")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Pharmacokinetics, pharmacodynamics, and tolerability of verinurad, a selective uric acid reabsorption inhibitor, in healthy adult male subjects
Authors Shen Z, Gillen M, Miner JN , Bucci G, Wilson DM , Hall JW
Received 28 April 2017
Accepted for publication 30 May 2017
Published 7 July 2017 Volume 2017:11 Pages 2077—2086
DOI https://doi.org/10.2147/DDDT.S140658
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Zancong Shen,1 Michael Gillen,2 Jeffrey N Miner,1 Gail Bucci,1 David M Wilson,1 Jesse W Hall1
1Ardea Biosciences, Inc., San Diego, CA, 2AstraZeneca, Gaithersburg, MD, USA
Purpose: Verinurad (RDEA3170) is a selective uric acid reabsorption inhibitor in clinical development for the treatment of gout and asymptomatic hyperuricemia. The aim of this study was to evaluate the pharmacokinetics, pharmacodynamics, and tolerability of verinurad in healthy adult males.
Subjects and methods: This was a Phase I, randomized, double-blind, placebo-controlled, single and multiple ascending dose study. Panels of eight male subjects received a single oral dose of verinurad or placebo in either a fasted or fed state; panels of 10–12 male subjects received ascending doses of once-daily verinurad or placebo in a fasted state for 10 days. Serial blood and urine samples were assayed for verinurad and uric acid. Safety was assessed by adverse event (AE) reports, laboratory tests, vital signs, and electrocardiograms (ECGs).
Results: A total of 81 adult males completed the study. Following single doses of verinurad, maximum observed plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) increased in a dose-proportional manner; Cmax occurred at 0.5–0.75 hours and 1.25 hours in the fasted and fed states, respectively. Food decreased AUC by 23% and Cmax by 37%-53%. There was a modest accumulation of verinurad following multiple daily doses. Verinurad reduced serum urate levels by up to 62% (40 mg, single dose) and 61% (10 mg, multiple dose). The increase in urinary excretion of uric acid was greatest in the first 6 hours after dosing and was still evident ≥24 hours for verinurad doses ≥2 mg. Verinurad was well tolerated at all doses. No serious AEs, severe AEs, discontinuations due to AEs, or clinically significant laboratory or ECG abnormalities were reported.
Conclusion: Single and multiple doses of verinurad were well tolerated, absorption was rapid, and exposure was dose proportional. Verinurad increased urinary uric acid elimination and resulted in sustained reductions in serum urate. These data support further clinical evaluation of once-daily verinurad as a treatment for gout.
Keywords: fractional excretion of uric acid, selective uric acid reabsorption inhibitor, serum urate, urinary uric acid
Introduction
Chronic gout is a significant clinical problem, and many patients remain suboptimally treated with currently available therapies. Gout is caused by chronic hyperuricemia, where sustained elevation of serum urate (sUA) ≥6.8 mg/dL causes monosodium urate crystals to nucleate and aggregate in joints, tissues, and organs.1,2 The primary objective of chronic therapy for gout is to reduce sUA levels to <6.0 mg/dL or <5.0 mg/dL with severe disease,3,4 as reduction below the sUA saturation point leads over time to the dissolution of crystals and alleviation of gout symptoms.4,5
The first line of therapy for the treatment of gout is the xanthine oxidase inhibitors (XOIs), allopurinol, and febuxostat, which inhibit urate production to lower sUA.3,4 However, the majority of patients with gout fail to achieve the target goal of sUA <6 mg/dL using an XOI alone.6,7 In those circumstances, management guidelines recommend switching to another treatment or combining treatments with complementary mechanisms of action.3,4
Lesinurad, a selective uric acid reabsorption inhibitor, was recently approved in the USA and Europe in combination with an XOI for the treatment of hyperuricemia associated with gout in patients who fail to achieve target sUA levels on an XOI alone.8 Lesinurad inhibits the uric acid transporter URAT1, which is responsible for most reabsorption of urate from the renal tubule.9,10 By inhibiting URAT1, lesinurad increases the excretion of uric acid and lowers sUA.
Verinurad (RDEA3170) is a novel, orally active, selective URAT1 inhibitor in clinical development for the treatment of gout and asymptomatic hyperuricemia. Although verinurad has the same mechanism of action as lesinurad, it is structurally distinct from lesinurad.11 This first human study of verinurad evaluated the pharmacokinetic (PK), pharmacodynamic (PD), and tolerability following single and multiple ascending doses in healthy adult male subjects.
Subjects and methods
The study was conducted according to the Good Clinical Practice and the Declaration of Helsinki and with institutional review board (Welwyn Clinical Pharmacology Ethics Committee, Hatfield, UK) approval. Written informed consent was obtained from all subjects before starting the study.
Subjects
Healthy male subjects, aged ≥18 years and ≤55 years, with body weight ≥50 kg, body mass index ≥18 kg/m2 and ≤30 kg/m2, and sUA ≥4.5 mg/dL, were eligible for the study. All subjects were in good health with laboratory parameters within normal limits, with no clinically significant diseases, relevant abnormalities in vital signs, or history of cardiac abnormalities. Subjects who had a history of kidney stones were excluded.
Study design
RDEA3170-101 was a Phase I, randomized, double-blind, placebo-controlled, single and multiple ascending dose study. Panels of eight male subjects were randomized 3:1 to receive a single oral dose of verinurad (six subjects) or placebo (two subjects) in either a fasted (2 mg, 5 mg, 20 mg, or 40 mg dose) or fed (5 mg or 20 mg dose) state. Panels of 12 male subjects were planned to receive ascending doses of once-daily verinurad (nine subjects: 1 mg, 5 mg, or 10 mg doses) or placebo (three subjects) for 10 days in a fasted state. Verinurad or placebo was administered in tablet form for doses ≥5 mg or as an oral solution for the 1 mg and 2 mg doses.
