")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Pharmacokinetics of conivaptan use in patients with severe hepatic impairment
Authors Marbury T , Fox J, Kaelin B , Pavliv L
Received 22 October 2016
Accepted for publication 5 January 2017
Published 13 February 2017 Volume 2017:11 Pages 373—382
DOI https://doi.org/10.2147/DDDT.S125459
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Thomas Marbury,1 Jerry Fox,2 Byron Kaelin,2 Leo Pavliv2
1Orlando Clinical Research Center, Inc., Orlando, FL, 2Department of Research and Development, Cumberland Pharmaceuticals, Inc., Nashville, TN, USA
Purpose: Conivaptan is an intravenous dual V1A/V2 vasopressin antagonist approved for the treatment of euvolemic and hypervolemic hyponatremia. Earlier studies showed that patients with moderate liver disease could be safely treated with conivaptan by reducing the dose by 50%, whereas patients with mild hepatic impairment needed no dose adjustment. The objective of this Phase 1, open-label study was to assess the pharmacokinetics, protein binding, and safety of 48 h of conivaptan infusion in individuals with severe hepatic impairment.
Patients and methods: Eight subjects with severe hepatic impairment (Child–Pugh score 10–15) and nine subjects with normal liver function were enrolled. Intravenous conivaptan (20 mg) was given as a 30 min loading dose on Day 1 followed by two consecutive 20 mg continuous infusions over 24 h each. Subjects were monitored for adverse events and changes in clinical laboratory parameters. Plasma and urine pharmacokinetic samples were collected at defined times. Subjects were followed through Study Day 5.
Results: Hepatically impaired individuals exhibited higher concentrations of plasma conivaptan throughout the treatment period. Overall exposure, as measured by area under the plasma conivaptan concentration-time curve from time zero through infinity (AUCINF), was ~60% higher in impaired individuals compared to those with normal liver function. Terminal elimination half-life was slightly longer in impaired subjects (12 h) as compared to normal subjects (9 h), and clearance was 65% higher in subjects with normal liver function, while urinary excretion was higher in impaired individuals. Albumin levels directly, and alkaline phosphatase inversely, correlated with conivaptan clearance.
Conclusion: A 20 mg conivaptan loading dose given >30 min followed by two daily infusions of 20 mg each was well tolerated by patients with severe hepatic impairment as monitored by adverse events and clinical laboratory values. Based on pharmacokinetic data, however, a 50% reduction in the conivaptan dose is recommended for patients with severe liver impairment.
Keywords: liver, hyponatremia, AVP antagonist, cirrhosis
Introduction
Hyponatremia (defined as serum sodium <135 mEq/L) is the most common electrolyte disorder encountered in clinical practice, estimated to be present in 15%–20% of hospital admissions.1 It is associated with significant morbidity and mortality.2 Some of the common etiologies that result in hyponatremia include liver cirrhosis, heart failure, syndrome of inappropriate antidiuretic hormone (SIADH), various drug treatments, and the presence of malignancies.3 Euvolemic hyponatremia is classified by a normal amount of total body sodium with increased body water, resulting in a dilution of sodium in the plasma. In hypervolemic hyponatremia, the total body sodium is increased; however, the total body water is elevated by an even greater amount relative to the sodium. This category is commonly found in liver cirrhosis and cardiac disease. Edema or ascites are often a presenting symptom. Other symptoms of general hyponatremia include fatigue, confusion, muscle weakness, seizures, and coma.
Treatment for hyponatremia is not standardized across the medical community. Often, no treatment is ordered despite the likelihood of treatment benefits to patient well-being and outcomes. Intervention for hospitalized hyponatremic patients has historically included fluid restriction, hypertonic saline, and demeclocycline. Vaprisol® (conivaptan HCl) (Cumberland Pharmaceuticals, Inc., Nashville, TN, USA) was first approved in 2005 and represented a more targeted treatment option. Conivaptan is a nonpeptide antagonist of the two main arginine vasopressin (AVP) receptors, V1A and V2. It is approved in the USA as an intravenous treatment for euvolemic and hypervolemic hyponatremia. Inhibition of V2 receptor activity in the kidney promotes aquaresis (solute-free excretion) of water, which increases the relative concentration of sodium in the plasma, contributing to the correction of hyponatremia. Conivaptan is extensively bound to human plasma proteins after intravenous infusion. After metabolism primarily by the CYP3A enzyme in the liver, 83% of conivaptan is eliminated through the feces.4 The use of the vaptan class of drugs represents a potentially better treatment option for hyponatremia, given the control over free water excretion without direct modification of electrolytes. In addition, in >90% of hyponatremia cases, the patients’ level of AVP is inappropriately elevated, beyond that which is expected in the presence of negative feedback mechanisms, further contributing to the low sodium levels.5 Therefore, an AVP antagonist is a targeted treatment for this syndrome.6
Conivaptan is approved for use in patients who are hospitalized. Conivaptan is predominantly metabolized by the liver so an understanding of the drug kinetics in a patient with impaired liver metabolism is important to prescribing treatment. This is especially of value given that liver cirrhosis is a contributing factor for hyponatremia.
