lesions.
Phase III studies
The efficacy and safety of oral BG-12 have been further evaluated in two large Phase III studies (DEFINE and CONFIRM).45,46 Both studies were designed to investigate the clinical efficacy and risk–benefit profile of BG-12, were scheduled for 24 months, and enrolled in each case over 1,200 adults with RRMS.
The DEFINE study was a three-arm trial over 96 weeks evaluating BG-12 in two dosages against placebo.45 Patients were randomly assigned to receive oral BG-12 at a dose of 240 mg twice daily, BG-12 at a dose of 240 mg three times daily, or placebo. The primary end point was the proportion of patients who had a relapse by 2 years. Other end points included the ARR, the time to confirmed progression of disability defined as an increase in EDSS score of 1.0 point from baseline and sustained for 12 weeks, and findings on MRI, such as the number of Gd+ lesions and the number of new or enlarging T2 lesions.45 A total of 1,237 patients were randomly assigned to a treatment or placebo group, and 952 patients completed the study.45 The estimated proportion of patients who had a relapse was significantly lower in the two BG-12 groups than in the placebo group (27% with BG-12 twice daily and 26% with BG-12 thrice daily versus 46% with placebo; P<0.001 for both comparisons). The annualized relapse rate at 2 years was 0.17 in the twice-daily BG-12 group and 0.19 in the thrice-daily BG-12 group, as compared with 0.36 in the placebo group, representing relative reductions of 53% and 48% with the two BG-12 regimens, respectively (P<0.001 for the comparison of each BG-12 regimen with placebo).45 The estimated proportion of patients with confirmed progression of disability was 16% in the twice-daily BG-12 group, 18% in the thrice-daily BG-12 group, and 27% in the placebo group, with significant relative risk reductions of 38% with BG-12 twice daily (P=0.005) and 34% with BG-12 thrice daily (P=0.01). BG-12 also significantly reduced the number of Gd+ lesions and of new or enlarging T2-weighted hyperintense lesions (P<0.001 for the comparison of each BG-12 regimen with placebo).45
The CONFIRM study had a similar trial design to DEFINE, with an additional, open fourth arm rater-blinded, and aimed to compare daily subcutaneous injections of 20 mg GA, or BG-12 at the dosage of 240 mg two or three times daily and placebo.46 The primary end point was the ARR at 2 years. Secondary end points were the proportion of patients with a relapse within 2 years, disability progression, the number of new or enlarging hyperintense T2 lesions, and other MRI parameters.46 BG-12 and GA were not directly compared in the CONFIRM trial, as the study was not powered to make such a comparison. In this study, a total of 1,430 patients were randomly assigned to receive treatment or placebo, and 1,417 were included in the intent-to-treat population.46 The ARR at 2 years was 0.40, 0.22, 0.20, and 0.29 for placebo, BG-12 twice daily, BG-12 thrice daily, and GA, respectively, translating into ARR reductions relative to placebo of 44%, 51%, and 29% for 480 mg BG-12, 720 mg BG-12, or GA, respectively (P<0.001 for both BG-12 dosages, P=0.01 for GA differences between drugs nonsignificant). Interestingly, the reduction of disease progression was not significant for BG-12 or GA; however, as compared with placebo, twice-daily BG-12, thrice-daily BG-12, and GA significantly reduced the numbers of new or enlarging T2-weighted hyperintense lesions (all P<0.001) and new T1-weighted hypointense lesions (P<0.001, P<0.001, and P=0.002, respectively).46 In post hoc comparisons of BG-12 versus GA, differences were not significant except for the annualized relapse rate (thrice-daily BG-12), new or enlarging T2-weighted hyperintense lesions (both BG-12 doses), and new T1-weighted hypointense lesions (thrice-daily BG-12) (nominal P<0.05 for each comparison).46
Safety and tolerability
The safety and tolerability of FAEs has been well known on the basis of the acquired experience using Fumaderm® in psoriasis patients. With FAE treatment of psoriasis, cases of acute renal failure have been reported, though other literature seems to refute this risk.26 No serious infections or neoplasm have occurred with any frequency with FAE treatment. Evidence-based guidelines47 for the treatment of psoriasis report very good long-term safety with FAEs, with more than 50,000 patient-years on file. In a retrospective safety study in patients treated with FAEs for severe psoriasis for up to 14 years, the most frequently reported adverse events (AEs) were flushing, diarrhea, and nausea. Excluding these, clinically relevant AEs included gastrointestinal complaints, flushing, lymphopenia, and eosinophilia.47
On the basis of such evidence, a new oral formulation of BG-12 was developed to address the gastrointestinal complaints seen with FAE treatment in psoriasis. In Phase II and III clinical trials,43,45,46 FAEs were well tolerated, especially after using BG-12 containing only DMF in enteric-coated microtablets to improve gastrointestinal tolerability.
