Back to Journals » Infection and Drug Resistance » Volume 16
Pharmacokinetics and Pharmacodynamics of a Novel Vancomycin Derivative LYSC98 in a Murine Thigh Infection Model Against Staphylococcus aureus
Authors He P, Li X, Guo X, Bian X, Wang R, Wang Y, Huang S, Qi M, Liu Y, Feng M
Received 1 December 2022
Accepted for publication 14 February 2023
Published 18 February 2023 Volume 2023:16 Pages 1019—1028
DOI https://doi.org/10.2147/IDR.S399150
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Peng He,1 Xin Li,2 Xiaohan Guo,1 Xingchen Bian,1 Rui Wang,1 Yue Wang,1 Sijing Huang,1 Mengya Qi,1 Yuanxia Liu,3 Meiqing Feng1
1Department of Biological Medicines & Shanghai Engineering Research Center of Immunotherapeutics, Fudan University School of Pharmacy, Shanghai, 201203, People’s Republic of China; 2Institute of Antibiotics, Huashan Hospital, Fudan University, Shanghai, People’s Republic of China; 3Shanghai Municipal Hospital of Traditional Chinese Medicine, Shanghai University of Traditional Chinese Medicine, Department of Pathology, Shanghai, People’s Republic of China
Correspondence: Meiqing Feng, Department of Biological Medicines & Shanghai Engineering Research Center of Immunotherapeutics, Fudan University School of Pharmacy, Shanghai, 201203, People’s Republic of China, Tel +86 21 51980035, Email [email protected] Yuanxia Liu, Shanghai Municipal Hospital of Traditional Chinese Medicine, Shanghai University of Traditional Chinese Medicine, Department of Pathology, Shanghai, People’s Republic of China, Email [email protected]
Introduction: LYSC98 is a novel vancomycin derivative used for gram-positive bacterial infections. Here we compared the antibacterial activity of LYSC98 with vancomycin and linezolid in vitro and in vivo. Besides, we also reported the pharmacokinetic/pharmacodynamic (PK/PD) index and efficacy-target values of LYSC98.
Methods: The MIC values of LYSC98 were identified through broth microdilution method. A mice sepsis model was established to investigate the protective effect of LYSC98 in vivo. Single-dose pharmacokinetics of LYSC98 was studied in thigh-infected mice and liquid chromatography–tandem mass spectrometry (LC-MS/MS) method was used to determine LYSC98 concentration in plasma. Dose fractionation studies were performed to evaluate different PK/PD indices. Two methicillin-resistant Staphylococcus aureus (MRSA) clinical strains were used in the dose ranging studies to determine the efficacy-target values.
Results: LYSC98 showed a universal antibacterial effect in Staphylococcus aureus with a MIC range of 2– 4 μg/mL. In vivo, LYSC98 demonstrated distinctive mortality protection in mice sepsis model with an ED50 value of 0.41– 1.86 mg/kg. The pharmacokinetics results displayed maximum plasma concentration (Cmax) 11,466.67− 48,866.67 ng/mL, area under the concentration–time curve from 0 to 24 h (AUC0– 24) 14,788.42− 91,885.93 ng/mL·h, and elimination half-life (T1/2) 1.70– 2.64 h, respectively. Cmax/MIC (R2 0.8941) was proved to be the most suitable PK/PD index for LYSC98 to predict its antibacterial efficacy. The magnitude of LYSC98 Cmax/MIC associated with net stasis, 1, 2, 3 and 4 - log 10 kill were 5.78, 8.17, 11.14, 15.85 and 30.58, respectively.
Conclusion: Our study demonstrates that LYSC98 is more effective than vancomycin either in killing vancomycin-resistant Staphylococcus aureus (VRSA) in vitro or treating S. aureus infections in vivo, making it a novel and promising antibiotic. The PK/PD analysis will also contribute to the LYSC98 Phase I dose design.
Keywords: LYSC98, vancomycin derivative, pharmacokinetics and pharmacodynamics, Staphylococcus aureus, murine thigh infection model
Introduction
Staphylococcus aureus is a Gram-positive human commensal which inhabits the anterior nares of approximately 30% of the healthy people.1,2 As a leading cause of hospital-associated (HA) and community-associated (CA) bacterial infections, S. aureus is associated with numerous mild skin and soft tissue infections, as well as life-threatening pneumonia, bacteremia, osteomyelitis, endocarditis, sepsis and toxic shock syndrome.3,4
Penicillin remains the therapeutic choice if the isolate is sensitive.5 But once bacterial resistance occurs, such as methicillin-resistant Staphylococcus aureus (MRSA) infections, vancomycin is the advanced drug to treat.6 Other categories of antibiotics, like fluoroquinolones, trimethoprim–sulfamethoxazole, clindamycin, and minocycline, may also be effective in cases where sterilization is required.7 While they are not as effective as vancomycin, either because they have a less anti-staphylococcal activity or because drug resistance develops during treatment.6–8 Therefore, vancomycin was commonly considered as the last line of defense in the treatment of S. aureus. However, clinical isolates with reduced susceptibility or complete resistance to vancomycin have emerged within the past 20 years.9–11 Vancomycin-resistant S. aureus (VRSA) are generally associated with persistent infections, treatment failure, and poor clinical outcomes.11
LYSC98 is a new synthesized compound derived from vancomycin by chemical modification of its side chain (Figure 1). This compound has never been reported before and its antibacterial effect was still unclear. Besides, there were also no studies identifying the PK/PD index and efficacy-target values of LYSC98. While PK/PD study is becoming an indispensable part of antibacterial drug development, PK/PD targets are used to support the dosing regimen design in phase I clinical trials and are essential data for the susceptible breakpoints establishment.12 Animal models are frequently employed in the determination of PK/PD targets because of the consistent results to that in humans, as well as the flexibility dosing regimen design to analyze exposure–response relationships.