A clinical study of the pharmacokinetics of conivaptan in patients with either mild or moderate hepatic impairment was completed and published.7 In that study, the degree of liver impairment was defined by each subject’s Child–Pugh score, which is based on the presence or absence of encephalopathy and ascites together with measures of serum bilirubin, albumin, and prothrombin time. Mild hepatic impairment was defined, as is the clinical convention, as a Child–Pugh score of 5–6, whereas a score of 7–9 indicated a patient with moderate impairment. Patients in either category were treated with a 30 min loading dose of 20 mg conivaptan IV followed by two continuous infusions, each totaling 20 mg, given over a 24 h period for a cumulative treatment period of 48 h. The medication was well tolerated in both populations; hence, this dose was chosen for the current study. Urine levels of conivaptan detected a small signal where excretion increased with the severity of hepatic impairment; this evaluation was continued in the current study. Results from the previous study concluded that the pharmacokinetics of conivaptan in patients with mild liver impairment were similar to those of healthy patients, whereas the overall exposure of conivaptan in patients with moderate dysfunction was ~80% higher than normal patients.6 This suggested the need for reduced dosing in patients with moderate liver impairment.
The current study allowed the first detailed review of conivaptan pharmacokinetics in patients with severe liver impairment, defined per clinical convention, as a Child–Pugh score of 10–15. Safety assessments were also recorded and analyzed.
Patients and methods
Study drug
Conivaptan HCl is represented by the molecular formula C32H26N4O2·HCl and has a molecular weight of 535.04 g/mol. It is an antagonist of AVP at both the V1A and V2 receptors in humans.
Patient population
The study recruited male and female subjects between 30 and 75 years of age, inclusive. A total of 16 subjects were intended to be enrolled into two groups of eight subjects each based on their baseline level of hepatic function. Group 1 patients had a Child–Pugh score from 10 to 15, thereby categorizing them with severe hepatic impairment. Subjects with normal liver function comprised Group 2 for comparison purposes; they were selected to approximately match the Group 1 subjects with respect to race, sex, age, and body mass index (BMI).
Eligible subjects in both groups weighed at least 45 kg and had a BMI between 18 and 40 kg/m2. Their baseline serum sodium was between 115 and 140 mEq/L, inclusive, and systolic blood pressure was under 140 mmHg while diastolic was >56 mmHg. Subjects were negative for human immunodeficiency virus (HIV)-1 and HIV-2 antibodies and must not have routinely consumed >15 units of alcohol per week. Female subjects could not be pregnant or nursing. All subjects of reproductive potential were required to use effective contraception during the study and for 28 days following. Due to the need to give multiple blood samples for pharmacokinetic assessments, subjects were excluded if they donated or lost >550 mL of blood, or received a transfusion of blood products, in the 8 weeks prior to screening. Conivaptan is metabolized by CYP3A4 but no patients were taking concomitant drugs that induce or inhibit CYP3A4 activity.
Specific to the Group 1 patients with severe hepatic impairment, additional eligibility criteria included a lack of any history of clinically relevant illness other than their liver disease. Biliary obstruction was excluded as an etiology of hepatic impairment. Other characteristics of liver disease that were exclusionary included the following: rapidly deteriorating liver function; hepatic encephalopathy greater than Grade 1 within the past 3 months; tense ascites; variceal bleeding within the past 6 months; severe portal hypertension; past shunt surgeries; or platelet counts <50,000×109/L and/or prothrombin time >18 s. Patients with Grade 1 hepatic encephalopathy were allowed to enroll if their condition had not deteriorated in the past 3 months.
Study design
This study was designed in adherence to Good Clinical Practice principles. The study was reviewed and approved by the following Institutional Review Boards (IRBs): Schulman Associates IRB, Cincinnati, OH, USA, and Aspire IRB, La Mesa, CA, USA. Written informed consent was fully obtained from all patients or their legally authorized representative prior to enrollment in the study. This open-label pharmacokinetic study also evaluated the safety and tolerability of a loading dose, followed by continuous infusions of intravenous conivaptan HCl (Vaprisol; Cumberland Pharmaceuticals, Inc., Nashville, TN, USA) in patients with severe hepatic impairment as defined by their baseline Child–Pugh score. Patients with normal liver function served as the comparator group. The study was executed at investigative sites in the USA (ClinicalTrials.gov, NCT01370148).
The dosing regimen mimicked that used in a previous pharmacokinetic study of patients with mild or moderate liver impairment.7 On the first treatment day, subjects received a 20 mg/100 mL premixed bag of intravenous conivaptan >30 min (loading dose) through a large arm vein. Immediately following the loading dose, an additional 20 mg/100 mL dose of conivaptan was infused >23.5 h in tandem with 300 mL of sterile water with 5% dextrose (D5W). At the beginning of the second day, another 20 mg/100 mL dose of conivaptan was infused, also with 300 mL D5W, >24 h and using a vein in the opposite arm from that which was used on Day 1.