In a Phase IIb study in patients with RRMS, the most commonly reported AEs besides MS relapses were flushing and headache.43 Generally, flushing with BG-12 started within 30 minutes of drug administration, subsided within 90 minutes, and did not lead to discontinuation. Gastrointestinal events were more commonly seen with BG-12 than with placebo (30%–41% with BG-12 versus 25% with placebo); however, these were mainly mild to moderate and were reported mostly in the early administration of treatment.43 While no head-to-head studies have been conducted, these events also appeared to occur less frequently and with a lower severity than seen with FAEs in psoriasis treatment, although it is not known if these observed reductions were due to the oral formulation of BG-12 or differences in the patient population. Infections were reported in similar proportions of BG-12 and placebo patients (34% in both groups).43
In Phase III studies, the overall incidence of AEs was similar across all treatment groups, highlighting the favorable safety profile of BG-12.45,46 AEs that occurred more frequently in patients receiving BG-12 than in patients receiving placebo included flushing, gastrointestinal events (eg, diarrhea, nausea, upper abdominal pain, abdominal pain, and vomiting), proteinuria, and pruritus.45,46 The incidences of flushing and gastrointestinal events were highest in the first month of the study and decreased thereafter. Overall, the incidence of AEs leading to discontinuation of the study drug was similar across the groups in both studies, although discontinuations due to flushing and overall gastrointestinal events occurred more frequently in patients who received BG-12 than in patients who received placebo.45,46 The incidence of infections was similar across the study groups with the most common infections detected being nasopharyngitis, upper respiratory tract infection, urinary tract infection, and influenza. No opportunistic infections were observed in the BG-12 groups, and no serious infections were reported in patients with lymphocyte counts of less than 0.5 × 109 per liter.45,46 There were no cases of malignancies associated with the study drug. The incidence of serious adverse events was similar across the study groups and there were no cases of renal failure classified by the investigator as serious AEs.45,46 As expected, the mean white cell count and lymphocyte count decreased in patients treated with BG-12. Usually, the mean white cell count and lymphocyte count decreased over the first 12 months and then plateaued, remaining within normal range. Overwhelming decreases of the white cell count and lymphocyte count (3.0 × 109/L and 0.5 × 109/L, respectively) occurred in 4%–10% of patients treated with BG-12. Decreases of the mean white cell count and lymphocyte count were not associated with an increase of infections.45,46 In both trials, researchers observed an increased incidence of elevations in liver aminotransferase levels, primarily between months 1 and 6. No such elevations of aminotransferase levels were concurrent with increases in bilirubin that were more than two times the upper limit of the normal range. There were no reports of hepatic failure.45,46
Recently, four cases of PML treated with Fumaderm® or self-compounded DMF for psoriasis have emerged.48,49 Only two cases48,49 may be classified as directly related to fumarates. In these cases, there was a clear violation of prescription information for patients with a severely reduced lymphocyte count or severely impaired immune system.50 Regarding the other two cases,51 one of the patients had a history of treatment with efalizumab, a monoclonal antibody against lymphocyte function-associated antigen 1 that has been withdrawn from the market because of the risk of developing PML,51 and the other patient had neurosarcoidosis, which primarily predisposes to PML.
The safety and tolerability of BG-12 has been studied within the clinical setting for up to 2 years, in randomized controlled studies, and will be further studied in the 5-year extension study of the ENDORSE Phase III clinical trials.52
During the ENDORSE phase extension, an interim safety result reported 14 malignancies (13 patients), diverse in type/location: six in patients who continued on DMF and eight in patients who switched to DMF. There were three deaths, none considered to be related to the study drug.52
Regarding pregnancy issues, the FDA classification for dimethyl fumarate is “pregnancy category C”.53
Future perspectives
On the basis of the results of the CONFIRM and DEFINE Phase III studies, the sponsors submitted regulatory approval to the FDA and European Medicines Agency in 2012. In March 2013, the FDA gave its approval to use of DMF as first-line therapy in patients affected by RRMS.53 Further, the Committee for Medicinal Products for Human Use adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal drug, intended for the treatment of adult patients with RRMS.54
Further information on long-term safety and efficacy of DMF treatment, including its suitability for use in pregnancy, will be obtained via implementation of respective registries for pregnancy and the ENDORSE study.