Subjects were confined to the research facility from 2 days before dose administration until 72 hours after the last dose on day 4 (single dose) or day 13 (multiple doses). Prior to dosing, all subjects fasted overnight for at least 10 hours. Under fasting conditions, no food was allowed for at least 4 hours after the administration of study medication. Under fed conditions, subjects received study medication 30 minutes after completing a standard moderate fat (approximately 30%–40% of calories) and calorie (approximately 642–800 calories) breakfast. Tablets were administered with approximately 240 mL of water, while administration of the oral solution was followed by enough water to give a total volume of 240 mL. Additional water was allowed as desired except for 1 hour before and 1 hour after the administration of study medication. Subjects returned for post-study assessments on day 8 (±1 day) following a single dose and on day 17 (±1 day) following multiple doses.
In the single ascending dose regimen, serial PK plasma samples were collected on day 1 up to 72 hours post dose. Urine samples (total catch) for PK/PD were collected at every 6- or 12-hour interval prior to dosing and up to 72 hours post dose. In the multiple ascending dose regimen, serial PK plasma samples were collected over 24 hours post dose on days 1, 5, and 10; additional samples were collected prior to dosing (days 3, 4, 7, 8, and 9) and up to 72 hours following the last dose. Serial PD serum and urine samples were collected on day −1 prior to dosing and days 1, 5, and 10 for 24 hours post dose; additional samples were collected up to 72 hours following the last dose on day 10. Both plasma and urine samples were stored at −70°C until analyzed for verinurad concentrations by Ardea Biosciences, Inc. (San Diego, CA, USA). Serum samples were analyzed for sUA and creatinine and urine samples for uric acid and creatinine by Covance Central Laboratory Services (Indianapolis, IN, USA).
Analytical methods
For quantitative determination of verinurad, 0.040 mL aliquots of K3EDTA plasma or urine were extracted by protein precipitation (plasma) or dilution (urine) with acetonitrile containing [2H6]verinurad as internal standard. An aliquot of each plasma extract was further diluted with water, while urine samples were further diluted with injection solvent (acetonitrile:water, 1:2, v/v). Both plasma and urine samples were injected into an Agilent 1100 high-performance liquid chromatography (HPLC) system, where the analytes were chromatographically separated by gradient HPLC at a flow rate of 0.9 mL/min using a Zorbax-SB-C18 4.6 mm×50 mm, 3.5 μm pore column (Agilent Technologies, Santa Clara, CA, USA) and introduced into an API 5000 (plasma samples) or API 4000 (urine samples) triple quadrupole mass spectrometer (AB Sciex, Framingham, MA, USA). The mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile. When introduced into the mass spectrometer, the chromatographic effluent was positively ionized using Turboionspray mode and mass analysis was performed using selected reaction monitoring (unit/low resolution) for the precursor→product ion transitions of m/z 349.1→263.0 (verinurad) and 355.0→264.0 ([2H6]verinurad) with a 150 ms dwell for each transition (an additional dummy ion transition was added with a 30 ms dwell time to prevent cross talk). The methods for plasma and urine were validated according to the US Food and Drug Administration Bioanalytical Method Validation guidelines (2001)12 with a 0.100 ng/mL and 10.0 ng/mL lower limit of quantitation, respectively, and showed %Theoretical (% CV) during study analyses of 96.1%–103% (4.31%–6.98%) and 96.5%–104% (2.09%–4.69%), respectively.
PK/PD assessments
PK parameters were derived using WinNonlin Professional, version 5.2 (Pharsight Corporation, St Louis, MO, USA). Plasma PK parameters, calculated using noncompartmental analysis, included maximum observed plasma concentration (Cmax), time to Cmax (Tmax), and area under the plasma concentration–time curve (AUC) from 0 to 24 hours (AUC0–24) or to infinity (AUC0–∞), calculated using the linear trapezoidal rule. Other plasma PK parameters were apparent plasma terminal half-life (t1/2), apparent total clearance, corrected by bioavailability (CL/F), and volume of distribution at equilibrium, corrected by bioavailability (Vss/F). The fraction of the verinurad dose (% dose) excreted unchanged in urine from time 0 to 24 hours post dose (fe0–24) was also calculated.
The PD parameters were determined using SAS® software (SAS Institute Inc., Cary, NC, USA). The maximum percent change from baseline and the time of the maximum percent change in sUA were obtained directly from the concentration versus time data. The amount of uric acid recovered in urine (AeUR) was calculated as the urine concentration of uric acid multiplied by the urine volume. Renal clearance of uric acid (CLUR) was obtained from the amount of uric acid recovered in the urine (AeUR) divided by plasma urate AUC over the same time interval, while CLUR divided by creatinine clearance (CrCl) ×100 yielded the fractional excretion of uric acid (FEUA).
Safety assessment
Subjects were observed during the course of the study, and any adverse events (AEs) or remedial actions were documented in the clinical report form of the subject. Rheumatology Common Toxicity Criteria (RCTC) version 2.0 was used by the investigator to assess the severity of an AE. Clinical laboratory evaluations, supine blood pressure, pulse rate, respiratory rate, and oral body temperature were measured at specific times during the study. A physical examination and standard digital 12-lead electrocardiograms (ECGs) were recorded at screening and follow-up.
Statistical analyses
The sample size is typical for this type of study. No formal statistical power assessment was performed. Early data on PK variability suggested that this sample size is sufficient to provide reasonable characterization of the target PK parameters.
