Back to Journals » Drug Design, Development and Therapy » Volume 8
Pharmacokinetics and tolerability of the new second-generation nonnucleoside reverse- transcriptase inhibitor KM-023 in healthy subjects
Authors Cha Y, Lim KS, Park MK ![]() , Schneider S, Bray B, Kang M, Chung J
, Schneider S, Bray B, Kang M, Chung J ![]() , Yoon SH, Cho J
, Yoon SH, Cho J ![]() , Yu K
, Yu K ![]()
Received 6 April 2014
Accepted for publication 6 May 2014
Published 26 September 2014 Volume 2014:8 Pages 1613—1619
DOI https://doi.org/10.2147/DDDT.S65596
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Yu-Jung Cha,1,* Kyoung Soo Lim,2,* Min-Kyu Park,1 Stephen Schneider,3 Brian Bray,3 Myung-Chol Kang,3 Jae-Yong Chung,1 Seo Hyun Yoon,1 Joo-Youn Cho,1 Kyung-Sang Yu1
1Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, Seoul, South Korea; 2Department of Clinical Pharmacology and Therapeutics, CHA University School of Medicine and CHA Bundang Medical Center, Seongnam, South Korea; 3Kainos Medicine USA Inc., Morrisville, NC, USA
*These authors contributed equally to this work
Background: KM-023 is a new second-generation nonnucleoside reverse-transcriptase inhibitor that is under development for the treatment of human immunodeficiency virus (HIV) type 1 infection.
Objective: This study determined KM-023 tolerability and pharmacokinetic characteristics in healthy subjects.
Materials and methods: A randomized, double-blinded, placebo-controlled, dose-escalation study was conducted in 80 healthy South Korean male volunteers. The subjects were allocated to single- or multiple-dose (once daily for 7 days) groups that received 75, 150, 300, or 600 mg drug or placebo in a 4:1 ratio. Safety and pharmacokinetic assessments were performed during the study. Plasma and urine concentrations were quantified using liquid chromatography–tandem mass spectrometry.
Results: The average maximum concentration (Cmax) and area under the concentration–time curve from time 0 to infinity (AUC∞) values of KM-023 for the 75–600 mg doses in the single-dose study ranged from 440.2 ng/mL to 1,245.4 ng/mL and 11,142.4 ng • h/mL to 33,705.6 ng • h/mL, respectively. Values of the mean Cmax at a steady state and AUC within the dosing interval ranged from 385.1 ng/mL to 1,096.7 ng/mL and 3,698.9 ng • h/mL to 10,232.6 ng • h/mL, respectively, following 75–600 mg doses in the multiple-dose study. Dose proportionality was not observed for KM-023. KM-023 showed a 0.6-fold accumulation after multiple doses in the 600 mg dose group. The mean half-life values ranged between 20.7 and 31.2 hours. KM-023 was generally well tolerated without serious adverse events.
Conclusion: KM-023 demonstrated dose- and time-dependent nonlinear pharmacokinetic characteristics after single or multiple doses over a dose range (75–600 mg) in healthy subjects. KM-023 showed favorable tolerability in this study. This Phase I clinical trial information can be used to design further clinical studies appropriately to evaluate KM-023 in patients with HIV-1 infection.