Serial blood sampling for PK began immediately before the start of the loading dose and continued until 48 h after the end of the two-day continuous infusion. All samples were drawn from the arm opposite that used for the infusion on any given day. Samples were drawn at the following time points: up to 15 min prior to dose 1, within 2 min of the end of the loading dose, and at Study Hours 1, 1.5, 2, 4, 8, 12, 24, 36, and 48. After the end of the final infusion, additional blood samples were drawn for PK analysis at hours 1, 2, 7, 12, 24, 36, and 48 for a total of 96 h of PK evaluation. Samples were immediately cooled following blood draw and centrifuged at 4°C within 30 min of collection for 10 min at 2,000 ×g, and then stored at −70°C until analysis.
Urine samples for the assessment of the cumulative urinary excretion of conivaptan were collected for the following time intervals: 0–4, 4–8, 8–12, 12–24, 24–36, 36–48, 48–72, and 72–96 h after the start of the loading dose infusion. Samples were immediately frozen and stored at −70°C prior to analysis at the bioanalytical lab using a validated liquid chromatography tandem mass spectrometry (LC–MS/MS) assay.
The primary pharmacokinetic endpoints included area under the plasma conivaptan concentration–time curve from time zero through infinity (AUCINF), the plasma conivaptan concentration at hour 48 (C48) and the plasma conivaptan concentration at the end of the loading dose on Study Day 1 (CLD). Plasma concentrations and protein binding, assessed once at hour 48 immediately following the end of the study drug infusion, were determined using a validated LC–MS/MS assay. Additional pharmacokinetic variables were also calculated.
Safety assessments were monitored throughout the 4-day period, including physical exams, vital signs, hematology and biochemical analyses, urinalysis, electrocardiogram monitoring, and specific examinations for infusion site reactions (ISRs). An ISR was defined as any local event other than isolated pain, bleeding, or bruising at the site of conivaptan administration. Reactions were classified as 1 of 10 terms: erythema, pain/discomfort/tenderness, warmth, edema, phlebitis, venous induration, thrombophlebitis, venous thrombosis, infection, or cellulitis.
To control confounding factors related to the study objectives, concomitant treatments were restricted. Herbal supplements and vitamins were not allowed from Study Day 7 through Study Day 5. Medications used for the treatment of SIADH were discontinued at least 1 week prior to study start. Food and beverages containing alcohol, caffeine, grapefruit, or blood orange were disallowed from Study Day 2 through Study Day 5. Intake was tightly controlled and monitored, whereas subjects were confined to a Phase 1 research unit during the treatment period. Subjects were permitted to continue pre-existing, stable doses of other therapeutic medications at the discretion of the investigators, except those metabolized by CYP3A4.
Statistics
A total of 16 subjects were planned for enrollment. Subjects who received any exposure to study drug were included in the safety analysis set while those that produced adequate PK data to allow calculation of AUCINF, C48, and CLD were included in the PK analysis set. For subjects with missing data or protocol violations, inclusion in the PK analysis set was considered by the pharmacokineticist on a case-by-case basis. Continuous variables for baseline characteristics and safety were summarized descriptively. Laboratory values were summarized descriptively, as change from baseline.
Results
Disposition and baseline comparability
Seventeen subjects were enrolled at two clinical sites. One subject with normal liver function did not receive all doses of study drug due to an adverse event; their enrollment was replaced with another subject according to protocol directives. However, it was ultimately determined that the data from the withdrawn patient was applicable to both data sets. The subject received one dose of study drug in compliance with the protocol so was ultimately included in the safety analysis and for the PK analyses applicable for the single dose. Nine subjects with normal liver function and eight with severe hepatic impairment were enrolled. All 17 subjects completed the study observations.
Demographic characteristics of both treatment groups are summarized in Table 1. No significant differences were noted between normal and hepatic-impaired subjects.
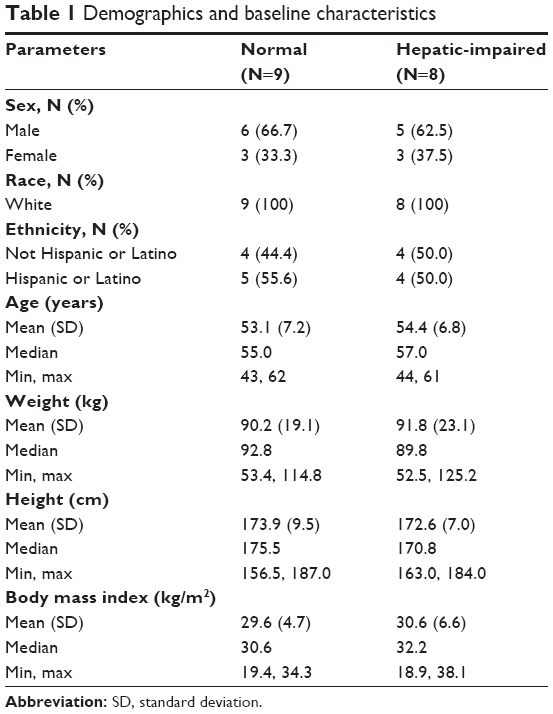 | Table 1 Demographics and baseline characteristics |
Baseline vital signs and serum biochemistries were obtained at screening for all enrollees (Table 2). Vital signs did not differ between the treatment groups. As expected, the means of many analytes differed with respect to baseline groups, reflecting the expected biochemical signs of those patients with liver disease: total bilirubin, alkaline phosphatase, amylase, and aspartate aminotransferase were elevated in the liver impairment group; albumin was decreased. The average Child–Pugh score for the hepatic-impaired group was 10.9±1.0 (mean ± standard deviation) with a range of 10–12. This complied with the eligibility requirement for patients with severe liver impairment (Child–Pugh score between 10 and 15).