 |
Figure 1 The chemical structure of vancomycin and LYSC98. The red boxes represent the sites of modification. |
In this study, the antibacterial activity of LYSC98 was compared with vancomycin and linezolid in vitro and in vivo. We identified the pharmacokinetic and pharmacodynamic characteristics of LYSC98 by using an immunosuppressed murine thigh infection model against S. aureus. The PK profile, exposure–response relationships and PK/PD targets were determined. Our research provided a basis for the clinical administration plan and drug-sensitive breakpoint of this novel and promising antibiotic.
Materials and Methods
Bacterial, Media, and Antibiotic
Totally 67 clinical strains [Institute of Antibiotics, Huashan Hospital, Shanghai], including 47 MRSA and 20 VRSA strains, and 2 reference S. aureus strains ATCC29213 and ATCC25923 were used in this study. Bacteria were cultured and quantified on a LB agar plate and grown for 16 h at 37 °C. LB agar plate consists of 1% Peptone (OXOID), 0.5% yeast powder (OXOID), 1% sodium chloride (Sinopharm), and 1.5 AGAR powder (Meilunbio, Dalian). LYSC98 (Purity: 92.02, Lot no: 20200322) was supplied by Shanghai Laiyi Center for Biopharmaceutical R&D (Shanghai, China). The compound was reconstituted and diluted to appropriate concentrations with 5% glucose solution.
In vitro Susceptibility Testing
The minimum inhibitory concentration (MIC) of vancomycin, linezolid and LYSC98 against all isolates were determined in triplicate with broth microdilution method according to Clinical and Laboratory Institute (CLSI, 2018) guidelines.
MIC values were identified through microdilution in sterilized 96-well polypropylene microtiter plates. The test medium was Mueller–Hinton broth (MHB), and the strain concentrations were adjusted to 5×105 CFU/mL. After 20 h of incubation at various concentrations of LYSC98 or other drugs at 37 °C, MIC was defined as the lowest concentration of antibiotic with no visible bacteria growth. ATCC29213 served as a quality control strain.
In vivo Protective Effect
A sepsis model was established through intraperitoneal administration of 3×106 CFU S. aureus strains 18-W27-73 in 0.1 mL LB. After bacterial challenge, the mice were treated with different concentrations of LYSC98 or other compounds. The survival of eight mice in each group was monitored for 7 days after infection.
Murine Thigh Infection Model
The animal studies were approved by the Experimental Animal Ethics Committee of Pharmacy in Fudan University (2019–08-WY-FMQ-01) and followed the Experimental Animal Welfare Review Guide. Six-week-old, specific-pathogen-free, male ICR mice (SLAC Laboratory Animal Co., Ltd, Shanghai) weighing 18–22 g were used in all studies. The neutropenic murine thigh infection model was established as previously described.13 Animals were rendered neutropenic by intraperitoneal injections of 150 and 100 mg/kg cyclophosphamide (Sigma–Aldrich, St Louis, MO, USA), on days −4 and −1 respectively prior to infection. Two hours prior to treatment (−2 h), 200 μL LB containing about 1–2×106 CFU bacterial inoculum was administered into the bilateral gastrocnemius muscle via an intramuscular injection. At 0 h, a cohort of animals was sacrificed via CO2 to determine the bacterial levels at the start of treatment. The remaining animals were euthanized 24 h after treatment.
Thigh muscle of the infected area was taken and weighed, sterile grinding beads and 200 μL sterile saline were added. Homogenized with XHF-1 type low-temperature tissue homogenizer (Ningbo Xinzhi Biotechnology Co., LTD.) at 75 Hz for 15 seconds, 3 times, until the tissue was completely homogenized. The homogenates were serially diluted in sterile saline before dilutions were plated on LB Agar plates, incubated overnight at 37 °C and the colonies counted. The CFU/thigh was calculated and transformed to log10 value. The efficacy of each group was measured by comparing the bacterial load to the untreated group 24 hours later.
