Back to Journals » Drug Design, Development and Therapy » Volume 15
Pharmacokinetic Study of Frovatriptan Succinate Tablet After Single and Multiple Oral Doses in Chinese Healthy Subjects
Authors Zheng H, Xia Y, Qu S ![]() , Fan L, Zhang J, Ma Z, Chen Y, Fan H
, Fan L, Zhang J, Ma Z, Chen Y, Fan H ![]()
Received 2 March 2021
Accepted for publication 4 June 2021
Published 7 July 2021 Volume 2021:15 Pages 2961—2968
DOI https://doi.org/10.2147/DDDT.S308958
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Hongquan Zheng,1,2 Yan Xia,3 Shengjun Qu,4 Lin Fan,2 Jingjing Zhang,1 Zhixiang Ma,1 Yangsheng Chen,4 Hongwei Fan1,2
1Department of Clinical Pharmacology Lab, Nanjing First Hospital, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China; 2School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing, Jiangsu, People’s Republic of China; 3Jiangsu Leeway Biological Technology Co., Ltd, Nanjing, Jiangsu, People’s Republic of China; 4CP Pharmaceutical Qingdao Co., Ltd, Qingdao, Shandong, People’s Republic of China
Correspondence: Hongwei Fan
Nanjing First Hospital, Nanjing Medical University, 68 Changle Road, Nanjing, 210006, People’s Republic of China
Tel +86 25 5288 7030
Fax +86 25 5288 7031
Email [email protected]
Yangsheng Chen 3601 Tuanjie Road, Economic and Technological Development Zone, Qingdao, People’s Republic of China
Tel +86 18766396996
Email [email protected]
Purpose: The present report describes findings from a Phase I clinical study that evaluated the single- and multiple-dose pharmacokinetics of frovatriptan succinate tablet in Chinese healthy subjects.
Methods: A total of 24 healthy subjects were enrolled. In single-dose study, 2.5, 5, and 10 mg oral doses of frovatriptan succinate tablet were administrated. A 2.5 mg frovatriptan succinate tablet was administrated 12 times in 7 days in the multiple-dose study. Blood samples were collected at scheduled time points.
Results: The results in single-dose study indicated that the blood levels were proportional to the administered dose, with the mean Cmax and AUClast ranging from approximately 6.27 ng/mL– 17.35 ng/mL and 92.52 h⋅ng/mL – 287.40 h⋅ng/mL over the dose range. In the multiple-dose study, moderate drug accumulation was noted, which was attributable to forvatriptan’s long t1/2 of about 26.47 to 30.63 h. Gender differences were noticed in both single- and multiple-dose study; exposure PK parameters were consistently higher in female than in male.
Conclusion: These pharmacokinetic evaluations in healthy Chinese subjects found that frovatriptan succinate tablet has an acceptable pharmacokinetic profile in Chinese subjects.
Keywords: frovatriptan succinate, pharmacokinetics, single-dose, multiple-dose
Introduction
Triptans are a class of selective serotonin 5-hydroxytryptamine (5-HT) 1B/1D agonists that is recommended as a first-line treatment for moderate to severe migraine.1,2 Despite the similar molecular structures, individual triptans differ in pharmacokinetic and pharmacodynamic properties and can be categorized into 2 main groups. Some triptans, such as sumatriptan, have a faster onset of action, belong to group 1. On contrast, other triptans, such as frovatriptan, have a slower onset of action and belong to group 2.3
Frovatriptan was developed to provide a long duration of action and reduced potential for side effects and drug interactions.4 It is reported that frovatriptan results in good efficacy 2 and 4 hours after administration comparing to rizatriptan and lower relapse rate as compared with rizatriptan, zolmitriptan, and almotriptan.5 Frovatriptan is also supported to be used for the acute and short-term prophylaxis of menstrual migraine.6,7 In clinical practice, the recommended dose of frovatriptan is 2.5 mg per time.8 The pharmacokinetic profile of multiple oral doses of frovatriptan in healthy subjects is characterized by long t1/2 (about 26 h), with a Tmax of 2 to 4 hours. Steady state is reached in about 4 to 5 days with linear pharmacokinetics over a dose range of 1 to 40 mg. The CYP1A2 isoenzyme of cytochrome P-450 is the primary metabolic pathway for frovatriptan. Frovatriptan reversibly distributes into blood cells, especially erythrocytes, with the binding rate of about 60%.9
The present report is based on findings from a Phase I clinical study that was conducted in Chinese healthy male and female subjects, which add to our understanding of the clinical pharmacokinetics (PK) after single- and multiple-dose administration of frovatriptan succinate tablet treatment.