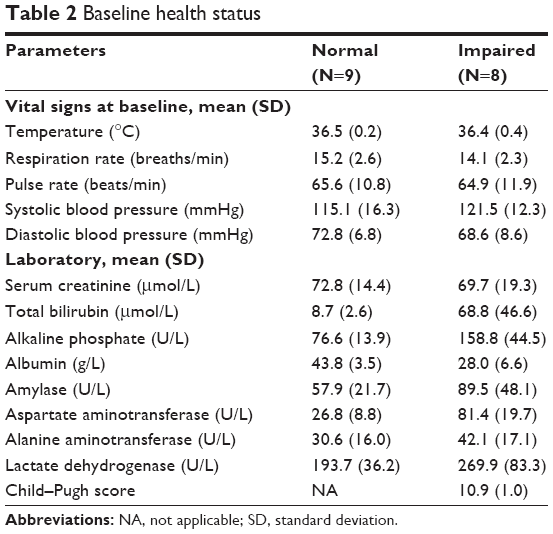 | Table 2 Baseline health status |
Pharmacokinetics
Plasma concentration
In both treatment groups, the maximum plasma conivaptan concentration was achieved at the end of the loading dose. Subjects with normal liver function attained a higher plasma concentration at the end of the loading dose (mean of 814 ng/mL) compared to hepatic-impaired patients (519 ng/mL) but, beginning 60 min later, normal patients maintained lower levels of conivaptan throughout the remainder of the infusion period and the 48 h following cessation of treatment (Table 3; Figure 1).
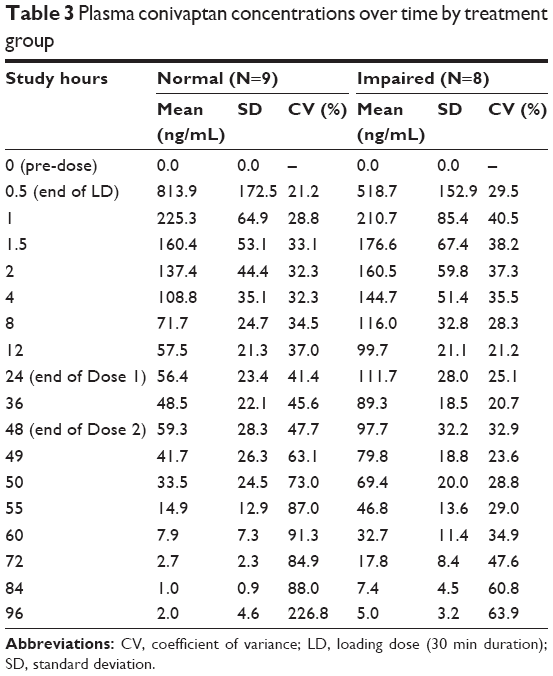 | Table 3 Plasma conivaptan concentrations over time by treatment group |
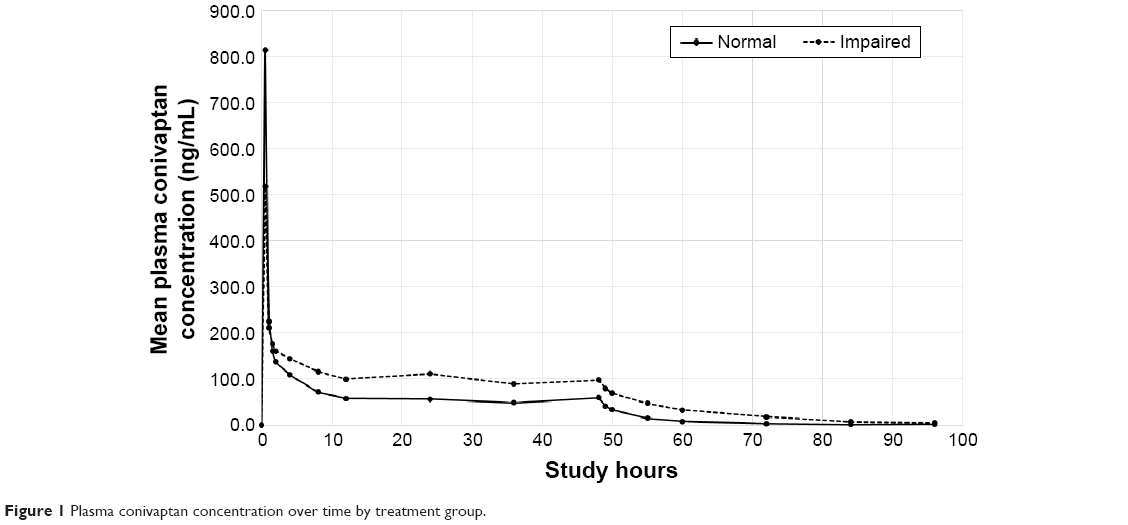 | Figure 1 Plasma conivaptan concentration over time by treatment group. |
Pharmacokinetic parameters
Total exposure, as measured by AUCINF, was ~60% higher in the impairment group compared with those with normal liver function (6,573.6±1,462.9 h·ng/mL vs 4,111.3±1,107.0 h·ng/mL). The C48 also showed higher levels in the impairment group (97.7±32.2 ng/mL vs 59.3±28.3 ng/mL). An inverse trend was observed for the CLD (813.9±172.5 in normal; 518.7±152.9 ng/mL in impaired).