Conclusion
The therapeutic pipeline for MS is enriched with novel agents that have the potential to provide improved efficacy and ease of administration for patients. The development of drugs with easier administration, such as oral agents, would further promote adherence and could increase the number of patients with MS in treatment, reducing discomfort and inconvenience and making it easier for patients to adhere to their treatment regimen, thus improving efficacy. In view of these requests, the most recent therapeutic strategies have been developed with the intent to target specific steps in the pathogenesis of MS. Recent understanding of the pathogenic mechanisms underlying MS have helped to extend the research field to different new drugs suitable for treating MS. Among these new treatment options, BG-12 seems to be at least equally effective as, if not more effective than, established injectable therapies. Moreover, the ability of DMF to activate Nrf2 underpins the cytoprotective modality that further augments the natural antioxidant responses in MS tissues. The neuroprotective effect of Nrf2 is not yet targeted by other MS therapies and renders the compound a candidate for the treatment of progressive forms of MS. In addition to trials on secondary or primary progressive MS forms, the substance may also be an effective option for the treatment of neurodegenerative disease.
The most challenging aspect for physicians will be making their patients understand the need to balance the pros and cons of new oral treatments, where serious side effects may outweigh benefits in certain individuals. With gold standard agents such as IFN and GA, the main complaints are injection-site reactions and flu-like symptoms; compared with the higher stakes of new agents, these side effects seem to be more acceptable. Safety is likely to become the most important factor in prescribing MS drugs in future clinical practice, while certainly more research is also needed to compare escalation, different treatment combinations, and induction treatment strategies.
The safety profile of BG-12 is well known from its use in thousands of patients with psoriasis, and undoubtedly qualifies the drug for long-term use. Tolerability could, however, become an issue, as about 15% of patients initially experience diarrhea and up to 30% experience flushing. For this reason, a Phase IV postmarketing study is focusing purely on tolerability, with the primary objective of evaluating the effect of symptomatic therapies on gastrointestinal-related events reported by patients with relapsing forms of MS by initiating therapy with DMF in the clinical practice setting. Patients will have to be educated about AEs and be closely supported to avoid early discontinuation of treatment. BG-12 looks set to be very successful, as it combines a degree of clinical efficacy at least equal to, and probably higher than, established injectable therapies, with an excellent long-term safety profile.
The four PML cases recently reported, highlight the importance of close monitoring when patients undergo a new compound. More frequent complete blood count analyses may be advisable, especially after the onset of therapy, and an accurate evaluation of patients eligible to receive the drug. Similar to treatment with natalizumab, prior treatment with other immunosuppressants may have increased one of the patient’s risk of PML.
Depending on the exact licensing and on acceptance by both patients and neurologists, clinical routine will eventually determine where DMF treatment will be positioned in terms of MS therapy. On the basis of all available data, a widespread first-line use in treatment-naïve MS patients can be expected. In summary, it is anticipated that DMF will soon become an integral part of MS immunotherapy.
The choice between these new therapies will most likely be based on an overall assessment of efficacy, safety, tolerability, and adherence over the postmarketing period.
Disclosure
Carla Tortorella has received honoraria for speaking from Biogen Idec Inc, Sanofi-Aventis, Merck Serono, Novartis, Bayer Schering, Genzyme, and Teva Pharmaceutical. Claudio Gasperini has received fees as invited speaker from Bayer-Schering, Biogen Dompé, Merck Serono, and Novartis. The authors report no conflicts of interest in this work and received no payment in preparation of this manuscript.