Methods
Study Information
The study was performed at Phase I Clinical Research Center of Nanjing First Hospital, Nanjing, China, approved by the Ethics and Research Committee of Nanjing First Hospital (Ethic Approval Number YW20180921-03) and the Clinical trial registration number is CTR20190462. All research processes were in line with the requirements of International Conference on Harmonization (ICH) and Good Clinical Practice (GCP), the existing Declaration of Helsinki regarding medical research in humans and relevant laws and regulations of National Medical Products Administration. All participants were informed of the study’s aim, procedures and potential risks and voluntarily signed the informed consent forms prior to the initiation of the study.
Subjects
All 24 healthy subjects were included in the study with 12 in the single-dose format study and 12 in the multiple-dose format study. Eligible subjects in the study included healthy Chinese men and women aged from 18 to 45 years, having a body mass index (BMI) varied from 19 to 26 kg/m2 (including the boundary value). The weight of male subjects should not be less than 50 kg, whereas female subjects should weigh no less than 45 kg. All of the subjects involved in the studies underwent a strict screening process, including the collection of demographic data (birth date, gender, nation), the measure of height and weight, a detailed review of their medical and surgical histories and complete medical examinations, such as physical examinations, 12-lead electrocardiogram (ECG), chest X-ray and clinical laboratory tests. None of the subjects were smokers or alcoholics. Anyone who had a history of drug abuse was excluded. Women with childbearing potential were required to take a pregnancy test at screening.
Study Design
The single-dose format study was conducted according to a randomized, open-label, three-period, and three crossover design. Twelve healthy subjects (six men and six women) were randomly divided into three groups (two men and two women per group). Subjects of each group received each of the following 3 treatment procedures in a randomized sequence: 2.5-5.0-10.0 mg, 5.0-10.0-2.5 mg and 10.0-2.5-5.0 mg, according to a computer-generated randomization schedule (SAS 9.4). There was a 10-day washout time between each period. Prior to drug administration, all subjects were required to keep fasting (only water was allowed) for at least 10 hours and blank blood samples were collected within 60 minutes before dosing. Water was permitted 1 h before and 1 h after administration and standard meals were served until 4 hours after administration.
In the multiple-dose format study, twelve subjects were arranged to take 2.5 mg frovatriptan succinate tablet on day 1 morning and 2.5 mg every 12 h from day 2 morning to day 7 morning for 11 times. On day 1 and day 7, keeping subjects fasted overnight for at least 10 hours prior to administration. No subjects were allowed to drink water 1 hour before and after administration, and standard meals were not given until 4 hours after administration. On day 2 to 6, drinking water is prohibited for 30 minutes before and after administration, and breakfast was provided 30 minutes after administration.
Frovatriptan succinate tablet (2.5 mg per tablet, batch No.170923) was supplied by CP Pharmaceutical Qingdao Co. Ltd. Subjects were required to take the tablets with 240mL warm water and stay in upright position of the upper body and avoid strenuous exercise 4 hours after administration each time.
Blood Sample Collection
Blood samples (2 mL each time) in the single-dose study were collected from upper extremity vein into the precooled anticoagulant vacutainers containing K2-EDTA (K2-ethylenediaminetetraacetic acid) at the time points as follows: 0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 5.0, 6.0, 8.0, 12.0, 24.0, 36.0, 48.0, 72.0 and 96.0 h.
In the multiple-dose study, pharmacokinetic sampling was implemented at 0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 5.0, 6.0, 8.0, 12.0 and 24.0 h on day 1. On day 4, 5, and 6 morning, 2 mL venous blood before drug administration was drawn to observe the minimum value of trough plasma drug concentration. Additional plasma samples were obtained at 0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 5.0, 6.0, 8.0, 12.0, 24.0, 36.0, 48.0, 72.0 and 96.0 h on day 7. Other conditions were the same as the single-dose study.
The well-mixed blood samples placed in an ice bath were separated into sample and backup tubes (no less than 0.8 mL respectively) within 1 h. Within 1 h after collected, the separated samples were stored at −20°C temporarily and transferred to −70°C within 12 h, or stored at −70°C directly.