Keywords: KM-023, HIV-1, nonnucleoside reverse-transcriptase inhibitor, pharmacokinetics, tolerability, healthy subjects
Introduction
Current practice guidelines recommend a combination of two nucleoside reverse-transcriptase inhibitors (NRTIs) plus a non-NRTI (NNRTI), such as efavirenz or a ritonavir-boosted protease inhibitor, as first-line therapy for the treatment of human immunodeficiency virus (HIV) type 1 infection,1,2 which has reduced the mortality and morbidity of HIV-1-infected patients.3,4 Among these drugs, the use of efavirenz with two NRTIs as an initial therapy regimen resulted in higher overall virological efficacy compared with lopinavir–ritonavir plus two NRTIs in Phase III clinical trials.1,5
Despite its advantages, the use of efavirenz has been limited by the emergence of both central nervous system toxicities and viral resistance.6–9 Second-generation NNRTIs, such as rilpivirine and etravirine, were developed to overcome these shortcomings. These drugs showed improved viral-resistance profiles with better efficacy and less toxicity compared with first-generation NNRTIs.9,10
KM-023 (3-[3-ethyl-5-isopropyl-2,6-dioxo-1,2,3,6-tetrahydro-pyrimidine-4-carbonyl]-5-methyl benzonitrile) has been under development as a novel second-generation NNRTI (Kainos Medicine USA Inc., Morrisville, NC, USA). KM-023 has displayed favorable safety profiles and pharmacological characteristics in preclinical studies. KM-023 exhibited antiviral activity in the low-nanomolar range against laboratory strains and clinical HIV-1 isolates, and it showed activity against K103N (unpublished data), which is the major efavirenz-resistant mutation.11 There were no significant findings in in vivo safety pharmacology studies conducted in mice and dogs. The oral bioavailability and terminal elimination half-life (t1/2) of KM-023 were 64%–69% and 5.09–5.75 hours in preclinical studies in dogs and monkeys, respectively. KM-023 moderately induced cytochrome P450 (CYP)-3A4 in in vitro studies using human hepatocytes (unpublished data). This study assessed the tolerability and pharmacokinetic characteristics of KM-023 following single or multiple oral doses in healthy subjects.
Materials and methods
Subjects
Eligibility criteria included adults aged 20–45 years with a body mass index of 18.5–25 kg/m2 who had no clinically significant abnormalities upon clinical laboratory evaluation or physical examination. Volunteers who had clinically significant medical conditions or clinical laboratory test abnormalities, history of drug addiction or alcoholism, or positive serological tests (eg, hepatitis B surface antigen, hepatitis C antibodies, or HIV) were excluded. Written informed consent was obtained from each subject prior to participation. Subjects who discontinued prior to completing the study were replaced.
The Seoul National University Hospital Institutional Review Board approved the study protocol. This study was performed according to the principles described in the Declaration of Helsinki and Good Clinical Practice (ClinicalTrials.gov identifier: NCT01348516).12
Study design
This study was conducted as a first-in-human, randomized, double-blind, placebo-controlled, single- or multiple-dose, dose-escalation clinical trial. Eligible subjects in the single-dose study were randomly allocated into one of the following KM-023 dose groups: 75 mg, 150 mg, 300 mg, or 600 mg (each n=10). These subjects also randomly received a single oral dose of KM-023 or placebo at a ratio of 4:1.
Subjects in the multiple-dose study were also randomly assigned to one of four dose groups, which were the same as the single-dose study in doses and number of subjects. Subjects received KM-023 or placebo once daily for 7 days.
Determining KM-023 concentration
Blood and urine samples were collected to determine the pharmacokinetic characteristics of KM-023. Blood samples (5 mL) in the single-dose study were collected in heparinized tubes predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 60, and 72 hours after dosing to determine the KM-023 concentrations. Urine samples (20 mL) were collected at 0–4, 4–8, 8–12, 12–24, 24–48, and 48–72 hours after dosing.
Blood samples (5 mL) in the multiple-dose study were collected in heparinized tubes predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 16 hours after dosing on days 1 and 7. Samples for trough KM-023 concentration were obtained before dosing on days 2–6. Additional samples were collected at 0 hours on days 8, 9, and 10. Urine samples were collected over time intervals of 0–4, 4–8, 8–12, and 12–24 hours after dosing on days 1 and 7.
Blood samples were centrifuged at 2,000 g for 10 minutes at 4°C. Separated plasma samples were stored at −70°C prior to analysis. Urine volume was measured, and 20 mL urine samples were stored at −70°C before analysis. KM-023 concentrations in plasma and urine were quantified using a validated liquid chromatography–tandem mass spectrometry method. The internal standard was GS-9503 (3-[3-butyl-5-isopropyl-2,6-dioxo-1,2,3,6-tetrahydro-pyrimidine-4-carbonyl]-5-methyl benzonitrile). Mass spectrometric detection was conducted using multiple-reaction monitoring transition at mass-to-charge ratios 326.3:298.2 for KM-023 and 354.3:298.3 for GS-9503 in both plasma- and urine-sample analyses. The assays were validated over a range of 0.5–500 ng/mL (plasma) and 5–5,000 ng/mL (urine). Assay accuracy and precision ranged from 94.2% to 97.1% and ≤7.5% in plasma and 88.6% to 97.0% and ≤3.6% in urine, respectively.