The PK and PD populations consisted of all subjects who received verinurad and had evaluable PK data and PD data, respectively. The safety population consisted of all subjects who received verinurad.
A fixed-effect model with food as a fixed effect was used to assess the effect of food on the PK of verinurad. The natural log-transformed PK parameters for the 5 mg and 20 mg single dosing groups under fed conditions were compared against those under fasted conditions. An estimate of the geometric mean ratios (GMRs) of the PK parameters with the corresponding 90% CI was generated.
Dose proportionality assessments were performed on Cmax and AUC for the single doses of 1 mg, 2 mg, 5 mg, 10 mg, 20 mg, and 40 mg using a power model:
|
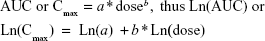
where the slope b measures the proportionality between dose and AUC or Cmax. When slope b is close to unity (1.0) with 95% CI within (0.8, 1.25), the relationship between dose and the PK parameter is concluded to be dose proportional for the dose range studied.
Results
Subject disposition and characteristics
A total of 81 adult males enrolled and completed the study. Mean age ranged from 26 years to 32 years and mean body mass index from 24.3 kg/m2 to 26.1 kg/m2 across the pooled placebo and pooled verinurad treatment groups (Table 1). Overall, the treatment groups appeared to be balanced with regard to demographics and baseline characteristics.
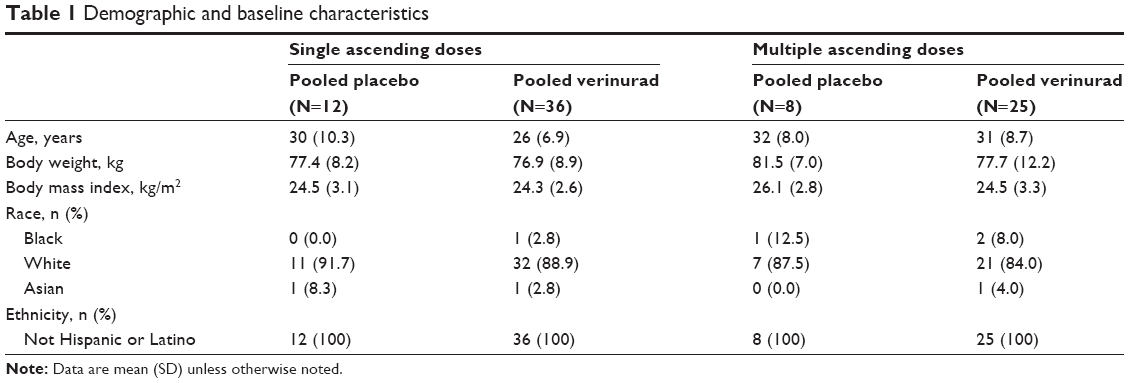 | Table 1 Demographic and baseline characteristics |
Verinurad PK