The elimination half-life was longer in the hepatic impaired group (12.0±2.4 h vs 9.1±3.4 h). A full display of pharmacokinetic parameters is displayed in Table 4. The end of the loading dose represented the Tmax for both groups (Tmax=0.5 h). Clearance (CL) was ~65% lower in subjects with severe hepatic impairment.
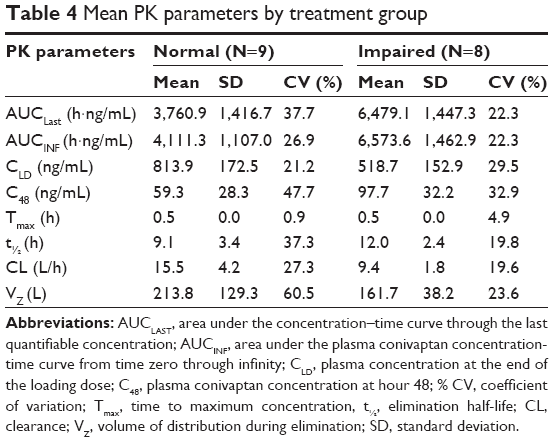 | Table 4 Mean PK parameters by treatment group |
Protein binding
Unbound concentrations were computed from total concentration and protein binding data. The mean unbound and total plasma conivaptan concentrations at 48 h were 0.29±0.17 and 38.49±25.98 ng/mL in the normal hepatic function group. In the severe hepatic impairment group, mean unbound conivaptan concentrations were 220% higher (0.93±0.43 ng/mL), and the total plasma conivaptan concentrations were 63% higher (62.88±27.63 ng/mL).
Relationship of baseline laboratory parameters to conivaptan clearance
The relationship between baseline bilirubin, albumin, and alkaline phosphatase, and the rate of conivaptan clearance were investigated by creating scatter plots (Figure 2) and also using linear regression models. Based on these results, no relationship was found between a subject’s baseline bilirubin value and their conivaptan clearance. Alternatively, high baseline albumin levels correlated with an increased rate of clearance, whereas high baseline alkaline phosphatase levels correlated with a decrease in the rate of conivaptan clearance.
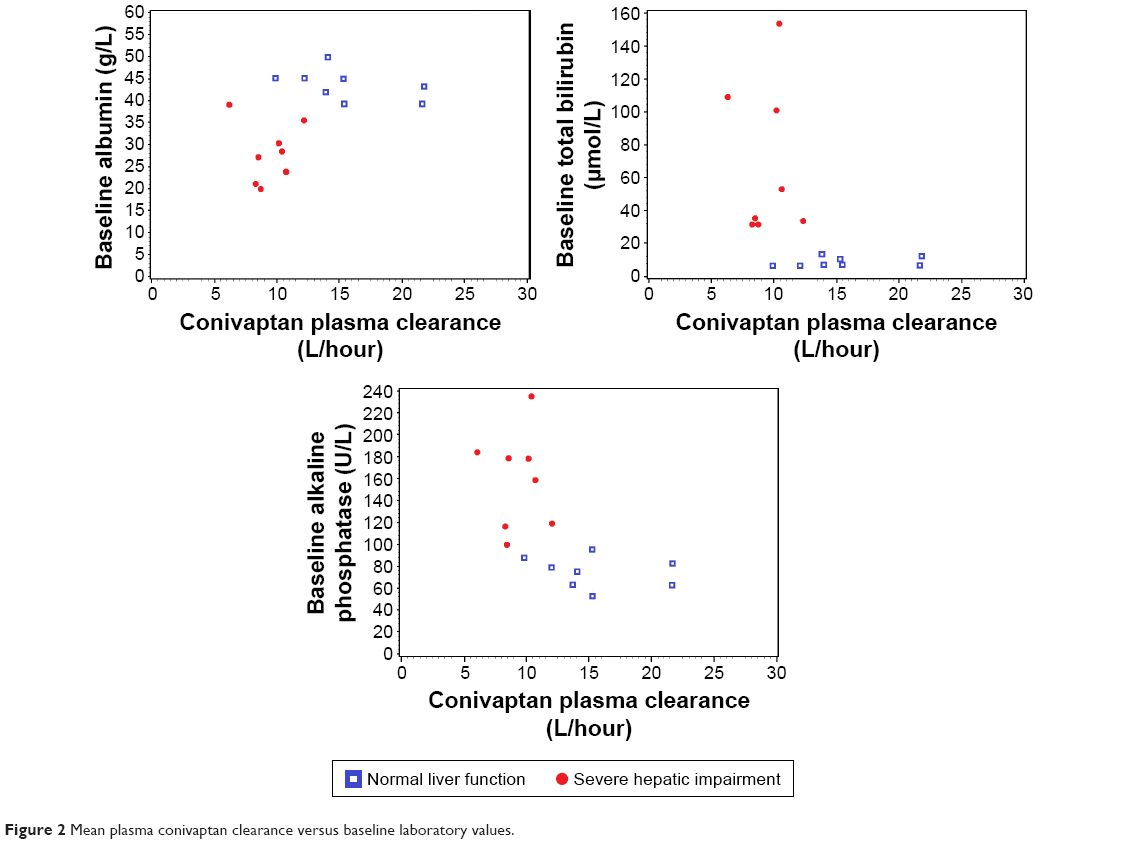 | Figure 2 Mean plasma conivaptan clearance versus baseline laboratory values. |
Urinary excretion of conivaptan
Mean urine conivaptan concentrations over time are presented in Table 5. Concentrations in the urine are higher at each respective time point for the hepatic impairment patients as compared to those without liver disease.