Pharmacokinetic analysis
Individual pharmacokinetic parameters were assessed using noncompartmental analysis with Phoenix® software (version 1.0; Certara, St Louis, MO, USA). Maximal plasma concentrations (Cmax) and time to Cmax (Tmax) were obtained from the observed values. The area under the time-versus-concentration curve (AUC) from time 0 to the last available measurement (AUClast) and the AUC within a dosing interval (AUCτ) were calculated using the linear up/log down method. The AUC from time 0 to infinity (AUC∞) was the sum of AUClast and Ct/λz, where Ct is the last measurable plasma concentration and λz the elimination-rate constant, as determined by linear regression analysis associated with the terminal (log-linear) portion of the plasma concentration–time curve. The t1/2 was determined as ln2/λz. The apparent oral clearance (CL/F) was calculated as dose/AUC∞. The fraction of unchanged drug excreted in the urine (fe) was estimated as the amount of unchanged drug excreted in the urine (Ae) over the dose. The accumulation index was estimated by calculating AUC144–168h/AUC0–24h.
Tolerability assessments
Tolerability was evaluated by monitoring adverse events (AEs) throughout the study. Physical examination, vital sign measurements, 12-lead electrocardiography, computerized impedance cardiography, and clinical laboratory tests were performed periodically before and after dosing during the study period.
Statistical analysis
Statistical analysis was performed using SPSS software (version 17.0; SPSS, Chicago, IL, USA). All safety data and pharmacokinetic parameters were summarized as treatment and dose using descriptive statistics. Dose proportionality was assessed using the Kruskal–Wallis test on the dose-normalized Cmax and AUC values, and linear regression on the log-transformed Cmax and AUC values. Dose proportionality was also evaluated concerning whether the 95% confidence interval (CI) for the log-transformed Cmax and AUC values included 1.0 in the power model. Repeated-measures analysis of variance (RM-ANOVA) was used to investigate differences in the plasma trough concentrations in each treatment group to identify whether steady-state conditions were achieved by day 7. A P-value <0.05 was considered statistically significant.
Results
Study population
A total of 41 subjects were enrolled in the single-dose study. One subject who received 75 mg KM-023 discontinued the study because of a withdrawal of informed consent. A total of 41 subjects were enrolled in the multiple-dose study. Of these subjects, one subject in the placebo group discontinued due to an influenza infection. No significant differences were observed in the demographic characteristics between the treatment groups (Table 1).
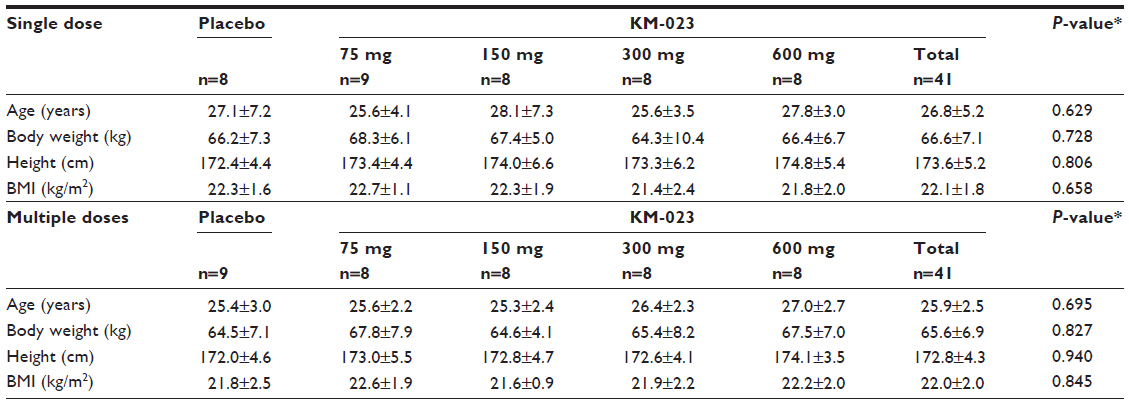 | Table 1 Demographic characteristics of participants |
Pharmacokinetics
The average KM-023 plasma concentration–time profiles for each treatment group after single or multiple doses are illustrated in Figure 1. The predose concentrations were compared in the multiple-dose study using RM-ANOVA depending on the KM-023 dose, and a steady state was attained after day 4 (75 mg) or day 6 (150–600 mg).