Pharmacokinetics
Single-dose pharmacokinetics of LYSC98 was studied in thigh-infected mice at doses level of 2, 4 and 8 mg/kg following intravenous administration made in the tail vein and by bolus (0.2 mL/dose). Anticoagulant tubes containing 100 U of low molecular weight heparin (LMWH) sodium was used for plasma recovery. Blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, 12, 18, 24 h at each dose. Three mice were used per time point. Plasma was separated by centrifugation at 4000 g for 10 min at 4 °C and stored at −80 °C until LYSC98 concentration analysis. The detection of compound concentration was entrusted to Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
A liquid chromatography–tandem mass spectrometry (LC-MS/MS) method was used to determine LYSC98 concentration in plasma. The lower limit of quantification of LYSC98 in plasma was 0.050 mg/L. The protein binding of LYSC98 in murine plasma was determined at spiked concentrations of 0.08–0.68 mg/L using an equilibrium dialysis method.
WinNonlin software (Version 6.3; Pharsight Inc., St. Louis, MO, United States) was employed to calculate the PK parameters using a noncompartmental model, including the elimination half-life (t1/2), the area under the concentration–time curve over 24 h (AUC0–24), and the peak drug concentration (Cmax). The PK parameter estimation for treatment doses was calculated based on a noncompartmental model.
Pharmacokinetic/Pharmacodynamic Index Determination
Neutropenic mice were infected with the standard strain of S. aureus ATCC29213 for a dose fractionation experiment. Dose-fractionation study is useful in reducing the interdependence among the PK/PD index and confirming which one is the most important to efficacy. The total daily doses included 2, 4, 8 mg/kg, divided evenly every 6, 8, 12 and 24 h. Groups of three mice and six thighs were included in each dosing regimen. Efficacy was calculated as the change in log10 CFU obtained after 24 h treatment with LYSC98.
To determine the dominant PK/PD index driving efficacy, the number of bacteria in the thigh muscles at the end of treatment was correlated with three parameters: the free drug peak level divided by the MIC (fCmax/MIC), the area under the free concentration–time curve over 24 h divided by the MIC (f AUC0–24/MIC) and the cumulative percentage of a 24 h period that the free drug concentration in plasma exceeds the MIC (%fT > MIC), for each of the dosage regimens studied. The mathematical model used was derived from the Sigmoid Emax model:
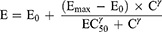
where E is the effect, in this case, the log10 change in CFU per gram thigh muscles between treated mice and untreated controls after the 24-h period of study, Emax is the maximum effect, C is the PK/PD index value, EC50 is the value of PK/PD index required to achieve 50% of the Emax, and γ is the slope of the dose-effect curve. The R2 value from non-linear regression analysis (WinNonlin 6.3) was used to assess the correlation of treatment efficacy with each of the PK/PD indices.
PK/PD Targets for Efficacy
Dose-ranging efficacy studies were then performed to determine the PK/PD targets for net stasis, 1-log10 CFU, 2-log10 CFU, 3-log10 CFU and 4-log10 CFU kill with two clinical S. aureus strains using the neutropenic murine thigh infection model. Increasing single dosing regimens of LYSC98 were administered varied from 1 to 16 mg/kg. A sigmoid dose–response model derived from the Hill equation was used to calculate the dose, and PK/PD targets of LYSC98 producing a net bacteriostatic effect, 1-log10 CFU, 2-log10 CFU, 3-log10 CFU and 4-log10 CFU kill over 24 h compared to the organism burden at the start of treatment.
Results
In vitro Susceptibility Testing
In 49 S. aureus strains consisting of standard strains and MRSA, LYSC98 showed a universal antibacterial effect with a MIC range of 2–4 µg/mL, which was weaker than vancomycin but comparable to linezolid (Figure 2A). However, among other 20 vancomycin-resistant S. aureus (VRSA), LYSC98 appeared to be more effective. The MIC value of vancomycin, the prototype drug of LYSC98, increased to 16–32 µg/mL due to drug resistance, while there was no significant difference in the MIC value of LYSC98 against these drug-resistant bacteria from normal strains (Figure 2B).
 |
Figure 2 (A and B) MIC distribution of LYSC98, linezolid and vancomycin in 49 S. aureus strains (A) and in 20 VRSA strains (B). (C) In vivo protective effect of LYSC98, linezolid and vancomycin on S. aureus-infected mice. |
In vivo Protective Effect
To investigate the antibacterial effect of LYSC98 in vivo, we used MASR 18-W27-73 (Table 1) to infect mice by intraperitoneal injection. Contrary to in vitro results, LYSC98 showed better mortality protection at all doses (Figure 2C). Its ED50 (0.41–1.86 mg/kg) was significantly smaller than vancomycin (2.32–5.84 mg/kg) and linezolid (3.07–7.60 mg/kg).
 |
Table 1 Minimum Inhibitory Concentration (MIC) Information in S. aureus Used in the Study |
Pharmacokinetics
Single-dose PK of LYSC98 in plasma are shown in Figure 3A. The protein binding of LYSC98 in murine plasma varied from 44.1% to 47.0% in the concentration range of 0.08–0.68 mg/L, with a mean binding rate of 46.1%. The elimination half-life in plasma ranged from 0.25 to 0.33 h. Cmax concentrations ranged from 11,466.67 to 48,866.67 ng/mL and were linear across 2–8 mg/kg dose range (R2 0.9994). AUC0–24 values ranged from 14,788.42 to 91,885.93 ng/mL and were linear across 2–8 mg/kg dose range (R2 0.981). Detailed PK parameters are listed in Table 2.
