Back to Journals » Drug Design, Development and Therapy » Volume 11
Pharmacokinetic interactions between glimepiride and rosuvastatin in healthy Korean subjects: does the SLCO1B1 or CYP2C9 genetic polymorphism affect these drug interactions?
Authors Kim CO, Oh ES, Kim H, Park MS ![]()
Received 12 December 2016
Accepted for publication 19 January 2017
Published 23 February 2017 Volume 2017:11 Pages 503—512
DOI https://doi.org/10.2147/DDDT.S129586
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Choon Ok Kim,1 Eun Sil Oh,2 Hohyun Kim,3 Min Soo Park1,4
1Department of Clinical Pharmacology, Severance Hospital, Yonsei University College of Medicine, Seoul, 2Department of Pharmaceutical Medicine and Regulatory Sciences, College of Medicine and Pharmacy, Yonsei University, Incheon, 3Korea Medicine Research Institute, Inc., Seongnam, 4Department of Pediatrics, Yonsei University College of Medicine, Seoul, Korea
Abstract: To improve cardiovascular outcomes, dyslipidemia in patients with diabetes needs to be treated. Thus, these patients are likely to take glimepiride and rosuvastatin concomitantly. Therefore, this study aimed to evaluate the pharmacokinetic (PK) interactions between these two drugs in healthy males and to explore the effect of SLCO1B1 and CYP2C9 polymorphisms on their interactions in two randomized, open-label crossover studies. Glimepiride was studied in part 1 and rosuvastatin in part 2. Twenty-four participants were randomly assigned to each part. All subjects (n=24) completed part 1, and 22 subjects completed part 2. A total of 38 subjects among the participants of the PK interaction studies were enrolled in the genotype study to analyze their SLCO1B1 and CYP2C9 polymorphisms retrospectively (n=22 in part 1, n=16 in part 2). Comparison of the PK and safety of each drug alone with those of the drugs in combination showed that both glimepiride and rosuvastatin did not interact with each other and had tolerable safety profiles in all subjects. However, with regard to glimepiride PK, the SLCO1B1 521TC group had a significantly higher maximum plasma concentration (Cmax,ss) and area under the plasma concentration–time curve during the dose interval at steady state (AUCt,ss) for glimepiride in combination with rosuvastatin than those for glimepiride alone. However, other significant effects of the SLCO1B1 or CYP2C9 polymorphism on the interaction between the two drugs were not observed. In conclusion, there were no significant PK interactions between the two drugs; however, the exposure to glimepiride could be affected by rosuvastatin in the presence of the SLCO1B1 polymorphism.
Keywords: glimepiride, rosuvastatin, pharmacokinetics, SLCO1B1, CYP2C9
A Letter to the Editor has been received and published for this article.
Introduction
According to the International Diabetes Federation, ~8.3% of the adult population worldwide has diabetes, and it has been increasing in the last decade.1 Diabetes is one of the leading causes of mortality and morbidity.2 The major cause of mortality in diabetes is cardiovascular disease, and adults with diabetes have a two- to fourfold higher risk of cardiovascular disease than that among nondiabetic adults.3 Especially, diabetic dyslipidemia attributes to ~80% of deaths due to cardiovascular complications.4 It is necessary to treat dyslipidemia in patients with diabetes;3 therefore, these patients are likely to take lipid-lowering drugs and antidiabetic drugs simultaneously. Thus, it is important to consider potential drug interactions between lipid-lowering drugs and antidiabetic drugs.
For type 2 diabetic patients, sulfonylureas usually are the second-line therapy for increasing insulin secretion.5 Among them, glimepiride is a widely used third-generation sulfonylurea.4 Glimepiride is completely absorbed after oral administration, and its oral bioavailability is close to 100%.6 It is metabolized mostly by cytochrome P450 (CYP) 2C9.6 In addition, a previous in vitro study suggested that it could inhibit organic anion-transporting polypeptide (OATP) 1B1, which is a hepatic uptake transporter encoded by the solute carrier organic anion transporter 1B1 (SLCO1B1).7 Statins are the first-line drugs for the prevention and treatment of diabetic complications such as cardiovascular disease and diabetic dyslipidemia.8 Rosuvastatin is a relatively potent and tolerable statin and is commonly prescribed for dyslipidemia patients with diabetes.9 Its absorption from the gastrointestinal tract is ~50%, and its oral bioavailability is estimated to be 20%.10 Rosuvastatin undergoes substantial hepatic first pass extraction via OATP1B1.11 Although it is not extensively metabolized, CYP2C9 does play a minor role in its metabolism (<10%).11
Glimepiride and rosuvastatin are partially metabolized by the same metabolic pathway, and they may interact via hepatic OATP1B1. However, although glimepiride and rosuvastatin have been used concomitantly in clinical practice, it remains unclear whether a pharmacokinetic (PK) interaction between these two drugs exists in humans. In addition, there have been substantial studies showing that the genetic polymorphisms of CYP2C9 or SLCO1B1 have a certain functional or clinical significance in the PK of these drugs.7,12 Therefore, this study aimed to investigate the PK interaction between glimepiride and rosuvastatin in healthy subjects and to assess the influence of the genetic polymorphisms of CYP2C9 and SLCO1B1 on the PK interaction between these two drugs.