The verinurad plasma concentration–time profiles after single ascending doses in the fasted and fed states and after multiple ascending doses in the fasted state are shown in Figure 1. Absorption of a single dose was rapid, and exposure (Cmax and AUC) increased with dose up to the maximum dose tested (Table 2). Cmax was at 0.5–0.75 hours post dose in the fasted state, and was slightly delayed to 1.25 hours post dose in the fed state. The t1/2 was approximately 10–15 hours across doses. A moderate fat meal decreased Cmax approximately 37% with a 5 mg dose (GMR [90% CI]: 62.7% [46.8–84.1]) and approximately 53% with a 20 mg dose (GMR [90% CI]: 47.2% [27.5–81.1]). AUC0–∞ was approximately 23% lower with both a 5 mg dose (GMR [90% CI]: 76.8% [63.1–93.6]) and a 20 mg dose (GMR [90% CI]: 76.8% [57.5–103]).
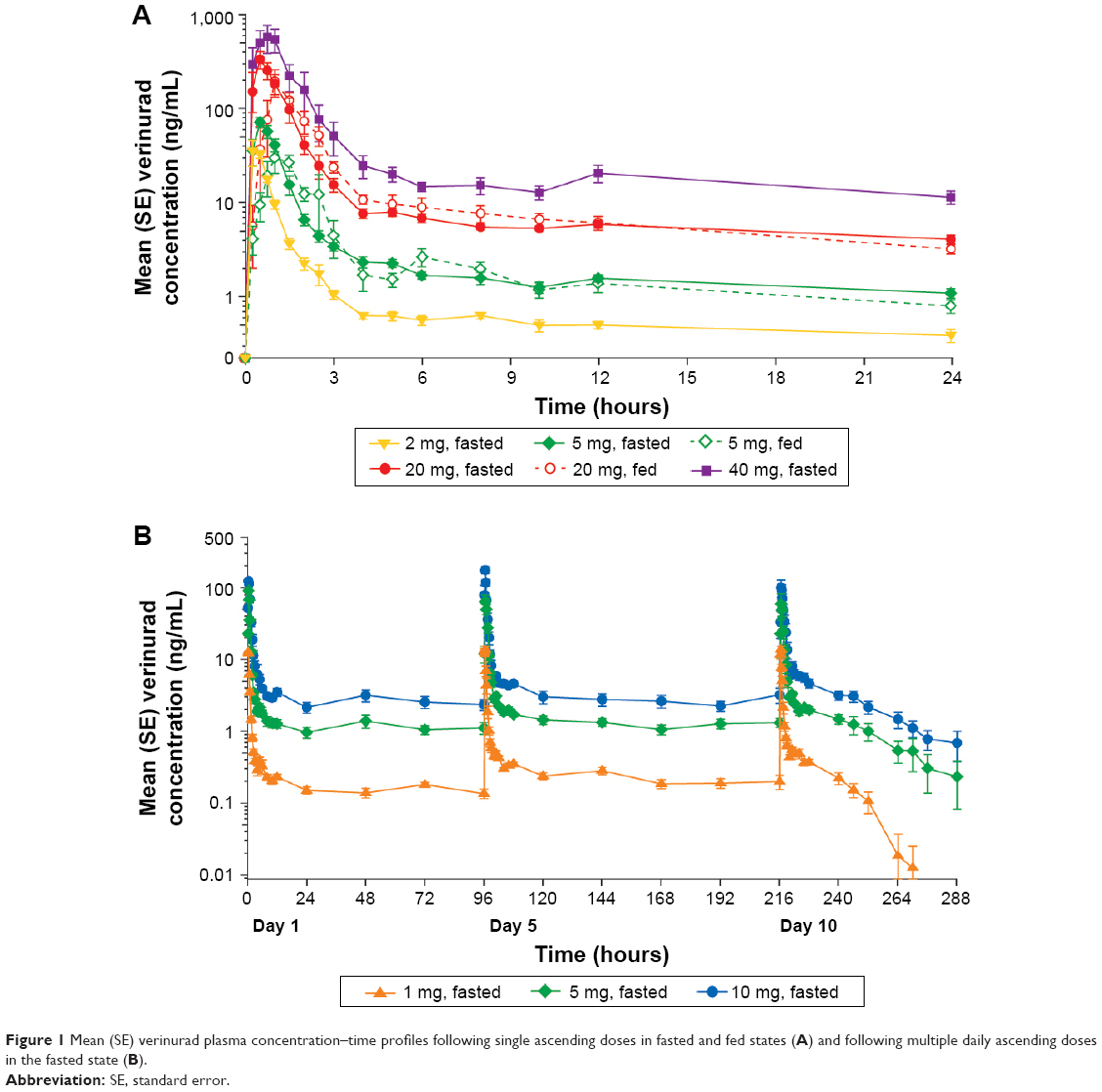 | Figure 1 Mean (SE) verinurad plasma concentration–time profiles following single ascending doses in fasted and fed states (A) and following multiple daily ascending doses in the fasted state (B). |
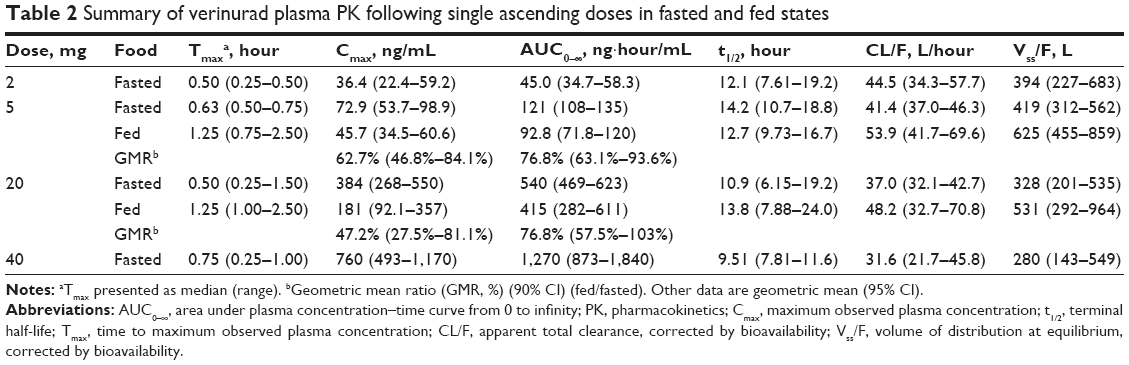 | Table 2 Summary of verinurad plasma PK following single ascending doses in fasted and fed states |
Following multiple daily doses, there was a modest increase in verinurad exposure (Table 3). Accumulation ratios ranged from 0.87 to 1.07 for Cmax and from 1.19 to 1.33 for AUC0–24. Urinary excretion of verinurad accounted for approximately 2%–3% of the administered dose (Table 3), suggesting that renal excretion is a minor elimination pathway for unchanged verinurad.
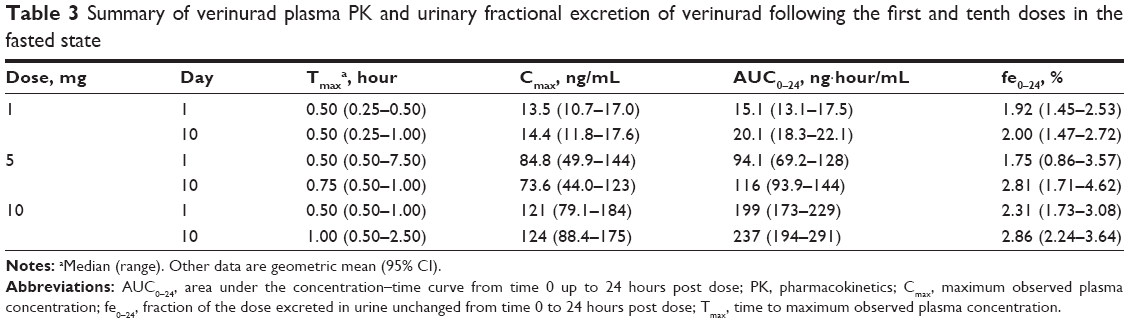 | Table 3 Summary of verinurad plasma PK and urinary fractional excretion of verinurad following the first and tenth doses in the fasted state |
The Cmax and AUC0–∞ of verinurad showed dose-proportional increases following a single oral dose in the 1–40 mg dose range under fasted conditions (Figure 2). The mean (95% CI) around the power model exponent for Cmax and AUC0–∞ was 1.06 (0.948-1.16) and 1.13 (1.07-1.19), respectively.