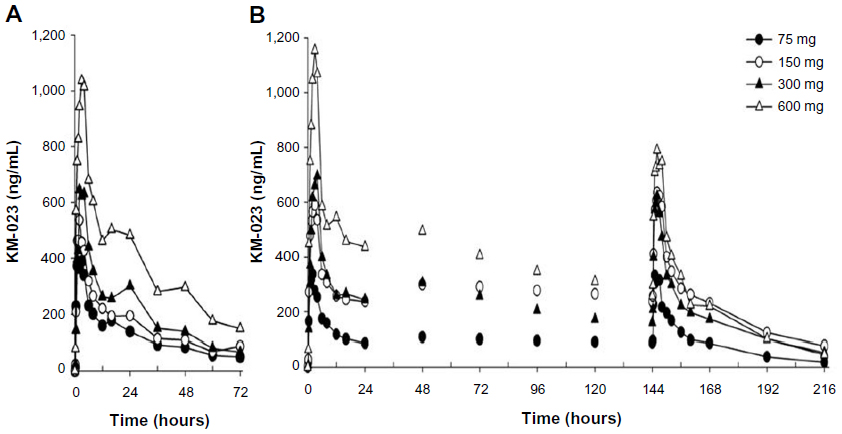 | Figure 1 (A and B) Mean plasma KM-023 concentration–time profiles after oral administration. (A) single dose; (B) multiple doses. |
Following single or multiple doses of KM-023, Tmax values were 0.5–6.0 hours, and mean t1/2 values were 20.7–31.2 hours. In the single-dose study, the mean KM-023 Cmax and AUC∞ values ranged from 440.2 to 1,245.4 ng/mL and 11,142.4 to 33,705.6 ng · h/mL, respectively. In the multiple-dose study, mean KM-023 Cmax,ss and AUCτ,ss values ranged from 385.1 to 1,096.7 ng/mL and 3,698.9 to 10,232.6 ng · h/mL, respectively (Table 2). Cmax and AUC values increased as the dose escalated, but neither Cmax nor AUC showed dose proportionality (Figure 2). In the single-dose study, the 95% CIs of the slope for Cmax and AUC∞ were 0.38–0.58 and 0.30–0.70, respectively. In the multiple-dose study, the 95% CIs of the slopes for Cmax,ss and AUCτ,ss were 0.27–0.61 and 0.14–0.62, respectively. After multiple doses of KM-023, the accumulation index ranged from 0.6 to 1.1. Notably, the accumulation index was 0.6 in the 600 mg dose group. The fe values accounted for less than 1% at all dose levels. Therefore, renal clearance was not evaluated (Table 2).