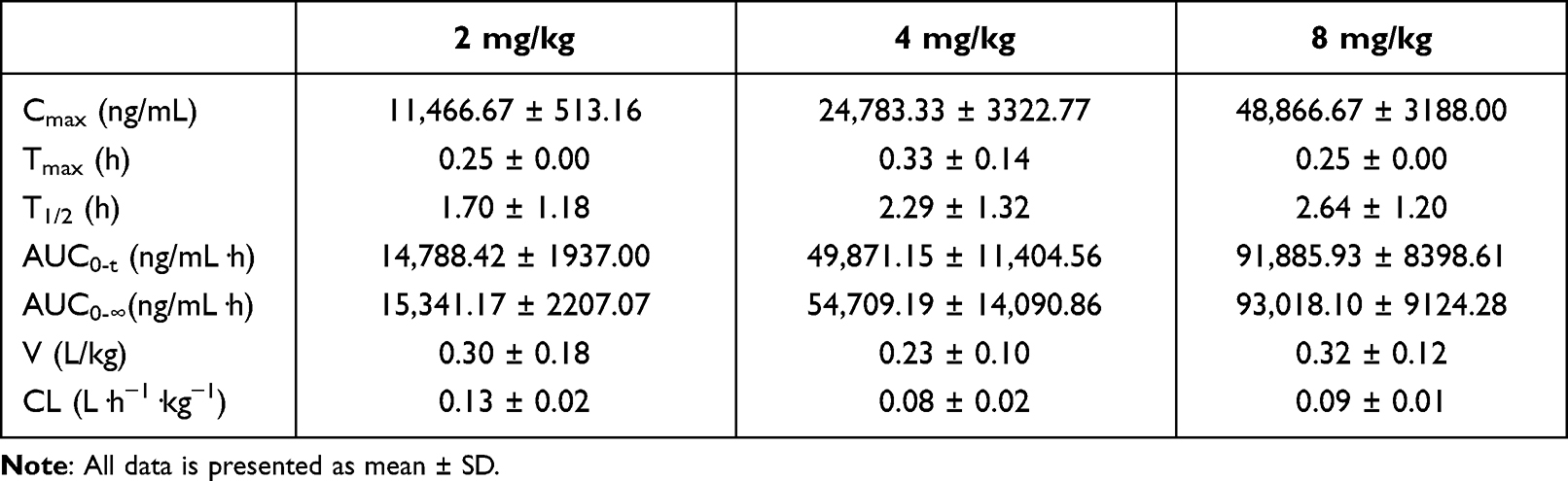 |
Table 2 PK Parameters Calculated Using Non-Compartment Model |
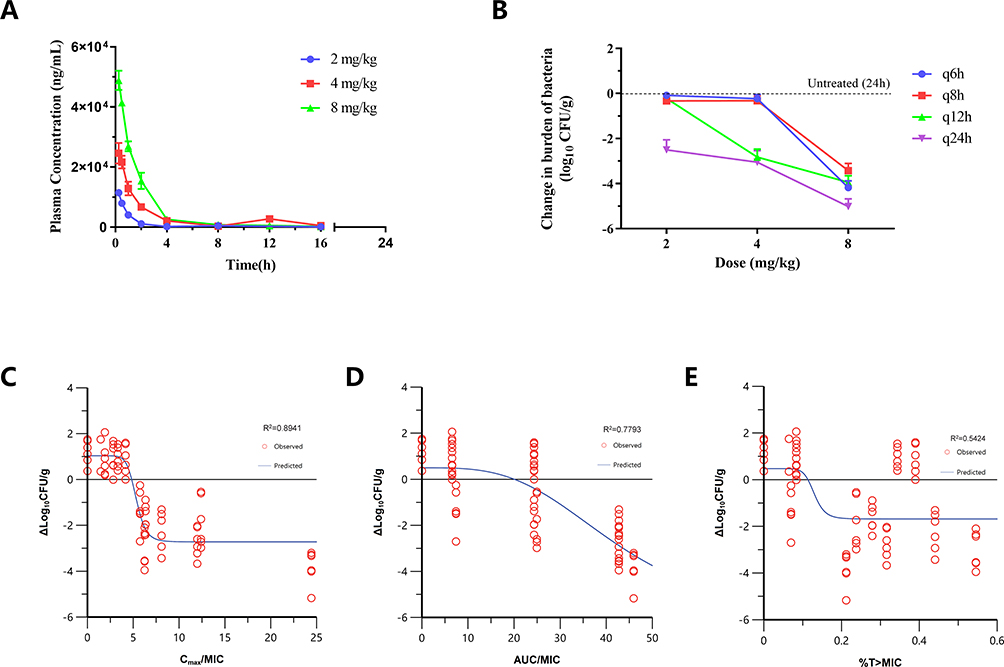 |
Figure 3 (A) Pharmacokinetics of LYSC98 in plasma following single intravenous doses at 2–8 mg/kg in neutropenic thigh-infected mice. Groups of three mice were sampled at each time point. Each symbol represents the mean value of plasma concentration in three mice. The error bar represents the standard deviations. (B) Regimens of LYSC98 treatment produced CFU burden reduction against S. aureus ATCC29213 in the dose fractionation experiment. Abscissa q6h, q8h, q12h, and q24h means LYSC98 were treated per 6,8,12 and 24 hours under constant total dose in each group. The efficacy of each group was measured by comparing the bacterial load with the untreated group 24 hours later. (C–E) Correlation of pharmacokinetic/pharmacodynamic (PK/PD) indices fCmax/MIC (C), fAUC0–24/MIC (D) and %fT > MIC (E) with efficacy. Treatment was initiated at 2 h post infection. LYSC98 was intravenously administered with a dosing range of 2–8 mg/kg, in once daily (q24h), twice daily (q12h), three times a day (q8h) and four times a day (q6h). Each circle represents data for each mouse. |
PK/PD Index Determination
LYSC98 treatment produced up to 5.02 log10 reduction of CFU burden against S. aureus ATCC29213 in the dose fractionation experiment (Figure 3B). The dose–response curves with different intervals showed the bactericidal effect was improved with the increased dose but not the decreased interval. The relationship between efficacy and three PK/PD indices, fCmax/MIC, fAUC0–24/MIC and %fT > MIC, is shown in Figure 3C–E, and the values of the square of the correlation coefficient (R2) were 0.8941, 0.7793 and 0.5424, respectively (Table 3). fCmax/MIC was considered as the PKPD index of LYSC98 since its best correlation.