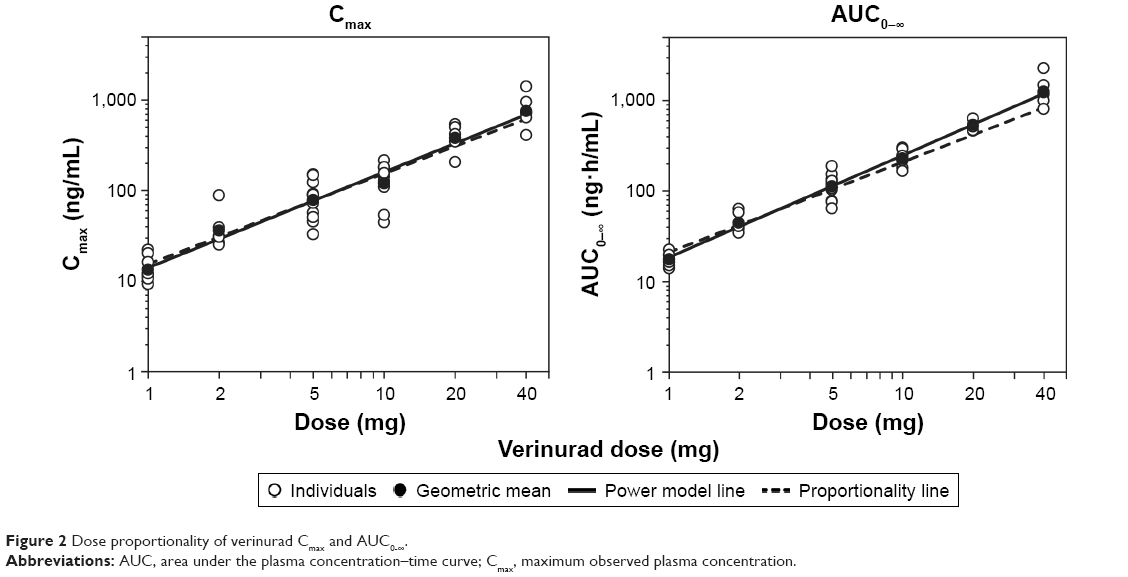 | Figure 2 Dose proportionality of verinurad Cmax and AUC0-∞. |
Verinurad PD
Reductions in sUA concentration occurred in a dose-dependent manner with both single and multiple doses of verinurad (Figure 3). Under fasted conditions, the maximum percent reduction following single-dose administration of verinurad 2 mg, 5 mg, 20 mg, or 40 mg was 16%, 24%, 48%, and 62%, respectively. The reductions at 24 hours post dose were 8%, 15%, 43%, and 58%, respectively, and apparent reductions were observed out to 72 hours post dose for doses ≥5 mg and out to 36 hours for the 2 mg dose. Food slightly enhanced the sUA lowering effect, increasing the maximum percent reduction to 29% with the 5 mg dose and to 53% with the 20 mg dose and the reductions at 24 hours to 17% and 46%, respectively.
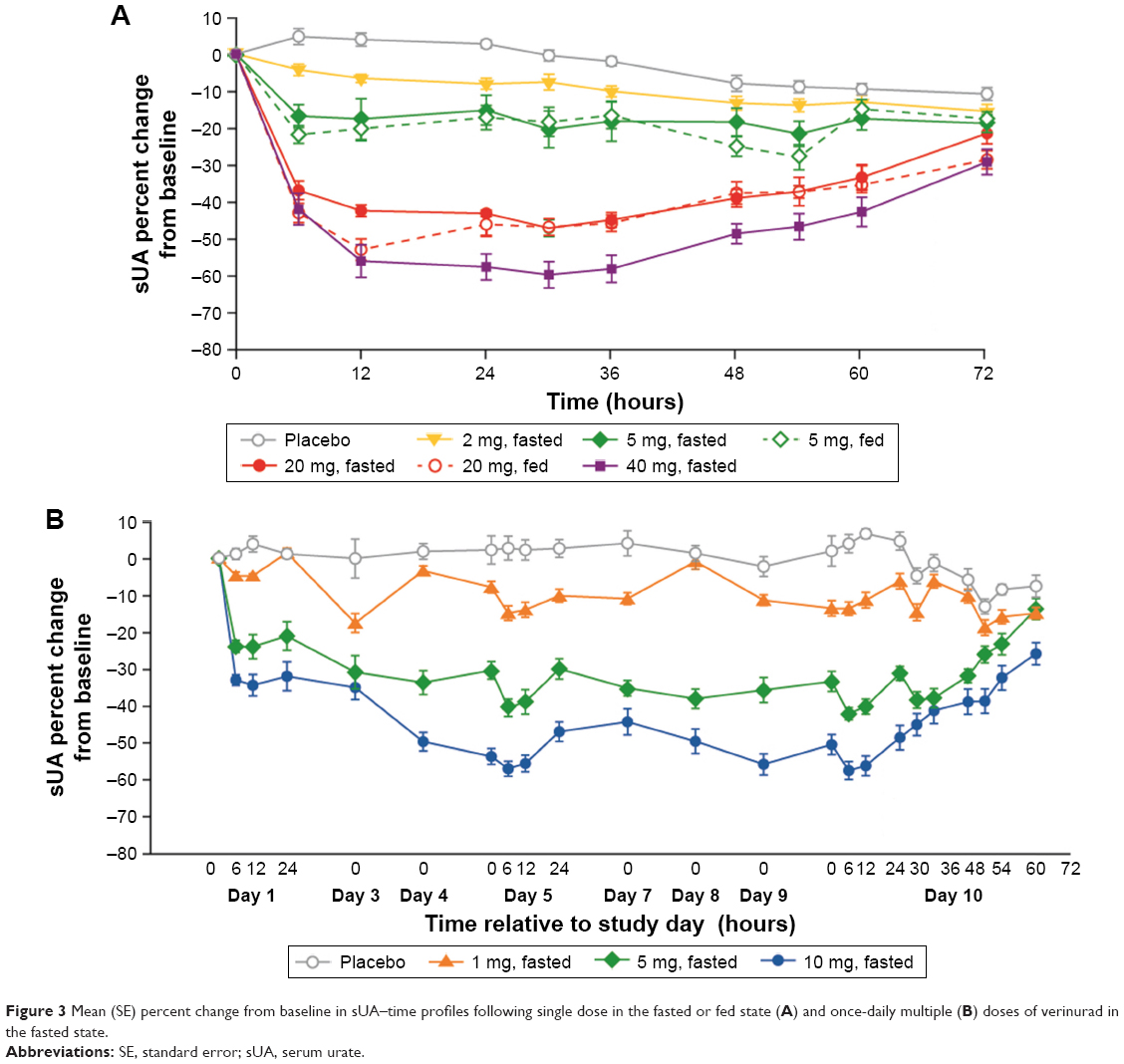 | Figure 3 Mean (SE) percent change from baseline in sUA–time profiles following single dose in the fasted or fed state (A) and once-daily multiple (B) doses of verinurad in the fasted state. |
Following multiple once-daily dosing for 10 days, the maximum percent reduction was 22%, 44%, and 61% for the verinurad 1 mg, 5 mg, and 10 mg doses, respectively, in the fasted state. Percent reductions in sUA at 24 hours post dose were 6%, 31%, and 49%, respectively.