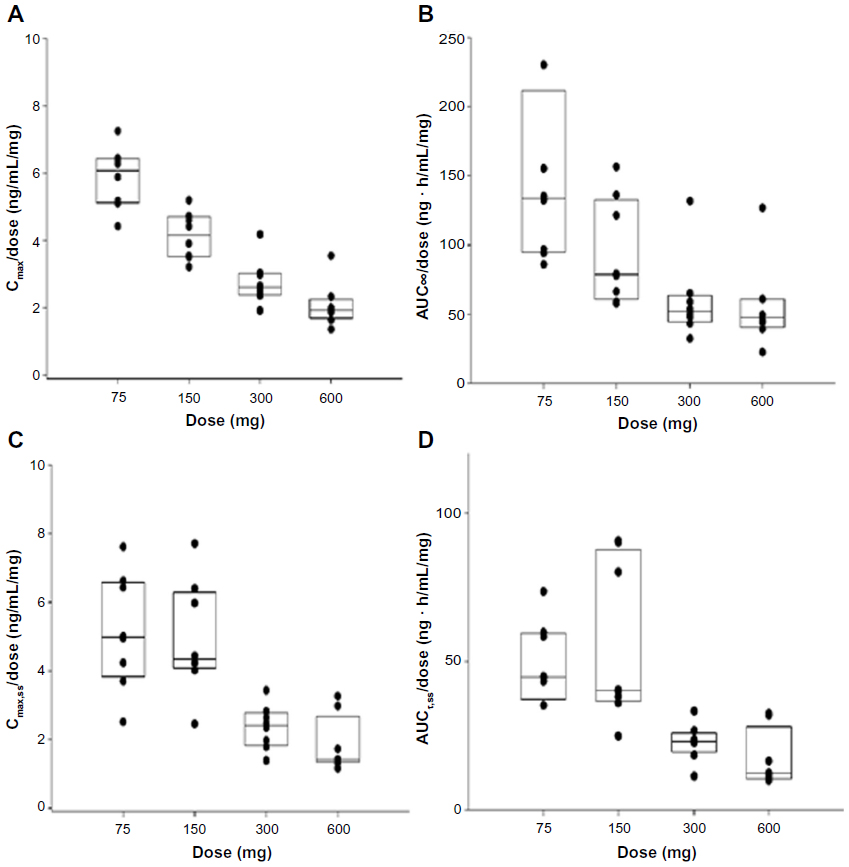 | Figure 2 Comparisons of Cmax/dose (A), AUC∞/dose (B), Cmax,ss/dose (C), and AUCτ,ss/dose (D) with respect to KM-023 doses. (A and B) single dose; (C and D) multiple doses. |
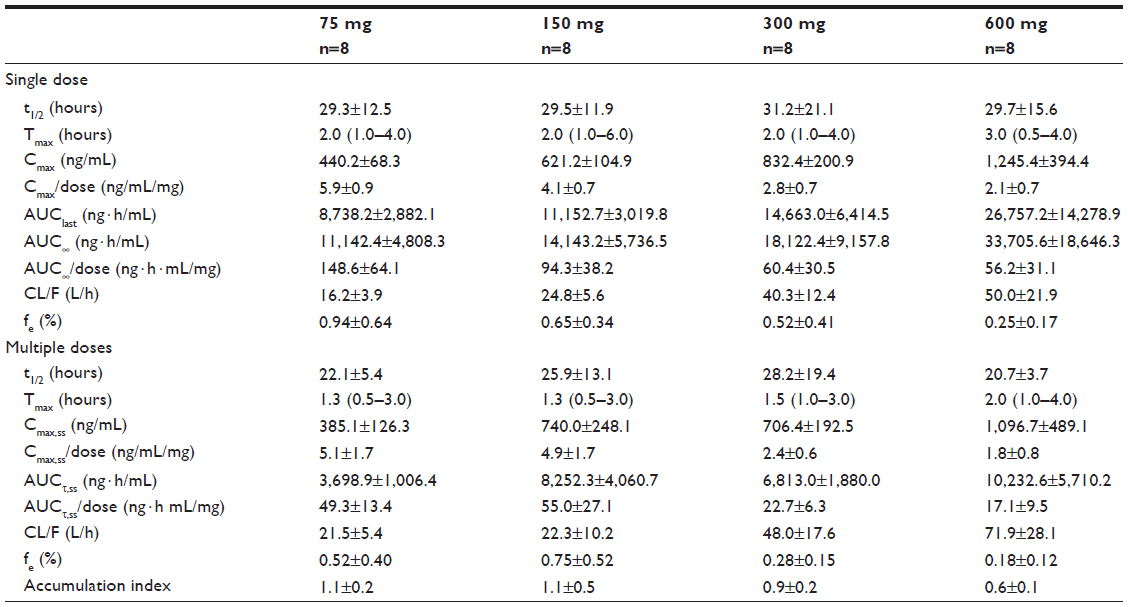 | Table 2 Pharmacokinetic parameters following single or multiple doses of KM-023 |
Tolerability
Fifteen subjects in the single-dose study experienced a total of 17 AEs throughout the study. Nine AEs that occurred in seven subjects were drug-related. Twenty-two subjects in the multiple-dose study experienced a total of 24 AEs during the study period. Seventeen AEs that occurred in 16 subjects were drug-related. Epistaxis (seven occurrences: two in one subject [single-dose study], five in four subjects [multiple-dose study]) was the most frequently reported AE. All seven occurrences were deemed to be drug-related AEs by the investigators. No serious AEs occurred in this study (Table 3). There were no clinically meaningful changes from baseline in clinical laboratory test results, vital signs, electrocardiography, computerized impedance cardiography, or physical examinations.