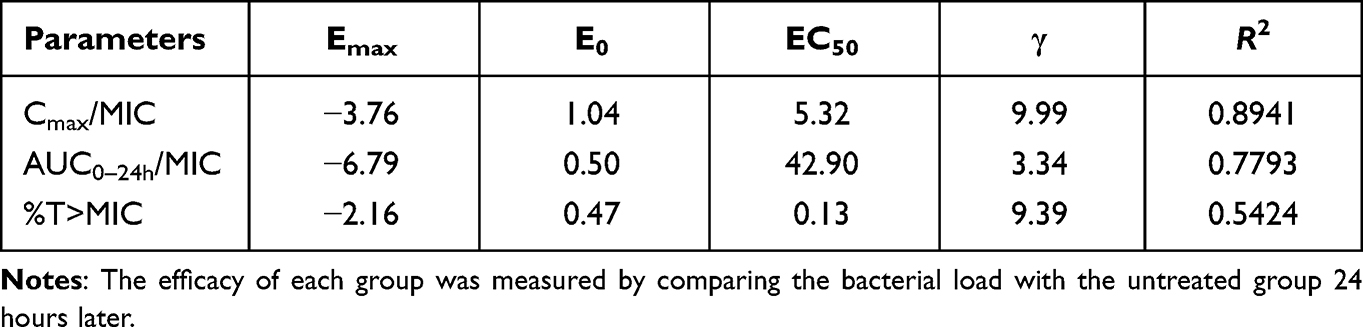 |
Table 3 Emax Model Parameters Characterizing the Relationship Between Efficacy and Different PK/PD Indices of LYSC98 |
PK/PD Targets for Efficacy
Two additional S. aureus strains 18-W26-14 (STRAINS MIC90) and 18-W27-73 (STRAINS MIC50) were used in the dose-escalation experiment to determine the PK/PD targets (Table 1). The dose–response data for the two strains are shown in Figure 4A.
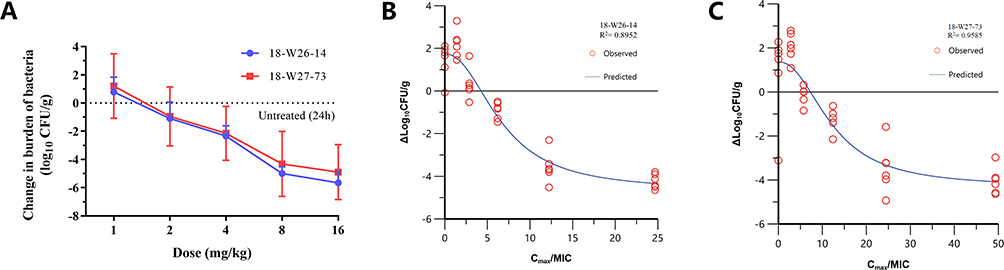 |
Figure 4 (A) In vivo dose–response curves for LYSC98 against two S. aureus strains over 24 h after a single-dose administration in the neutropenic murine thigh infection model. The efficacy was measured by comparing the bacterial load with the untreated group 24 hours later. The results are presented as mean ± standard deviation. The burden of organisms was measured at the start and end of treatment. The horizontal line at 0 represents no net change from baseline. (B and C) Correlation between fCmax/MIC and efficacy of LYSC98 in dose-escalation experiment. Treatment was initiated at 2 h post infection. LYSC98 was intravenous administered with single-dose range of 1–16 mg/kg. The efficacy of each group was measured by comparing the bacterial load with the untreated group 24 hours later. Each point represents data for each sample. R2 means square of the correlation coefficient. |
LYSC98 demonstrated potent efficacy against S. aureus strains in our study. The maximal effect reached a 4.9 to 5.6 log10 CFU killing compared with the initial bacterial burden. The dose–response data were modeled using the sigmoid Emax equation, showing fCmax/MIC was a strong predictor for treatment outcomes based on regression analysis (Figure 4B and C, R2 0.9585, R2 0.8952, respectively).