Determination of Drug Concentration
Frovatriptan concentrations in whole blood were determined by validated liquid chromatography-tandem mass spectrometry (LC-MS/MS). The LC-MS/MS system consists of a Nexera UHPLC LC-30A liquid chromatograph (SHIMADZU CORPORATION, Japan) and Triple Quad 5500 mass spectrometer (AB SCIEX, America) with an electrospray ionization source. For pretreatment, a volume of 100 μL blood sample was precipitated by 450 μL precipitant with internal standard (frovatriptan-d3 methanol solution: acetonitrile=1:1). An ACE-C8 column (50×2.1 mm, 3 μm) was used for chromatographic separation. The mobile phase for elution was a mixture of water with 0.1% formic acid as mobile phase A and methanol and acetonitrile (1:1) as mobile phase B. The gradient elution was as follows: 0–0.6 min, 4% B; 0.6–1.2 min, increase B to 95%, 1.2–1.8 min, 95% B, 1.8–1.85 min, reduce B to 4%, 1.85–2.5 min, 4% B. The elution was achieved at a flow rate of 0.55 mL/min with the column temperature of room temperature. The analyte was detected in positive mode and the optimized detecting conditions were summarized as follows: heater temperature 500°C; electrospray voltage 5500 V; curtain gas 20 Arb; collision gas 9 Arb; entrance potential 10 V; collision cell exit potential 10 V; ion source gas 1 70 Arb, ion source gas 2 80 Arb; collision energy 61 eV. The transitions m/z 244.14→213.1 for frovatriptan and m/z 247.13→168.09 for frovatriptan-d3 were monitored using multiple reaction monitoring mode.
The method is linear between the range of 0.0800 to 32.0 ng/mL. The inter-assay precision of quality control samples analyzed throughout the study was between 3.00% and 5.74%. Inter-assay accuracy varied between 95.9% and 109.19%.
Pharmacokinetic Analyses
Relevant pharmacokinetic parameters were analyzed based on a noncompartmental model using Phoenix WinNonlin 7.0 (Pharsight Corp, Mountain View, CA, USA). Analyte concentrations below the limit of quantification (BLQ) were set to zero. Values for Cmax of frovatriptan and Tmax were from the observed data by concentration-time curve. λz, the terminal-phase elimination rate constant, was equal to the slope calculated by a log-linear regression of the terminal-phase of the concentration-time curve. The half-life (t1/2 was calculated as (ln2)/ λz). For single- and multiple-dose pharmacokinetics, the AUC from time 0 to the time of the last quantifiable concentration (AUClast) or during 12 h (AUC12) was calculated by using the linear trapezoid method and AUC from time zero extrapolated to infinite time (AUCinf) was calculated as AUClast + Ct/λz (Ct, the last measurable concentration). AUC from time 0 to time tau (the dosing interval) at steady state (AUCtau,ss) was calculated, where tau = 12 hours. Apparent oral clearance CLz/F was calculated according to dose/AUCinf for single dose and dose/AUCtau,ss for multiple doses. The terminal-phase apparent volume of distribution after oral administration (VZ/F) was calculated by dividing clearance by λZ. The accumulation index RAUC and RCmax were calculated as day 7 AUCtau,ss/day 1 AUC12 and day 7 Cmax,ss/day 1 Cmax, respectively.
Statistical Analysis
SPSS version 26.0 was used for all statistical analysis and findings were considered statistically significant if p< 0.05. All experiments were expressed as mean ± SD (standard deviation). Nonparametric tests were used for Tmax in single- and multiple-dose study. Analysis of variance (ANOVA) was used for other parameters in single-dose study. Comparisons between gender and between the multiple-dose and single-dose data of 2.5 mg were performed using the Student’s t-test. All statistical tests used a 5% level of significance to determine significance. All P values cited were 2-tailed.
Safety Assessments
Safety was assessed in the whole clinical trial, including adverse event reports, results of physical examinations, vital signs, clinical laboratory evaluations, and ECGs.
Results
Demographics
Table 1 summarizes the baseline characteristics of subjects in the study.