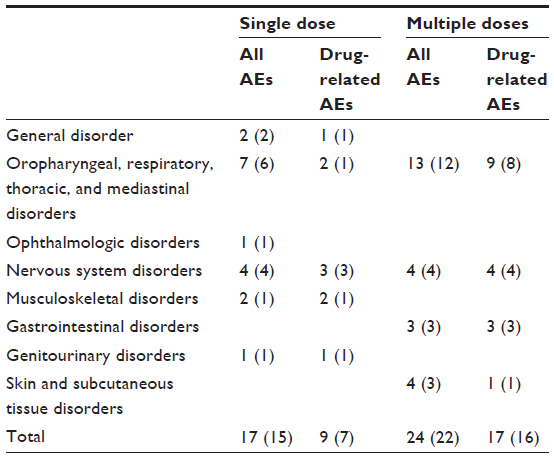 | Table 3 Summary of adverse events (AEs) after single or multiple KM-023 doses |
Discussion
The primary objective of this Phase I clinical study was to investigate the pharmacokinetic characteristics of KM-023, a newly developed second-generation NNRTI, and evaluate its tolerability in healthy volunteers. KM-023 showed dose- and time-dependent nonlinear pharmacokinetics. In addition, there was no laboratory or clinical evidence of clinically significant AEs to KM-023. The present study provides initial information on the tolerability and pharmacokinetic characteristics of KM-023 in humans.
We could not calculate renal clearance because the mean fe values were less than 1% in all dose groups. Our findings suggest that nonrenal pathways primarily eliminated KM-023. These results are consistent with other NNRTIs, which show negligible renal excretion and extensive hepatic metabolism.13 However, the detailed metabolites and metabolic pathways of KM-023 have not been characterized. Therefore, an evaluation of the metabolic pathways and a measurement of metabolite concentrations and KM-023 potency are needed.
KM-023 showed less than dose-proportional nonlinear pharmacokinetics over the 75–600 mg dose range. This pattern was also observed in first-generation NNRTIs. After a single oral administration of efavirenz (100–1,600 mg) and nevirapine (2.5–400 mg) in healthy subjects, Cmax or AUC increased less than dose-proportionally, which suggests diminished absorption or enhanced clearance at higher doses.14,15 The reduced absorption may be have been due to the saturation of influx transporters, and the enhanced clearance may have resulted from induced enzymes involved in hepatic/intestinal metabolism.16 Further studies are needed to investigate the mechanism of the less than dose-proportional nonlinear pharmacokinetics of KM-023.
The accumulation index was 0.6–1.1 over the range of the 75–600 mg doses. Interestingly, the average accumulation index decreased to 0.6 in the 600 mg dose group. These findings indicate that the metabolic enzymes related to KM-023 metabolism were induced by multiple KM-023 administrations. A similar autoinduction phenomenon has also been observed in efavirenz and nevirapine.13 KM-023 exhibited a potential for CYP3A enzyme activation and CYP3A4 messenger ribonucleic acid induction in primary human hepatocytes (Table 4). The markedly increased CL/F values after multiple doses of 600 mg, which could be partially explained by an autoinduction of CYP3A4 enzymes due to a sustained relatively high exposure of KM-023, and might be the reason for the apparently low accumulation. Therefore, it is anticipated that KM-023 exposure will be decreased when using high doses of KM-023 (particularly more than 600 mg) for clinical use.
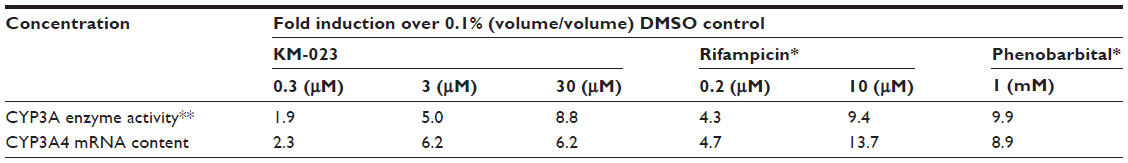 | Table 4 Changes in cytochrome P450 (CYP)-3A activities and CYP3A4 messenger ribonucleic acid (mRNA) contents in primary human hepatocytes treated with KM-023 or positive control compounds |
The most frequent AE after KM-023 administration in the present study was epistaxis. These symptoms were mild and resolved within 1–9 minutes, except in one subject, whose symptoms lasted intermittently for 10–24 hours. However, none of these AEs was clinically significant. In addition, there were no statistically significant changes in occurrence rates between the placebo and treatment groups, and the rate was not dose-dependent. Partially, epistaxis may be occured by the low humidity of the Clinical Trials Center during the winter.