The fCmax/MIC values essential to produce a treatment target are shown in Table 4. Briefly, the median fCmax/MIC targets needed for static, 1-log10 CFU, 2-log10 CFU, 3-log10 CFU and 4-log10 CFU were 5.78, 8.17, 11.14, 15.85, 30.58, respectively.
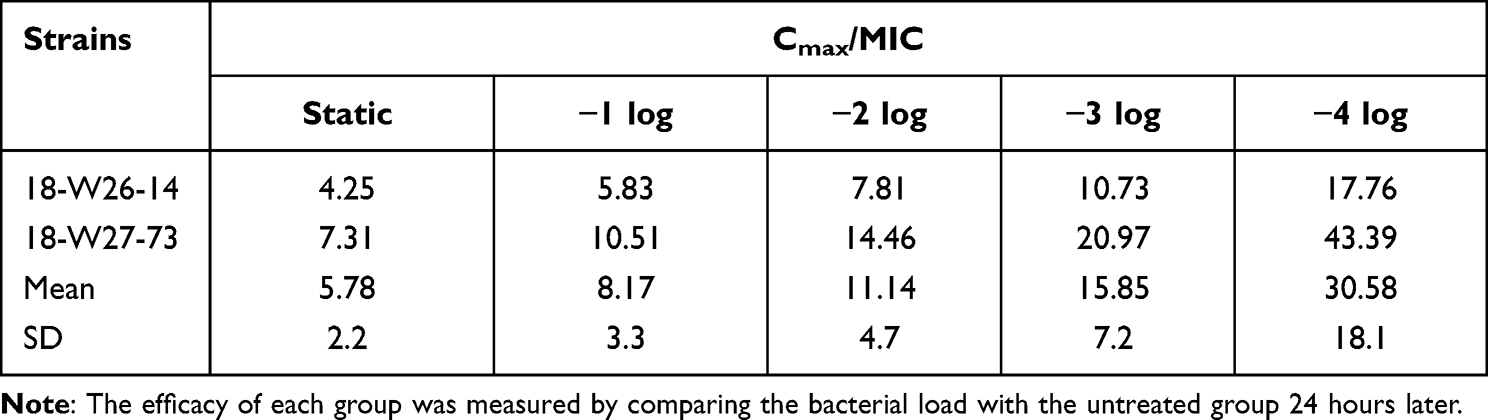 |
Table 4 In vivo Activity and PK/PD Analysis of LYSC98 Against Clinical Organisms |
Discussion
An ideal optimization requires a good knowledge of the mechanisms involved in the effect of antibiotics (pharmacodynamics, PD) and the change of antibiotic concentration in the patient body (pharmacokinetics, PK). Pharmacokinetic/Pharmacodynamic (PK/PD) analysis integrates them and studies the dosing required to enhance the success possibility of antibiotic therapy, as well as minimizes the side effects and the emergence of resistances.14,15 Using a neutropenic murine thigh infection model, our study determined the magnitudes of fCmax/MIC of LYSC98, a new antibacterial compound, associated with various bacterial reduction in S. aureus strains infection. Since there was no available PK data from human, we did not choose Monte Carlo Simulation to evaluated the clinical dosing regimens or PK/PD breakpoints. This work should be further completed in the future.
The inclusion of multiple bacterial isolates with various susceptibility should be considered in animal PK/PD experiment design to obtain a robust PK/PD target.16,17 In our study, totally 69 strains of Staphylococcus aureus were used for MIC test in vitro, and one of them, STRAIN 18-W27-73 (STRAIN MIC50), was used for murine infection and death protection test. Two clinical strains 18-W26-14 (STRAIN MIC90) and 18-W27-73 (STRAIN MIC50) were used in subsequent PK/PD study. According to the MIC results, two strains selected were STRAINS MIC50 and MIC90 of LYSC98, so we considered the results were fairly representative. Besides, MSSA and MRSA were included in our study which indicated the weak impact of penicillin resistance on LYSC98 activity. There was a difference in the MIC values of the strains while the bactericidal effect of LYSC98 were similar, it may be caused by the differences in virulence, tolerance and adaptive capacity to hosts of the strains. It also illustrates the importance to adopt a PK/PD approach combining in vivo and in vitro studies for dosing regimen evaluation rather than rely on MIC values alone.18
The PK/PD indices of vancomycin antibiotics are generally considered as f AUC0–24/MIC.19,20 Some other researchers think that there are no significant correlations between PK/PD indices and the clinical or microbiological efficacy of vancomycin.21 While in our study, Emax model analysis showed that for vancomycin derivate LYSC98, fCmax/MIC (R2 0.8941) correlated better with efficacy rather than f AUC0–24/MIC (R2 0.7793) and was only weakly correlated with %T>MIC (R2 0.5424).