Following single doses, AeUR (Figure 4A) and FEUA (Figure 4B) were increased following a single dose of verinurad ≥2 mg, with the largest increases occurring in the first 6 hours. Increased AeUR and FEUA were evident across a 24-hour interval with all doses. The mean percent change from baseline in AeUR over 24 hours was 45%, 51%, 109%, and 144% for the 2 mg, 5 mg, 20 mg, and 40 mg doses, respectively, administered in the fasted state, and 47% and 99% for the 5 mg and 20 mg doses, respectively, administered in the fed state. For FEUA, the mean percent change from baseline over 24 hours was 53%, 84%, 237%, and 385% for the 2 mg, 5 mg, 20 mg, and 40 mg doses, respectively, administered in the fasted state, and 82% and 283% for the 5 mg and 20 mg doses, respectively, administered in the fed state. Food does not appear to alter changes in AeUR or FEUA at the 5 and 20 mg doses of verinurad.
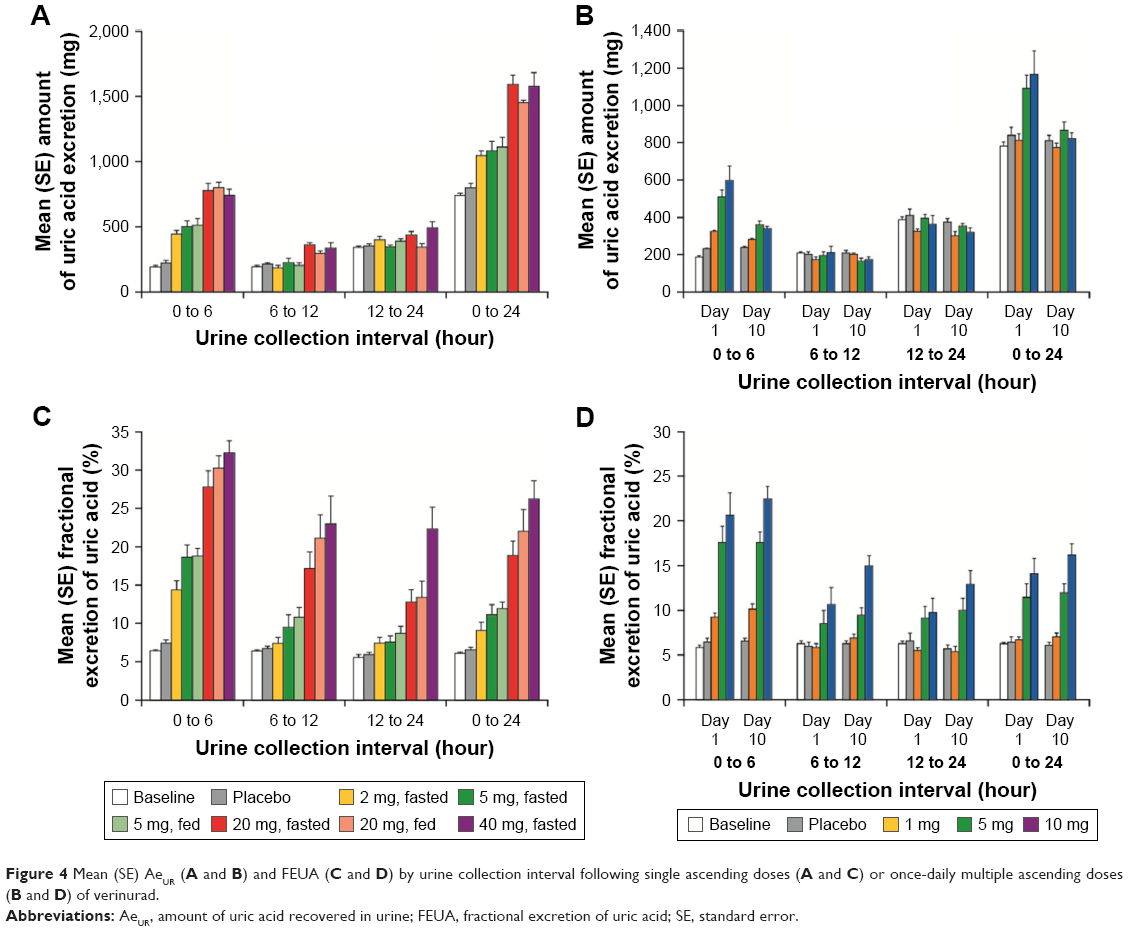 | Figure 4 Mean (SE) AeUR (A and B) and FEUA (C and D) by urine collection interval following single ascending doses (A and C) or once-daily multiple ascending doses (B and D) of verinurad. |
Following multiple once-daily doses on day 10, the increase in AeUR (Figure 4C) was diminished from baseline compared with the first dose on day 1. AeUR0–24 was generally unchanged, with mean percent change from baseline of 11%, 5%, and 8%, respectively, for the 1 mg, 5 mg, and 10 mg doses. The increase in FEUA0–24 (Figure 4D) was similar between day 1 and day 10. At the more active doses of 5 mg and 10 mg, mean percent changes from baseline were 72% and 120%, respectively.