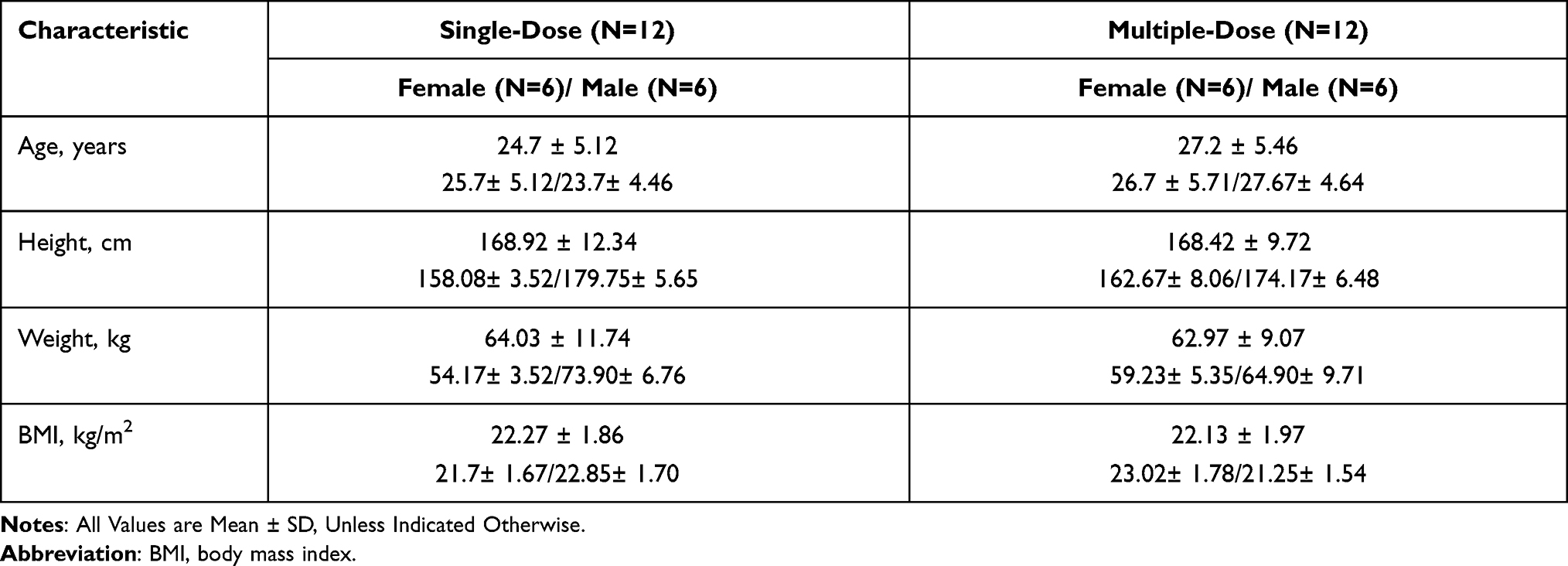 |
Table 1 Baseline Characteristics of Subjects Receiving Frovatriptan in the Study |
Pharmacokinetics
Single-Dose Study
The whole blood concentration – time curves after single oral doses of 2.5, 5 and 10 mg frovatriptan succinate tablet are illustrated in Figure 1. Pharmacokinetic parameters are presented in Table 2. Frovatriptan levels increased proportionally with increasing dose of frovatriptan succinate tablet. A regression of frovatriptan revealed a linear relationship with Cmax(r2= 0.9999), AUClast (r2=0.9996) and AUCinf (r2= 0.9994) proportional to dose between oral dose 2.5 mg to 10 mg.
 |
Table 2 Pharmacokinetic Parameters (Mean ± SD) of Frovatriptan in Whole Blood After Single-Dose Oral Administration |
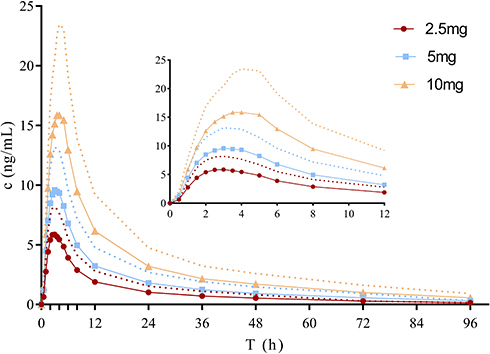 |
Figure 1 Mean (SD) blood concentration-time curve of frovatriptan after single-dose oral administration. (n = 12 per dose). Notes: The solid line shows the mean blood concentration-time curve of frovatriptan. The dotted curve denote the standard deviations (SD). Inset show the 0~12h interval blood concentration-time curve of frovatriptan after single-dose oral administration. |
PK parameters of frovatriptan succinate in whole blood by gender are summarized in Table 3. The Tmax was slightly longer in women (3.25–4.5 h) than in men (2.75–3.0 h). Systemic exposure was higher in female subjects compared with male subjects. AUClast and AUCinf were approximately 2-fold higher in female than male under doses 2.5 mg and 5 mg. Cmax under dose 2.5 mg were significantly (p< 0.01) higher in female compared with male, whereas Vz/F and CLz/F under dose 2.5 mg and CLz/F under dose 10 mg were significantly (p< 0.05) lower in female compared with male.