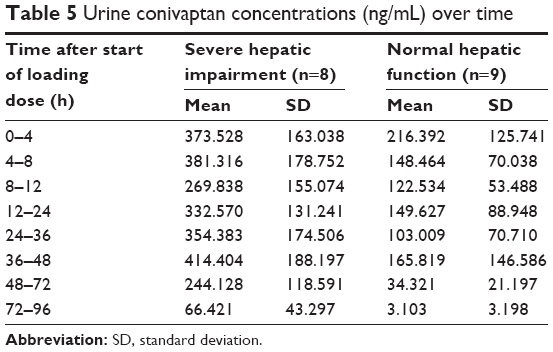 | Table 5 Urine conivaptan concentrations (ng/mL) over time |
Safety
There were no serious adverse events or deaths in this study. Adverse events reported during the study are displayed in Table 6. All subjects in the normal group and six of the eight subjects in the hepatic-impaired group experienced at least one AE. One subject in the normal group discontinued treatment with conivaptan due to an adverse event of increased serum sodium. This patient’s sodium levels increased by 14 mmol/L over a 2 h span, and then decreased spontaneously as the first 24 h infusion was ongoing. The most common AEs experienced by members of both treatment groups were related to ISRs with five of eight experiencing ISR in the severe hepatic impairment group and eight of nine in the normal hepatic function group. All other events were reported as unique occurrences. Only ISRs and increased serum sodium were deemed related to conivaptan treatment by the investigators.
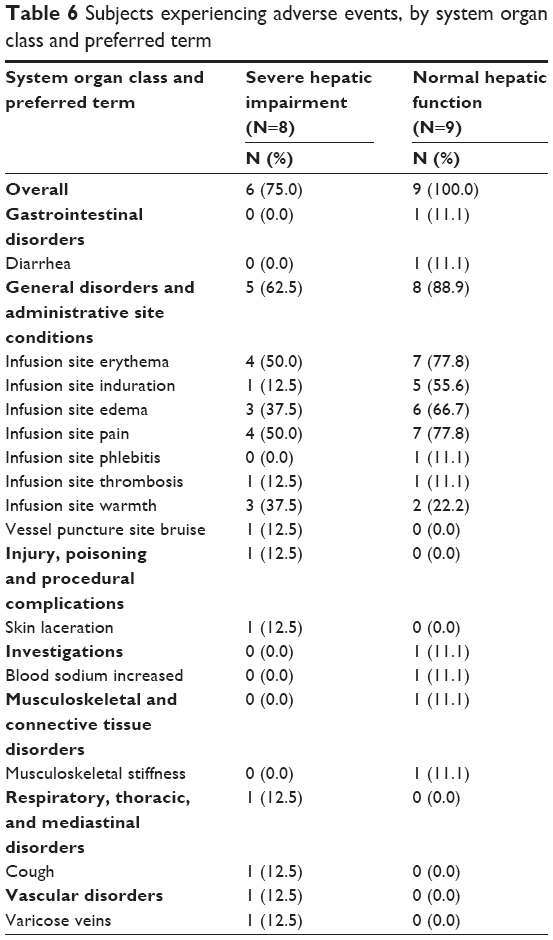 | Table 6 Subjects experiencing adverse events, by system organ class and preferred term |
No changes were seen in any vital sign trend over the course of the treatment period and follow-up. Hematology parameters were measured at baseline and Study Hour 72 (24 h after the cessation of Dose 2). No clinically significant changes were seen for any of the individuals enrolled in the study. Standard clinical biochemistry parameters were measured at baseline and every 12 h thereafter through Study Hour 96. No clinically significant changes were noted.
Discussion
With reported incidences of hyponatremia in hospitalized cirrhosis patients being >50%,8 the opportunity to treat hyponatremic patients inflicted with various degrees of hepatic impairment is high. There is much information and experience supporting the use of conivaptan as an efficacious and well-tolerated treatment in the general population,9,10 but it is important to understand any pharmacokinetic differences in populations with liver disease where low serum sodium is common.