Safety assessments
Verinurad was well tolerated at the doses studied. There were no deaths, serious AEs, or severe AEs reported during the study and no subject discontinued from the study due to an AE. Following single doses, one AE was reported in one (8.3%) subject in the pooled placebo group and 13 AEs in six (16.7%) subjects in the pooled verinurad group (Table 4). Seven of the 13 AEs occurred in two (33.3%) subjects in the verinurad 40 mg group, which included dizziness, the only AE that occurred in more than one subject. All but one AE was mild in severity. Constipation of moderate severity was reported for one subject in the verinurad 40 mg group. AEs possibly related to study drug were only reported by four (11.1%) subjects in the pooled verinurad treatment group and included dizziness.
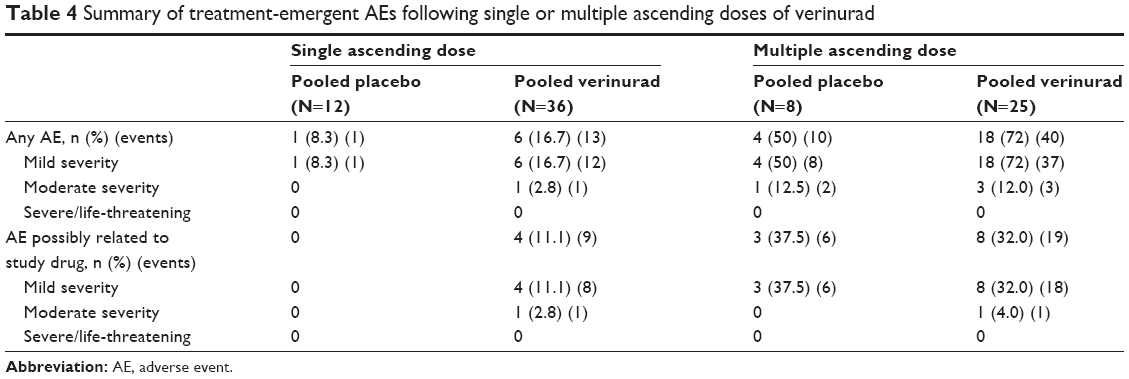 | Table 4 Summary of treatment-emergent AEs following single or multiple ascending doses of verinurad |
Following multiple dosing, ten AEs were reported in four (50.0%) subjects in the pooled placebo group and 40 AEs in 18 (72.0%) subjects in the pooled verinurad group. All but five AEs were mild in severity. One (12.5%) subject in the pooled placebo group reported two AEs of toothache with moderate severity. Three (12.0%) subjects in the pooled verinurad treatment group reported three AEs of moderate severity: fatigue, nausea, and nasopharyngitis. The most frequently reported AE in the pooled placebo group was headache (three [37.5%] subjects). All other AEs occurred in no more than one subject. The most common AEs reported in the pooled verinurad treatment group were headache and diarrhea (three [12.0%] subjects each). All other AEs occurred in no more than two subjects. The incidence of AEs possibly related to study drug was similar for the pooled placebo and pooled verinurad treatment groups (37.5% versus 32.0%, respectively) and included headache and diarrhea.
One subject in the single-dose 40 mg group and one subject in the multiple-dose 1 mg group experienced postdose increases in serum creatinine (sCr) >1.5× baseline value. The increase occurred 48 hours after the single dose and sCr returned to normal by the follow-up visit on day 7. The increase following multiple dosing occurred 36 hours after the dose on day 10 and sCr returned to normal at 48 hours post dose. Both events were not considered to be clinically significant by the investigator. Other individual creatinine changes were minimal and were seen in similar frequency in subjects receiving verinurad and placebo. None of the changes were reported as AEs.
Isolated changes in clinical safety laboratory parameters were observed in a small number of subjects, the majority of which were minor changes, and none were reported as AEs. There were no significant findings on physical examination or ECGs, and no treatment- or dose-related trends in vital signs.