PK/PD indices are the best descriptors of antibiotic efficacy depending on the activity pattern of each antibiotic.22 As a glycopeptide antibiotic, the mechanism of vancomycin is generally believed to be that it binds to alanine at the end of the precursor of the sensitive bacterial cell wall and blocks the synthesis of peptidoglycan, thus leading to cell wall defects and killing bacteria.23 In the common classification, vancomycin antibiotics belong to antibacterial agents with concentration-independent killing and long-term persistence.24 Due to the prolonged persistent effects that protect against regrowth when active drug concentration falls below the MIC, the best PK/PD indexes for these drugs are fCmax/MIC or the f AUC0–24/MIC, which is fairly consistent with our results.14,25
As for the choices between fCmax/MIC and f AUC0–24/MIC, we infer that the differences may be mainly considered from three aspects:
First, the half-life of different compounds needs to be considered. LYSC98 was engineered to have a significantly longer half-life in mice than vancomycin, which could explain its long-acting bactericidal effect.26 If the drug has a long half-life or post-antibiotic effect (PAE), since %fT > MIC is easy to reach a high level, increasing the drug concentration could improve the effect, which mainly shows the concentration-dependent characteristics. For instance, animal PK/PD studies of penicillin and amikacin, as well as some in vitro studies, showed an increased correlation of antibacterial effect with fCmax/MIC and f AUC0–24/MIC when the half-lives of the drug were prolonged, and a shortened half-life is associated with %fT > MIC better.20,27
Secondly, doses may also be an important factor. The drug may have different PK/PD indices at different dosage.28 In our study, we used a total of 3 doses of 2–8 mg/kg LYSC98 to conduct pharmacodynamic experiments. The data were well linear, but there was still a possibility that the maximum efficacy was not covered. This possibility may lead to a smaller overall dose of administration, a greater dependence of efficacy on concentration, and the PK/PD index will be more inclined to fCmax/MIC. It may be a limitation of our study.
Thirdly, we need to consider the impact of additional antimicrobial mechanisms. Based on previous study, vancomycin can also change the permeability of bacterial cell membrane, and selectively inhibit the synthesis of bacterial RNA.29 Ratio on different antibacterial mechanisms of LYSC98 may also have changed with specific structural modifications. One potent evidence is that it works well against vancomycin-resistant enterococcus (Figure 2). Certainly, this explanation remains hypothesis until further pharmacological confirmation.
Conclusion
LYSC98, a novel vancomycin derivative, showed effective bactericidal efficacy against Staphylococcus aureus both in vitro and in vivo. Different PK/PD indices were used to fit the pharmacodynamic parameters, and the results showed that fCmax/MIC fitting results were the most appropriate indices, which could better represent its bactericidal properties. The fCmax/MIC target values required to achieve static, −1 log, −2 log, −3 log and −4 log antibacterial activity in thigh-infected mice were 5.78, 8.17, 11.14, 15.85 and 30.58, respectively. Our study provides effective preclinical data for rational application and further research of LYSC98.
Acknowledgments
This work was financially supported by National Major Scientific and Technological Special Project for “Significant New Drugs Development” (grant number 2019ZX09721-001-009). We would also like to express our gratitude to Zhejiang Pharmaceutical Co., Ltd. and Wenzhou Haihe Pharmaceutical Co., Ltd. for their support in this research.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339(8):520–532. doi:10.1056/NEJM199808203390806
2. Rao Q, Shang W, Hu X, Rao X. Staphylococcus aureus ST121: a globally disseminated hypervirulent clone. J Med Microbiol. 2015;64(12):1462–1473. doi:10.1099/jmm.0.000185
3. David MZ, Daum RS. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev. 2010;23(3):616–687. doi:10.1128/CMR.00081-09
4. Zhou K, Li C, Chen D, et al. A review on nanosystems as an effective approach against infections of Staphylococcus aureus. Int J Nanomedicine. 2018;13:7333–7347. doi:10.2147/IJN.S169935
5. Drebes J, Kunz M, Pereira CA, Betzel C, Wrenger C. MRSA infections: from classical treatment to suicide drugs. Curr Med Chem. 2014;21(15):1809–1819. doi:10.2174/0929867320666131119122520
6. Lakhundi S, Zhang K. Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin Microbiol Rev. 2018;31(4). doi:10.1128/CMR.00020-18
7. Michel M, Gutmann L. Methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci: therapeutic realities and possibilities. Lancet. 1997;349(9069):1901–1906. doi:10.1016/s0140-6736(96)11192-2
8. Chambers HF. Methicillin resistance in staphylococci: molecular and biochemical basis and clinical implications. Clin Microbiol Rev. 1997;10(4):781–791. doi:10.1128/CMR.10.4.781
9. Howe RA, Bowker KE, Walsh TR, Feest TG, MacGowan AP. Vancomycin-resistant Staphylococcus aureus. Lancet. 1998;351(9102):602. doi:10.1016/S0140-6736(05)78597-4
10. Hidayat LK, Hsu DI, Quist R, Shriner KA, Wong-Beringer A. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med. 2006;166(19):2138–2144. doi:10.1001/archinte.166.19.2138
11. McGuinness WA, Malachowa N, DeLeo FR. Vancomycin resistance in Staphylococcus aureus. Yale J Biol Med. 2017;90(2):269–281.