Conivaptan is metabolized into low levels of active metabolites, which are not clinically relevant. However, 83% of conivaptan is ultimately cleared by the liver in healthy patients;4 therefore, it is ideal if the profile in patients with varying amounts of liver impairment is understood to allow meaningful treatment by the clinician. Patients with liver disease could potentially have lower drug levels due to low blood protein levels and increased volumes of distribution or they could have higher blood levels from decreased clearance due to impairment of the kidneys and liver. Therefore, it is important to study the pharmacokinetics of a drug in specific compromised patients. There are areas where the pharmacokinetic results from this relatively small study do not follow conventional findings so additional clinical data would be helpful. The variability of pharmacokinetic parameters in the normal population in this study actually exceeds that of the hepatic-impaired population, which is not typically expected. Also, while the peak concentration of conivaptan in impaired patients is lower than that in the normal group, possibly attributed to lower plasma protein binding, the volume of distribution is lower in the impaired group. This is contrary to expected results; however, conclusions can be drawn from this study to provide initial guidance in the treatment of euvolemic or hypervolemic hyponatremic patients with severe liver impairment.
Roy et al7 concluded that patients with mild hepatic impairment can generally be treated with conivaptan at the same dose as those without liver disease. However, those with moderate disease should reduce the dose by 50%. Therefore, a suggested dose in moderate hepatic impairment is 10 mg of intravenous conivaptan as a loading dose followed by 10 mg/day as an infusion for 2–4 days. The current study allowed the first known, controlled investigation of conivaptan in severe hepatic impairment.
Trends were found between baseline characteristics of patients with liver disease and the conivaptan clearance. Laboratory abnormalities that are considered expected in hepatic impairment include elevated alkaline phosphatase (ALKP) and total bilirubin, both of which are found in cases of low bile flow, and also low albumin levels. Comparing the subjects in this study that had severe hepatic impairment with those with normal liver function showed that patients presenting with either high ALKP or low albumin levels had a trend toward lower conivaptan clearance compared with patients without hepatic injury.
Specifically, in the case of high ALKP, it would be predicted to see lower conivaptan clearance because conivaptan is mainly eliminated through the liver, and bile flow is essential to this process. Elevated ALKP is typically seen in bile stasis. Alternatively, low albumin is a hallmark sign of liver impairment, so it can be postulated that patients with low albumin may be less efficient in clearing hepatic-metabolized drugs from their circulation.
The overall conivaptan exposure of patients with severe hepatic impairment in this study was higher than that in healthy subjects and similar to patients with moderate hepatic impairment that were studied in an earlier clinical trial.7 The mean AUCINF was 60% higher in patients with severe impairment (6,574±1,463 h·ng/mL) compared to those with normal liver function (4,111±1,107 h·ng/mL). In a more detailed review (Table 3), the maximum concentration in both populations occurred, as expected, at the end of the loading dose. Normal patients showed a 57% higher mean CLD of 814 ng/mL than impaired patients (519 ng/mL). However, after Study Hour 1, the mean concentration was higher in the severe hepatic function group from hour 1.5 through all time points in the remainder of the measurements. Concentrations also dropped more rapidly in the normal hepatic function group than in the severe hepatic impairment group with concentrations almost twice as high in the severe hepatic impairment group at the end of the 23.5 h infusion (111.7±28.0 ng/mL compared with 56.4±21.3 ng/mL). The difference was also observed at the end of the second 24 h infusion with the severe hepatic impairment group mean of 97.7±32.2 ng/mL compared with 59.3±28.3 ng/mL in the normal hepatic function group. Clearance was 65% higher in normal patients (15.5 L/h±4.2 SD) compared to patients with severe liver impairment (9.4 L/h±1.8 SD). Collectively, the pharmacokinetic study demonstrates the increased exposure and decreased plasma clearance of conivaptan in patients with severe liver impairment compared with normal patients. This finding supports the expectation that impaired liver function would result in slower metabolism and/or excretion of conivaptan by means of the elimination route of conivaptan.
Neither the current nor the past study evaluated efficacy since these were Phase I studies and patients did not have the indicated disease state. A goal of treating hyponatremia is a controlled rise in sodium levels, minimizing the risk of overly rapid correction.8 The current study supports a decreased initial dose, however, as a routine precaution, efficacy should be closely monitored in each individual patient in the clinical setting. Subjects in this study were dosed with the same regimen of conivaptan as those in a previous study of subjects with mild and moderate liver impairment.7 Also, criteria for the severity of liver impairment were based on the accepted Child–Pugh scores in both studies. The pharmacokinetic profile of subjects with severe hepatic impairment most closely resembles that of the subjects with moderate impairment in the previous study: Table 7 displays comparable mean values from the various groups in both studies.