References
Weinshenker BG. The natural history of multiple sclerosis. Neurol Clin. 1995;13:119–146. | |
Murray TJ. Diagnosis and treatment of multiple sclerosis. BMJ. 2006;332:525–527. | |
Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. | |
Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain. 1989;112(Pt 1):133–146. | |
Trojano M, Pellegrini F, Fuiani A, et al. New natural history of interferon-beta-treated relapsing multiple sclerosis. Ann Neurol. 2007;61(4):300–306. | |
Bergamaschi R, Quaglini S, Tavazzi E, et al. Immunomodulatory therapies delay disease progression in multiple sclerosis. Mult Scler. Epub May 31, 2012. | |
Batoulis H, Addicks K, Kuerten S. Emerging concepts in autoimmune encephalomyelitis beyond the CD4/T(H)1 paradigm. Ann Anat. 2010;192(4):179–193. | |
Frohman EM, Racke MK, Raine CS. Multiple sclerosis-the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. | |
Peterson LK, Fujinami RS. Inflammation, demyelination, neurodegeneration and neuroprotection in the pathogenesis of multiple sclerosis. J Neuroimmunol. 2007;184:37–44. | |
Witte ME, Bø L, Rodenburg RJ, et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol. 2009;219;193–204. | |
Hendriks JJA, Alblas J, Van der Pol SM, van Tol EA, Dijkstra CD, de Vries HE. Flavonoids influence monocytic GTPase activity and are protective in experimental allergic encephalitis. J Exp Med. 2004;200:1667–1672. | |
Vladimirova O, O’Connor J, Cahill A, Alder H, Butunoi C, Kalman B. Oxidative damage to DNA in plaques of MS brains. Mult Scler. 1998;4:413–418. | |
Miller DH, Thompson AJ, Morrissey SP, et al. High dose steroids in acute relapses of multiple sclerosis: MRI evidence for a possible mechanism of therapeutic effect. J Neurol Neurosurg Psychiatry. 1992;55(6):450–453. | |
Gallo P, Chiusole M, Sanzari M, et al. Effect of high-dose steroid therapy on T-cell subpopulations. A longitudinal study in MS patients. Acta Neurol Scand. 1994;89(2):95–101. | |
Goodkin DE. Interferon beta-1b. Lancet. 1994;344:1057–1060. | |
Schwid SR, Panitch HS. Full results of the Evidence of Interferon Dose-Response-European North American Comparative Efficacy (EVIDENCE) study: a multicenter, randomized, assessor-blinded comparison of low-dose weekly versus high-dose, high-frequency interferon beta-1a for relapsing multiple sclerosis. Clin Ther. 2007;29:2031–2048. | |
Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. | |
Hartung HP, Gonsett R, König N, et al; Mitoxantrone in Multiple Sclerosis Study Group (MIMS). Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind randomized multicenter trial. Lancet. 2002;360:2018–2025. | |
Martinelli V, Cocco E, Capra R, et al; Italian Mitoxantrone Group. Acute myeloid leukemia in Italian patients with multiple sclerosis treated with mitoxantrone. Neurology. 2011;77:1887–1895. | |
Polman CH, O’Connor PW, Havrdova E, et al; AFFIRM Investigators. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. | |
Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366:1870–1880. | |
Kappos L, Radue EW, O’Connor P, et al. FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. | |
Cohen JA, Barkhof F, Comi G, et al; TRANSFORMS Study Group. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. | |
Río J, Comabella M, Montalban X. Predicting responders to therapies for multiple sclerosis. Nat Rev Neurol. 2009;5:553–560. | |
Schweckendiek W. [Treatment of psoriasis vulgaris]. Med Monatsschr. 1959;13:103–104. German. | |
Mrowietz U, Christophers E, Altmeyer P. Treatment of severe psoriasis with fumaric acid esters: scientific background and guidelines for therapeutic use. The German Fumaric Acid Ester Consensus Conference. Br J Dermatol. 1999;141:424–429. | |
Mrowietz U, Rostami-Yazdi M, Neureither M, Reich K. [15 years of fumaderm: fumaric acid esters for the systemic treatment of moderately severe and severe psoriasis vulgaris]. J Dtsch Dermatol Ges. 2009; 7 Suppl 2:S3–S16. German. | |
Werdenberg D, Joshi R, Wolffram S, Merkle HP, Langguth P. Presystemic metabolism and intestinal absorption of antipsoriatic fumaric acid esters. Biopharm Drug Dispos. 2003;24:259–273. | |
Litjens NH, Burggraaf J, van Strijen E, et al. Pharmacokinetics of oral fumarates in healthy subjects. Br J Clin Pharmacol. 2004;58:429–432. | |
Nibbering PH, Thio B, Zomerdijk TP, Bezemer AC, Beijersbergen RL, van Furth R. Effects of monomethylfumarate on human granulocytes. J Invest Dermatol. 1993;101:37–42. | |
Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. | |
Scannevin RH, Chollate S, Jung MY, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341:274–284. | |