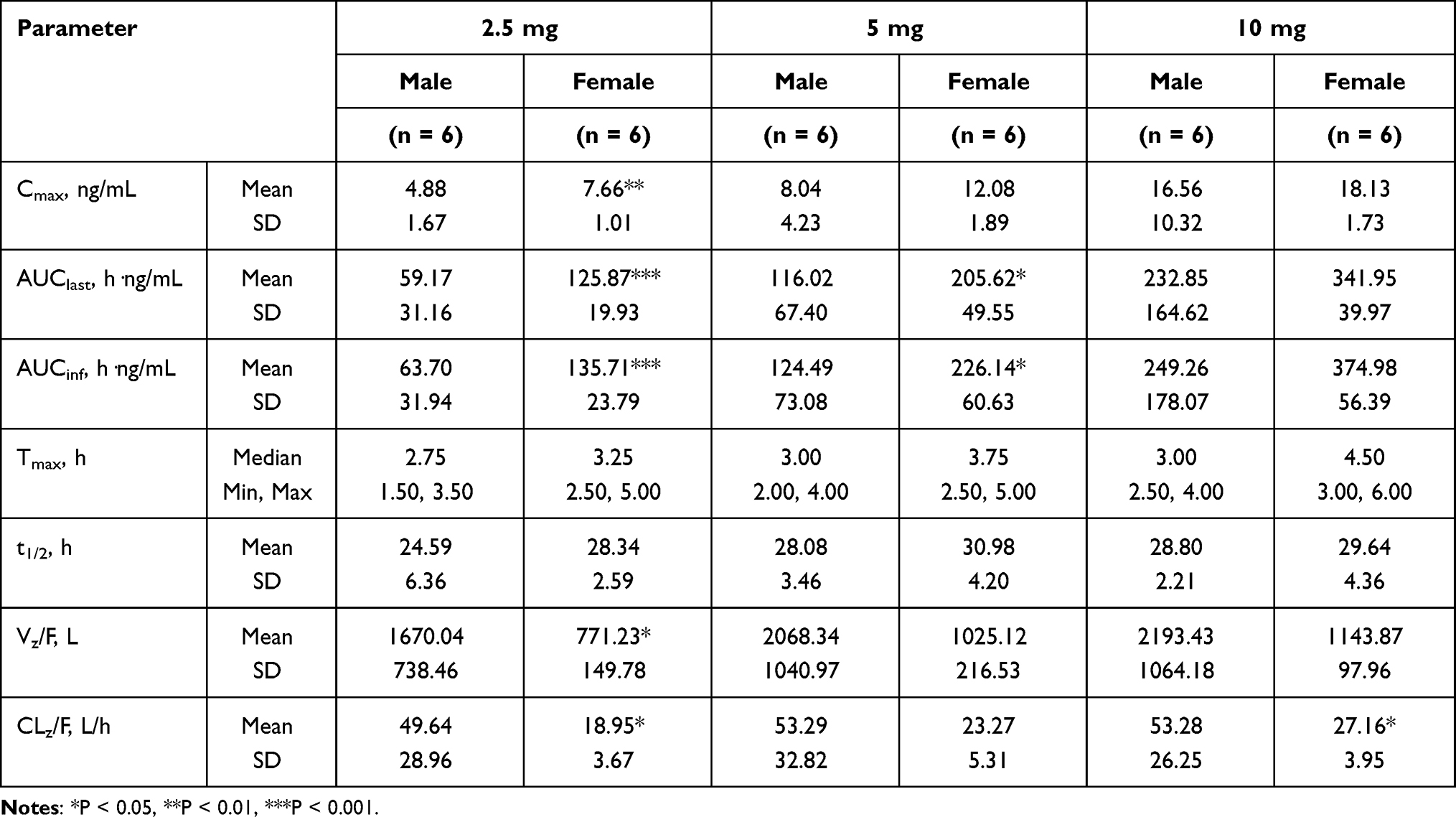 |
Table 3 Pharmacokinetic Parameters of Frovatriptan in Whole Blood by Gender After Ascending Single-Dose Oral Administration |
Multiple-Dose
One of the twelve subjects (random No. 019) refused to take administration on day 2 evening and withdrew from the study. Therefore, except the pharmacokinetic parameters of the 12 subjects after the first dose, the pharmacokinetic parameters of the stable states and the last dose were collected from only 11 subjects.