12. Mouton JW, Brown DFJ, Apfalter P, et al. The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: the EUCAST approach. Clin Microbiol Infect. 2012;18(3):E37–E45. doi:10.1111/j.1469-0691.2011.03752.x
13. Growcott EJ, Cariaga TA, Morris L, et al. Pharmacokinetics and pharmacodynamics of the novel monobactam LYS228 in a neutropenic murine thigh model of infection. J Antimicrob Chemother. 2019;74(1):108–116. doi:10.1093/jac/dky404
14. Asin-Prieto E, Rodriguez-Gascon A, Isla A. Applications of the pharmacokinetic/pharmacodynamic (PK/PD) analysis of antimicrobial agents. J Infect Chemother. 2015;21(5):319–329. doi:10.1016/j.jiac.2015.02.001
15. Gabrielsson J, Green AR, Van der Graaf PH. Optimising in vivo pharmacology studies—Practical PKPD considerations. J Pharmacol Toxicol Methods. 2010;61(2):146–156. doi:10.1016/j.vascn.2010.02.002
16. Bulitta JB, Hope WW, Eakin AE, et al. Generating robust and informative nonclinical in vitro and in vivo bacterial infection model efficacy data to support translation to humans. Antimicrob Agents Chemother. 2019;63(5). doi:10.1128/AAC.02307-18
17. Li X, Chen Y, Xu X, et al. Pharmacokinetics and pharmacodynamics of nemonoxacin in a neutropenic murine lung infection model against Streptococcus pneumoniae. Front Pharmacol. 2021;12:658558. doi:10.3389/fphar.2021.658558
18. Velkov T, Bergen PJ, Lora-Tamayo J, Landersdorfer CB, Li J. PK/PD models in antibacterial development. Curr Opin Microbiol. 2013;16(5):573–579. doi:10.1016/j.mib.2013.06.010
19. Moise-Broder PA, Forrest A, Birmingham MC, Schentag JJ. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin Pharmacokinet. 2004;43(13):925–942. doi:10.2165/00003088-200443130-00005
20. Nielsen EI, Cars O, Friberg LE. Pharmacokinetic/pharmacodynamic (PK/PD) indices of antibiotics predicted by a semimechanistic PKPD model: a step toward model-based dose optimization. Antimicrob Agents Chemother. 2011;55(10):4619–4630. doi:10.1128/AAC.00182-11
21. Shen K, Yang M, Fan Y, et al. Model-based evaluation of the clinical and microbiological efficacy of vancomycin: a prospective study of Chinese adult in-house patients. Clin Infect Dis. 2018;67(suppl_2):S256–S262. doi:10.1093/cid/ciy667
22. Reed MD. Optimal antibiotic dosing. The pharmacokinetic-pharmacodynamic interface. Postgrad Med. 2000;108(7 Suppl Contemporaty):17–24. doi:10.1016/S0140-6736(05)78597-4
23. Wilhelm MP. Vancomycin. Mayo Clin Proc. 1991;66(11):1165–1170. doi:10.1016/s0025-6196(12)65799-1
24. Rybak MJ. The pharmacokinetic and pharmacodynamic properties of vancomycin. Clin Infect Dis. 2006;42 Suppl 1(Supplement_1):S35–S39. doi:10.1086/491712
25. Holmes NE. Using AUC/MIC to guide vancomycin dosing: ready for prime time? Clin Microbiol Infect. 2020;26(4):406–408. doi:10.1016/j.cmi.2019.12.023
26. Knudsen JD, Fuursted K, Espersen F, Frimodt-Moller N. Activities of vancomycin and teicoplanin against penicillin-resistant pneumococci in vitro and in vivo and correlation to pharmacokinetic parameters in the mouse peritonitis model. Antimicrob Agents Chemother. 1997;41:1910–1915. doi:10.1128/AAC.41.9.1910
27. Craig WA, Redington J, Ebert SC. Pharmacodynamics of amikacin in vitro and in mouse thigh and lung infections. J Antimicrob Chemother. 1991;27 Suppl C:29–40. doi:10.1093/jac/27.suppl_c.29
28. Tam VH, Nikolaou M, Bourne PE. A novel approach to pharmacodynamic assessment of antimicrobial agents: new insights to dosing regimen design. PLoS Comput Biol. 2011;7(1):e1001043. doi:10.1371/journal.pcbi.1001043
29. Okano A, Isley NA, Boger DL. Total syntheses of vancomycin-related glycopeptide antibiotics and key analogues. Chem Rev. 2017;117(18):11952–11993. doi:10.1021/acs.chemrev.6b00820
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.