Lee DH, Gold R, Linker RA. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int J Mol Sci. 2012;13(9):11783–11803. | |
Altmeyer P, Hartwig R, Matthes U. [Efficacy and safety profile of fumaric acid esters in oral long-term therapy with severe treatment refractory psoriasis vulgaris. A study of 83 patients]. Hautarzt. 1996;47:190–196. German. | |
Treumer F, Zhu K, Gläser R, Mrowietz U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J Invest Dermatol. 2003;121:1383–1388. | |
de Jong R, Bezemer AC, Zomerdijk TP, van de Pouw-Kraan T, Ottenhoff TH, Nibbering PH. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur J Immunol. 1996;26:2067–2074. | |
Litjens NH, Rademaker M, Ravensbergen B, et al. Monomethylfumarate affects polarization of monocyte-derived dendritic cells resulting in down-regulated Th1 lymphocyte responses. Eur J Immunol. 2004;34:565–575. | |
Asadullah K, Schmid H, Friedrich M, et al. Influence of monomethylfumarate on monocytic cytokine formation – explanation for adverse and therapeutic effects in psoriasis? Arch Dermatol Res. 1997;289:623–630. | |
Vandermeeren M, Janssens S, Borgers M, Geysen J. Dimethylfumarate is an inhibitor of cytokine-induced E-selectin, VCAM-1, and ICAM-1 expression in human endothelial cells. Biochem Biophys Res Commun. 1997;234:19–23. | |
Mrowietz U, Asadullah K. Dimethylfumarate for psoriasis: more than a dietary curiosity. Trends Mol Med. 2005;11:43–48. | |
Schilling S, Goelz S, Linker R, Luehder F, Gold R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol. 2006;145:101–107. | |
Schimrigk S, Brune N, Hellwig K, et al. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baseline-controlled pilot study. Eur J Neurol. 2006;13:604–610. | |
Kappos L, Gold R, Miller DH, et al; BG-12 Phase IIb Study Investigators. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo controlled phase IIb study. Lancet. 2008;372:1463–1472. | |
MacManus DG, Miller DH, Kappos L, et al. BG-12 reduces evolution of new enhancing lesions to T1-hypointense lesions in patients with multiple sclerosis. J Neurol. 2011;258:449–455. | |
Gold R, Kappos L, Arnold DL, et al; DEFINE Study Investigators. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | |
Fox RJ, Miller DH, Phillips JT, et al; CONFIRM Study Investigators. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. | |
Hoefnagel JJ, Thio HB, Willemze R, Bouwes Bavinck JN. Long-term safety aspects of systemic therapy with fumaric acid esters in severe psoriasis. Br J Dermatol. 2003;149:363–369. | |
Ermis U, Weis J, Schulz JB. PML in a patient treated with fumaric acid. N Engl J Med. 2013;368:1657–1658. | |
van Oosten BW, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med. 2013;368:1658–1659. | |
Sweetser MT, Dawson KT, Bozic C. Manufacturer’s response to case reports of PML. N Engl J Med. 2013;368:1659–1661. | |
Schwab N, Ulzheimer JC, Fox RJ, et al. Fatal PML associated with efalizumab therapy: insights into integrin aLβ2 in JC virus control. Neurology. 2012;78:458–467. | |
Phillips JT, Fox R, Selmaj K, et al. Safety and tolerability of oral BG-12 (dimethyl fumarate) in relapsing–remitting multiple sclerosis (RRMS): interim results from ENDORSE Extension Study (P01.162). Neurology. 2013;80(Meeting Abstracts 1):P01.162. | |
US Food and Drug Administration. FDA approves new multiple sclerosis treatment: Tecfidera [press release]. Silver Spring, MD: US Food and Drug Administration; March 27, 2013 [updated March 28, 2013]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm345528.htm. Accessed August 19, 2013. | |
Tecfidera [webpage on the Internet]. London, UK: European Medicines Agency; 2013 [updated March 22, 2013]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002601/smops/Positive/human_smop_000498.jsp&mid=WC0b01ac058001d127. Accessed August 19, 2013. | |
O’Connor P, Wolinsky JS, Confavreux C et al; TEMSO Trial Group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;36514:1293–1303. | |
Vermersch P, Czlonkowska A, Grimaldi LM et al; for the TENERE Trial Group. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. November 21, 2013. [Epub ahead of print]. | |
Comi G, Jeffery D, Kappos L et al; ALLEGRO Study Group. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366(11):1000–1009. | |
Vollmer T, Soelberg Sorensen P, Arnold D on behalf of the BRAVO Study Group. A placebo-controlled and active comparator phase III trial (BRAVO) for relapsing-remitting multiple sclerosis. In: 5th Joint triennial congress of the European and Americas Committees for Treatment and Research in Multiple Sclerosis, Amsterdam, the Netherlands, October 19–22, 2011. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.