The whole blood concentration – time curves after multiple oral dose of 2.5 mg frovatriptan succinate tablet are illustrated in Figure 2 and PK parameters are presented in Table 4. Steady state (based on through blood concentrations) was achieved by day 4. Twice-daily dosing produced approximately 2-fold higher mean values for Cmax (p< 0.01) on day 7 than on day 1.
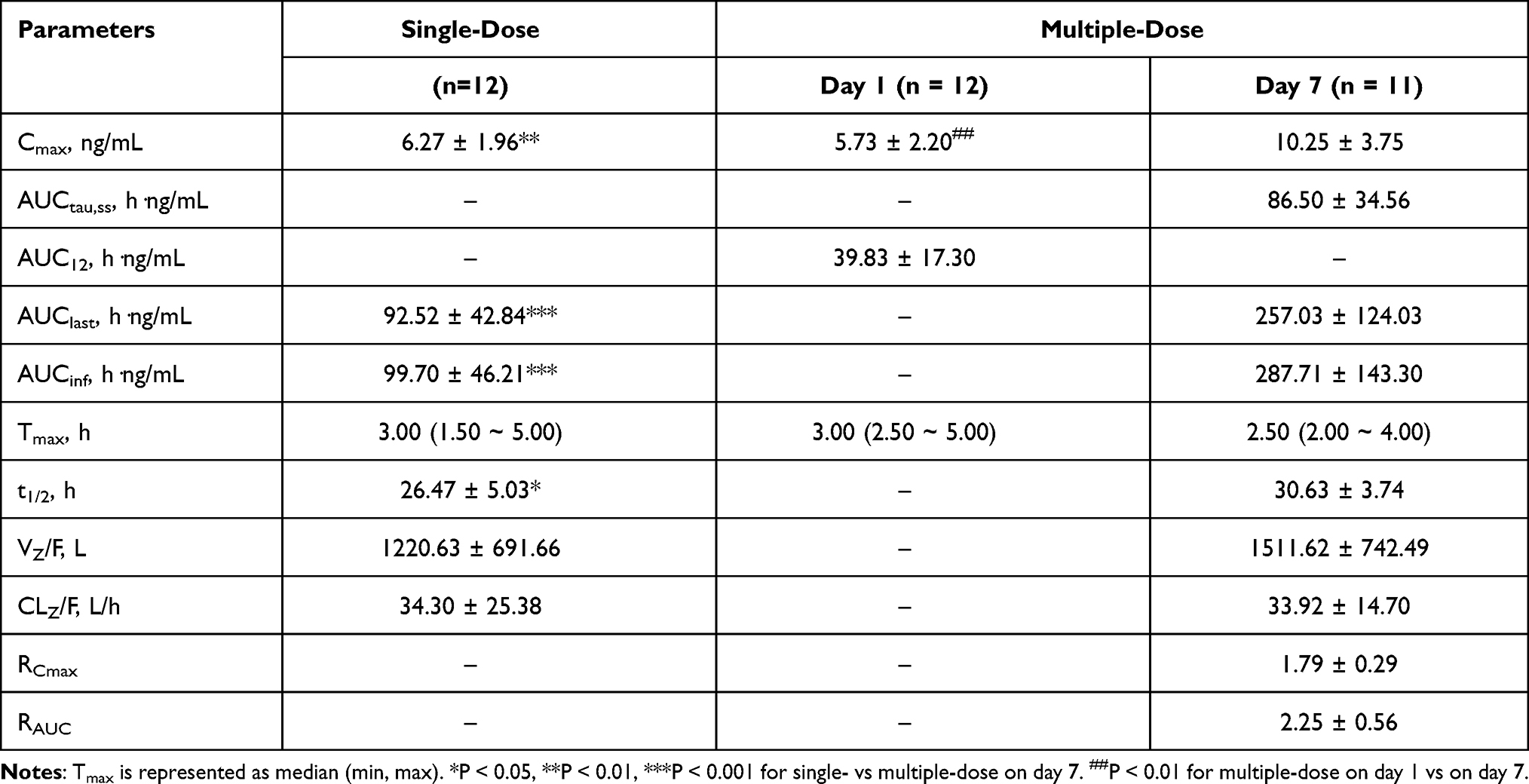 |
Table 4 Pharmacokinetic Parameters (Mean ± SD) of Frovatriptan in Whole Blood After Single-Dose (2.5 Mg) and Multiple-Dose Oral Administration |
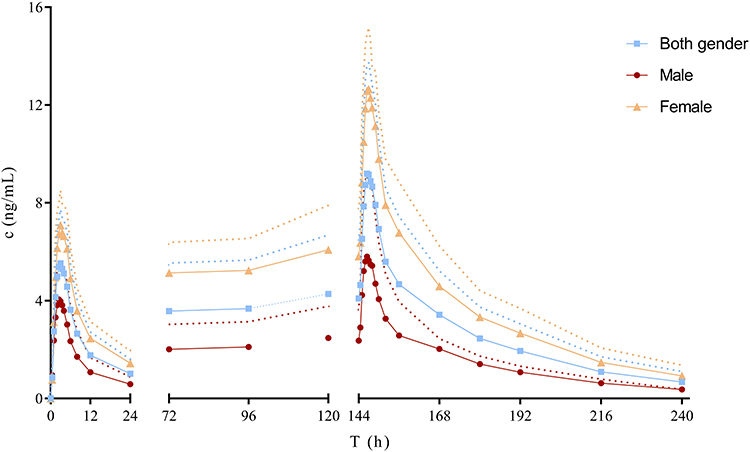 |
Figure 2 Mean (SD) blood concentration-time curve of frovatriptan after 2.5 mg multiple-dose oral administration. (0~24 h, n = 12; 72~240 h, n = 11). Notes: The solid line shows the mean blood concentration-time curve of frovatriptan. The dotted curve denote the standard deviations (SD). |
Systemic exposure was higher in female subjects than in male subjects (Figure 2 and Table 5). AUClast, AUCtau,ss and AUCinf were more than 2-fold higher in female than male. Cmax was significantly (p< 0.01) higher in female compared with male, whereas Vz/F and CLz/F were lower in female compared with male.
 |
Table 5 Pharmacokinetic Parameters (Mean ± SD) of Frovatriptan in Whole Blood by Gender After Ascending Multiple-Dose Oral Administration (Day 7) |
Safety Assessment