Discussion
The results of this study in healthy subjects are consistent with published data on another selective uric acid reabsorption inhibitor, lesinurad, where inhibition of URAT1 in the proximal tubule of the kidney increases urinary uric acid excretion and lowers sUA.13
Verinurad was quickly absorbed following oral administration. Plasma exposure of verinurad increased in a dose-proportional manner over the range of doses examined. In the systemic circulation, verinurad appeared to be a high clearance drug (CL/F ranged ~30–50 L/h) with extensive extravascular distribution. Renal excretion of verinurad is limited to only ~2%–3% in the urine, suggesting that the majority of verinurad is likely cleared in the liver via biotransformation to metabolites. A moderate fat meal slightly delayed the absorption of a single verinurad dose and decreased verinurad AUC by 23% and Cmax by 37%–53%. Modest accumulation of verinurad was observed following once-daily multiple doses.
sUA concentrations decreased in a dose-dependent manner following single and multiple once-daily dosing of verinurad over the dose range of 1–40 mg. Food slightly enhanced the maximum sUA lowering effect by an additional 5%, despite a reduction in verinurad plasma exposure. A similar effect was observed with lesinurad,13 as food produced up to a 10% greater reduction in sUA. The sUA lowering effect of verinurad was evident for at least 24 hours post dose, although most of the increased excretion into urine occurred during the first 6 hours after dosing. The sustained lowering of sUA supports the viability of a once-daily dosing regimen.
Consistent with its mechanism of action as a selective reabsorption inhibitor of URAT1, verinurad increased AeUR and FEUA in a dose-dependent manner following single doses. The increase in excretion of uric acid led to the observed reductions in sUA. Following repeated dosing, AeUR was comparable to baseline values. This is due to the fact that the amount of uric acid in the serum is lower, and therefore there is less available to be filtered in the kidney and less available to be blocked in reabsorption. However, the continued inhibition of reabsorption maintains sUA at low concentrations.
Verinurad appeared to be well tolerated by healthy male subjects when given either as single or multiple daily doses in either the fed or the fasting state. The incidence of AEs was low after single doses and did not appear to be dose related. Daily oral doses of verinurad over 10 days had no apparent effect on the safety and laboratory parameters measured.
A possible limitation of this study is the exclusive inclusion of healthy male subjects, as the drug is intended for use in both male and female subjects with gout or asymptomatic hyperuricemia. Healthy male subjects are most often used in Phase I studies, with both male and female patients included in subsequent Phase II and III studies. In addition, interpretation of the effects of food on the PK of verinurad may be limited due to the use of different panels of subjects each with a small sample size.
Conclusion
The selective uric acid reabsorption inhibitor verinurad induces sustained, dose-dependent decreases in sUA concentration, with concomitant increases in urinary urate concentrations in healthy subjects. These data support further clinical evaluation of once-daily verinurad as a treatment for gout and asymptomatic hyperuricemia.
Acknowledgments
Funding was provided by Ardea Biosciences/AstraZeneca. Editorial support was provided by Tom Claus, PhD, of PAREXEL and funded by AstraZeneca. Ardea Biosciences, Inc. is a member of the AstraZeneca group. This article was presented at the 2016 ACR/ARHP Annual Meeting, November 11–16, 2016, Washington, DC, USA, as a poster presentation. The poster’s abstract was published in “Poster Abstracts” in Arthritis Rheumatol. 2016;68(suppl 10): http://acrabstracts.org/abstract/pharmacokinetics-pharmacodynamics-and-tolerability-of-verinurad-a-selective-uric-acid-reabsorption-inhibitor-in-healthy-adult-male-subjects/.
Disclosure
Zancong Shen, Jeffrey N Miner, Gail Bucci, David M Wilson, and Jesse W Hall are employees or former employees of Ardea Biosciences, Inc. Michael Gillen is an employee of AstraZeneca. The authors report no other conflicts of interest in this work.
References
Neogi T. Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452. | ||
Terkeltaub R, Edwards NL. Gout: Diagnosis and Management of Gouty Arthritis and Hyperuricemia. 3rd ed. West Islip, NY: Professional Communications, Inc.; 2013. | ||
Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431–1446. | ||
Richette P, Doherty M, Pascual E, et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. 2017;76(1):29–42. | ||
Doherty M, Jansen TL, Nuki G, et al. Gout: why is this curable disease so seldom cured? Ann Rheum Dis. 2012;71(11):1765–1770. | ||
Becker MA, Fitz-Patrick D, Choi HK, et al. An open-label, 6-month study of allopurinol safety in gout: The LASSO study. Semin Arthritis Rheum. 2015;45(2):174–183. | ||
Singh JA, Akhras KS, Shiozawa A. Comparative effectiveness of urate lowering with febuxostat versus allopurinol in gout: analyses from large U.S. managed care cohort. Arthritis Res Ther. 2015;17:120. | ||
Zurampic (lesinurad). AstraZeneca. Available from: http://www.azpicentral.com/zurampic/zurampic.pdf#page=1. Accessed January 6, 2017. | ||
Reginato AM, Mount DB, Yang I, Choi HK. The genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2012;8(10):610–621. | ||
So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120(6):1791–1799. | ||
Girardet J-L, Miner JN. Urate crystal deposition disease and gout-new therapies for an old problem. Annu Rep Med Chem. 2014;49:151–164. | ||
U.S. Department of Heath and Human Services Food and Drug Administration. Guidance for Industry. Bioanalytical Method Validation. U.S. Department of Heath and Human Services Food and Drug Administration. Available from: http://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf. Accessed January 16, 2017. | ||
Shen Z, Rowlings C, Kerr B, et al. Pharmacokinetics, pharmacodynamics, and safety of lesinurad, a selective uric acid reabsorption inhibitor, in healthy adult males. Drug Des Devel Ther. 2015;9:3423–3434. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.