In conclusion, the pharmacokinetics of KM-023 showed dose- and time-dependent nonlinear pharmacokinetic profiles. Single or multiple doses of KM-023 at 75–600 mg were well tolerated in healthy subjects. Further clinical trials evaluating the efficacy and safety of KM-023 in HIV-1 positive patients are warranted.
Acknowledgments
Kainos Medicine, Inc. sponsored and funded the research described in this report. We thank Ms Hyewon Kim for assisting with the clinical trials. YJC received a training program grant from the Korea Healthcare Technology R&D Project at the Ministry for Health and Welfare (A070001).
Disclosure
SS, BB, and MCK were employees of Kainos Medicine USA Inc. The other authors have no conflicts of interest to disclose.
References
Riddler SA, Haubrich R, DiRienzo AG, et al. Class-sparing regimens for initial treatment of HIV-1 infection. N Engl J Med. 2008;358(20):2095–2106. | |
Thompson MA, Aberg JA, Hoy JF, et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society – USA panel. JAMA. 2012;308(4):387–402. | |
Palella FJ Jr, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338(13):853–860. | |
Pauwels R. New non-nucleoside reverse transcriptase inhibitors (NNRTIs) in development for the treatment of HIV infections. Curr Opin Pharmacol. 2004;4(5):437–446. | |
Arastéh K, Rieger A, Yeni P, et al. Short-term randomized proof-of-principle trial of TMC278 in patients with HIV type-1 who have previously failed antiretroviral therapy. Antivir Ther. 2009;14(5):713–722. | |
Shubber Z, Calmy A, Andrieux-Meyer I, et al. Adverse events associated with nevirapine and efavirenz-based first-line antiretroviral therapy: a systematic review and meta-analysis. AIDS. 2013;27(9):1403–1412. | |
Hammer SM, Eron JJ Jr, Reiss P, et al. Antiretroviral treatment of adult HIV infection: 2008 recommendations of the International AIDS Society – USA panel. JAMA. 2008;300(5):555–570. | |
Carr A, Cooper DA. Adverse effects of antiretroviral therapy. Lancet. 2000;356(9239):1423–1430. | |
Garvey L, Winston A. Rilpivirine: a novel non-nucleoside reverse transcriptase inhibitor. Expert Opin Investig Drugs. 2009;18(7):1035–1041. | |
Scholler-Gyure M, Kakuda TN, Raoof A, De Smedt G, Hoetelmans RM. Clinical pharmacokinetics and pharmacodynamics of etravirine. Clin Pharmacokinet. 2009;48(9):561–574. | |
Bacheler LT, Anton ED, Kudish P, et al. Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrob Agents Chemother. 2000;44(9):2475–2484. | |
Kainos Medicine Inc., Single Ascending Dose (SAD)/Multiple Ascending Dose (MAD) Safety/Pharmacokinetic (PK) Study of KM-023. [ClinicalTrials.gov identifier NCT01348516]. | |
Smith PF, DiCenzo R, Morse GD. Clinical pharmacokinetics of non-nucleoside reverse transcriptase inhibitors. Clin Pharmacokinet. 2001;40(12):893–905. | |
Adkins JC, Noble S. Efavirenz. Drugs. 1998;56(6):1055–1064. | |
Cheeseman SH, Hattox SE, McLaughlin MM, et al. Pharmacokinetics of nevirapine: initial single-rising-dose study in humans. Antimicrob Agents Chemother. 1993;37(2):178–182. | |
Ludden TM. Nonlinear pharmacokinetics: clinical Implications. Clinical pharmacokinetics. 1991;20(6):429–446. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.