In total, there were 44 adverse events (AE) in 11 subjects (91.7%) during single-dose format study and 11 AE in 5 subjects (41.7%) during multiple-dose format study. All the AE were related to neurological and gastrointestinal systems. During single- and multiple- dose study, dizziness (58.3% and 8.3%, respectively), headache (41.7% and 16.7%, respectively), nausea (25.0% and 8.3%, respectively) were the most frequently reported adverse events. No serious AEs were reported over the course of the studies.
Discussion
This report describes pharmacokinetic data from a phase I study of frovatriptan succinate tablet in healthy Chinese subjects. Frovatriptan succinate exhibited linear pharmacokinetics in single doses of 2.5 mg to 10 mg. Mean values for t1/2 (26.47–29.53 h) and CLz/F (34.30–40.22 L/h) were almost constant, regardless of administered dose. Frovatriptan has a high volume of distribution (Vz/F of approximately 1220–1668 L), which indicates that in addition to being distributed into erythrocytes,9 frovatriptan may also be concentrated into a wide range of tissues or organs.
Significant accumulation effect was observed over 7 days of administration, which is attributable to the long terminal half-life. After repeated doses of 2.5 mg frovatriptan succinate, the Cmax, AUClast and AUCinf were higher than the corresponding values obtained after 2.5 mg single-dose administration. RCmax and RAUC was 1.79 and 2.25 respectively. Patients who take long-term medication should pay attention to drug accumulation and receive plasma concentration monitoring if necessary.