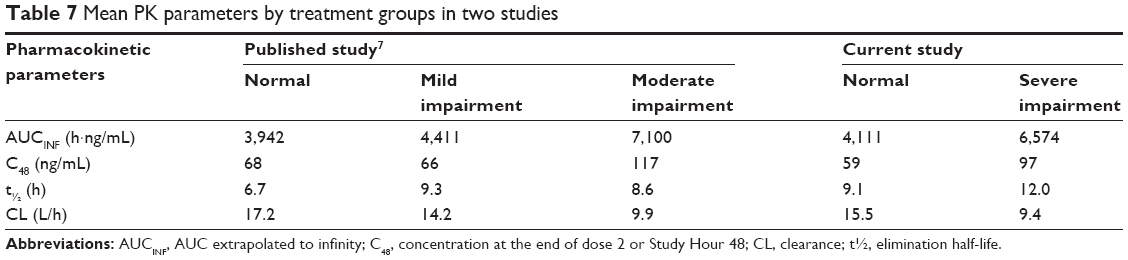 | Table 7 Mean PK parameters by treatment groups in two studies |
The fraction of bound conivaptan in both normal and impaired groups was >98%, indicating that the extent of conivaptan binding is not significantly affected by the degree of liver impairment. Levels of free plasma conivaptan in various patient populations seem to be more affected by the exposure of the drug than changes in protein binding.
The urine conivaptan measurements listed in Table 5 demonstrate the higher amount of conivaptan excreted in the urine of patients with severe hepatic impairment. The previous study presented urinary excretion by a different method but total amounts increased based on the level of hepatic impairment. Normal subjects excreted a mean of 957 μg through the observation period, whereas patients with mild and moderate hepatic impairment excreted 1,639 and 2,514 μg, respectively. Conivaptan is mainly excreted in the feces (83%); therefore, differences in urinary excretion are not expected to have a major effect on plasma and tissue levels. However, the higher urinary excretion in patients with increasing levels of liver impairment may suggest, in effect, a compensatory response where conivaptan clearance by the kidneys increases in patients that cannot effectively clear conivaptan hepatically. If supported, this finding may be a result of the higher plasma levels over time or the higher level of unbound conivaptan due to lower plasma protein levels, as seen in liver impaired patients. Additional study would be needed to investigate this and suggest if there is any clinical significance.
Conivaptan was generally well tolerated in patients with severe hepatic impairment. The incidence of adverse events in patients with liver disease was somewhat lower than those with normal liver function. One subject in the study needed to discontinue conivaptan treatment due to a large increase in serum sodium; this patient had normal liver function. Including ISRs, which are the most common adverse event generally seen with conivaptan, administration to patients with severe liver impairment does not seem to pose an increased safety concern as compared with administration to those with normal liver function. There were no deaths or serious adverse events on this study.
Conclusion
Treatment with intravenous conivaptan was well tolerated in a population of patients with severe hepatic impairment as defined by a baseline Child–Pugh score between 10 and 15, inclusive. The overall conivaptan exposure in the hepatic impaired group was 60% higher than that in a control group of patients with normal liver function. In addition, the pharmacokinetic data from the current population with severe hepatic impairment are similar to that of previous study in patients with moderate impairment, which concluded a reduction in dosing was advised for moderate impairment. Therefore, it is also advised to reduce conivaptan dosing in hyponatremic patients with severe hepatic impairment by 50%. Treatment is recommended to be initiated with a loading dose of 10 mg given intravenously >30 min, followed by infusions of 10 mg per day for 2–4 days. If serum sodium is not increasing at the desired rate, conivaptan may be titrated up to 20 mg per day.
Acknowledgments
This study was sponsored and funded by Astellas Pharma, Inc., and the study design and conduct was managed by the sponsor. Following the completion of enrollment, all commercial and development rights to Vaprisol were acquired by Cumberland Pharmaceuticals, Inc., who completed the analysis of the study and reviewed the manuscript.
Disclosure
Jerry Fox, Byron Kaelin, and Leo Pavliv are employees of Cumberland Pharmaceuticals, Inc. The authors report no conflicts of interest in this work.
References
Spasovski G, Vanholder R, Allolio B, et al; Hyponatraemia Guideline Development Group. Clinical practice guideline on diagnosis and treatment of hyponatraemia. Eur J of Endocrinology. 2014;170(3): G1–G47. | ||
Adrogué HJ. Consequences of inadequate management of hyponatremia. Am J Nephrol. 2005;25(3):240–249. | ||
Arieff AI. Management of hyponatraemia. BMJ. 1993;307(6899):305–308. | ||
Vaprisol® (conivaptan). Full prescribing information. Nashville, TN: Cumberland Pharmaceuticals Inc; 2014. | ||
Schrier RW, Bansal S. Diagnosis and management of hyponatremia in acute illness. Curr Opin Crit Care. 2008;14(6):627–634. | ||
Gross P. Treatment of hyponatremia. Intern Med. 2008;47(10):885–891. | ||
Roy MJ, Erdman KA, Abeyratne AT, et al. Pharmacokinetics of intravenous conivaptan in subjects with hepatic or renal impairment. Clin Pharmacokinet. 2013;52(5):385–395. | ||
Angeli P, Wong F, Watson H, Ginès P; CAPPS Investigators. Hyponatremia in cirrhosis: results of a patient population survey. Hepatology. 2006;44(6):1535–1542. | ||
Zeltser D, Rosansky S, van Rensvurg H, Verbalis JG, Smith N; Conivaptan Study Group. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447–457. | ||
Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159–168. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.