Gender differences were noticed in our previous study as well.9,10 Our work has shown that the exposure PK parameters were higher for female subjects compared with male subjects, while the distribution and clearance were higher in male than in female in both single- and multiple-dose studies. Estrogen is a well-known endogenous competitive inhibitor of CYP1A211 that metabolizes frovatriptan,9 which leads to higher CYP1A2 activity in men than in women,12 and may partly contribute to the gender differences of frovatriptan metabolism. Other possible reasons include the Gender differences of liver and kidney size, and blood flow between male and female.13
There is no evidence suggesting that frovatriptan succinate PK profiles differ based on race. However, our study suggests that Chinese subjects may have higher exposure than Caucasian. Buchan9 and Elkind10 assessed the pharmacokinetic parameters of 2.5 mg single-dose of frovatriptan by gender. The reported Cmax of male and female in Buchan’s study was 4.23 ng/mL and 7.00 ng/mL, respectively, the Tmax was 2.3 h and 3.0 h, respectively, and the AUCinf was 42.9 h·ng/mL and 94.0 h·ng/mL, respectively. The reported Cmax of adult male and female in Elkind’s study was 4.10 ng/mL and 6.98 ng/mL, respectively, the Tmax was 2 h and 3 h, respectively, and the AUCinf of adult female was 96.7 h·ng/mL. Whereas in our study, the AUCinf of male and female (63.70 h·ng/mL and 135.71 h·ng/mL, respectively) and Tmax (2.75 h and 3.25 h, respectively) were higher than the two previous studies in Caucasian. One in vitro study14 suggested that CYP1A2 activity is lower in Chinese than in Caucasians, which could possibly explain the differences between our study and the two previous studies. However, one limitation of our study is that the number of participants in each gender subgroup was small (n = 6 or 5), further studies are needed to evaluate the impact of ethnicity on frovatriptan succinate.
In conclusion, the results of our studies demonstrate the linear pharmacokinetics of frovatriptan succinate tablet in healthy Chinese volunteers. Based on our observation, frovatriptan succinate has an acceptable pharmacokinetic profile in healthy young volunteers.
Data Sharing Statement
The individual participant’s data that underlying the results reported in this article would be accessed with approval from the corresponding author after 6 months of publication. The study protocol, statistical analysis plan and clinical study report will also be available.
Funding
This study was sponsored by CP Pharmaceutical Qingdao Co., Ltd., Qingdao, Shandong, China.
Disclosure
There is no competing interests among the authors.
References
1. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: the American headache society evidence assessment of migraine pharmacotherapies. Headache. 2015;55(1):3–20. doi:10.1111/head.12499
2. Worthington I, Pringsheim T, Gawel MJ, et al. Canadian headache society guideline: acute drug therapy for migraine headache. Can J Neurol Sci. 2013;40(5 Suppl 3):S1–s80. doi:10.1017/S0317167100118943
3. Ong JJY, De Felice M. Migraine treatment: current acute medications and their potential mechanisms of action. Neurotherapeutics. 2018;15(2):274–290. doi:10.1007/s13311-017-0592-1
4. Sanford M. Frovatriptan: a review of its use in the acute treatment of migraine. CNS Drugs. 2012;26(9):791–811. doi:10.2165/11209380-000000000-00000
5. Evers S, Savi L, Omboni S, Lisotto C, Zanchin G, Pinessi L. Efficacy of frovatriptan as compared to other triptans in migraine with aura. J Headache Pain. 2015;(16):514. doi:10.1186/s10194-015-0514-8
6. Elkind AH, MacGregor EA. Frovatriptan for the acute treatment of migraine and prevention of predictable menstrual migraine. Expert Rev Neurother. 2008;8(5):723–736. doi:10.1586/14737175.8.5.723
7. MacGregor EA. A review of frovatriptan for the treatment of menstrual migraine. Int J Womens Health. 2014;6(1):523–535. doi:10.2147/ijwh.S63444
8. Frova. Drugs-professionals-FDA professional drug information; 2021, Available from: https://www.drugs.com/pro/frova.html.
9. Buchan P, Keywood C, Wade A, Ward C. Clinical pharmacokinetics of frovatriptan. Headache. 2002;42(s2):54–62. doi:10.1046/j.1526-4610.42.s2.3.x
10. Elkind AH, Wade A, Ishkanian G. Pharmacokinetics of frovatriptan in adolescent migraineurs. J Clin Pharmacol. 2004;44(10):1158–1165. doi:10.1177/0091270004268046
11. Landi MT, Sinha R, Lang NP, Kadlubar FF. Human cytochrome P4501A2. IARC Sci Publ. 1999;148:173–195.
12. Ou-Yang DS, Huang SL, Wang W, et al. Phenotypic polymorphism and gender-related differences of CYP1A2 activity in a Chinese population. Br J Clin Pharmacol. 2000;49(2):145–151. doi:10.1046/j.1365-2125.2000.00128.x
13. Anderson GD. Gender differences in pharmacological response. Int Rev Neurobiol. 2008;83:1–10. doi:10.1016/s0074-7742(08)00001-9
14. Yang J, He MM, Niu W, et al. Metabolic capabilities of cytochrome P450 enzymes in Chinese liver microsomes compared with those in Caucasian liver microsomes. Br J Clin Pharmacol. 2012;73(2):268–284. doi:10.1111/j.1365-2125.2011.04076.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.