Methods
Ethics
This study included a PK interaction study and a genotyping study. Each study protocol was approved by the Institutional Review Board (IRB) of Severance Hospital, Yonsei University College of Medicine (Seoul, Korea) and was conducted in accordance with the Declaration of Helsinki and the guidelines from the International Conference on Harmonization of Pharmaceuticals for Human Use-Good Clinical Practice (IRB for PK interaction study: 4-2013-0227 and IRB for genotyping study: 4-2013-0163). All participants were enrolled in the study after they provided written informed consent.
Subjects
Healthy Korean male subjects aged 20–45 years participated in the PK interaction study. Their body mass index was between 18.5 and 25.0 kg/m2 (body weight, kg/[height, m]2). All subjects were ascertained to be healthy by a review of their medical history, a physical examination, measurements of 12-lead electrocardiography (ECG), and laboratory tests. Volunteers who met the following criteria were not included: a medical history that might influence the PK of glimepiride or rosuvastatin, history of a clinically significant hypersensitivity to drugs or foods, systolic blood pressure >150 or <90 mmHg, diastolic blood pressure >100 or <50 mmHg, fasting blood glucose <60 mg/dL, or positive results in a serology test (hepatitis B surface antigen, anti-hepatitis C virus antibodies, and/or anti-HIV antibodies) or urine drug screening test. The study participants were not allowed to take any medications or herbals and to consume alcohol, caffeinated beverages, and grapefruit products as well as to smoke during the study. Only those participants in the PK interaction study who provided written informed consent for the genotyping study were included in the latter. Genotypes were assessed retrospectively after the PK study.
Study design
The PK interaction study consisted of two parts (part 1 and part 2). Each part was a randomized, open-label, multiple-dose, two-treatment, two-sequence crossover design, and volunteers were recruited separately. Part 1 was designed to evaluate the effect of rosuvastatin on the PK of glimepiride. The two treatments in part 1 were as follows: a 4 mg glimepiride tablet once daily for 7 days (treatment G), and coadministration of a 4 mg glimepiride tablet and a 20 mg rosuvastatin tablet once daily for 7 days (treatment GR). A total of 24 participants were randomly assigned to one of the two treatment sequence groups (G-GR or GR-G). Each treatment period was separated by a 14-day washout period. All participants received each treatment with 240 mL water under fasting conditions according to the assigned treatment sequence. Peripheral venous blood was collected in sodium heparin tubes prior to dosing on days 5, 6, and 7, and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 24, and 48 h after administration on day 7 in each period.
Part 2 was designed to determine the effect of glimepiride on the PK of rosuvastatin. There were two treatments: a 20 mg rosuvastatin tablet once daily for 7 days (treatment R) and treatment GR as described earlier. A total of 24 subjects were enrolled and were randomly assigned to one of the two treatment sequence groups (R-GR or GR-R). There was a 14-day washout period between the two sequence periods. Following the assignment to the respective treatment group and after administration of each study drug under fasting conditions, peripheral venous blood was collected in sodium heparin tubes on days 5, 6, and 7, and at 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 10, 12, 16, 24, and 48 h after dosing on day 7 in each period. The blood samples collected in part 1 and part 2 were centrifuged, and each aliquot was stored at or below −70°C until analysis.
A total of 38 subjects among the participants who were enrolled in the PK interaction study were recruited and provided written informed consent for the genotyping study (n=22 in part 1, n=16 in part 2). Whole blood was collected once before the first study drug administration and was stored at or below −70°C until analysis. After completion of the PK interaction study, the genotypes were analyzed retrospectively, and a potential influence of the genetic polymorphisms on the PK interaction between glimepiride and rosuvastatin was assessed.
Plasma glimepiride, rosuvastatin, and N-desmethyl rosuvastatin assay
The plasma concentrations of glimepiride, rosuvastatin, and N-desmethyl rosuvastatin were measured using a validated high-performance liquid chromatography assay (Acquity UPLC system; Waters, Milford, MA, USA) coupled with tandem mass spectrometry (MS/MS, Xevo TQ-S; Waters). For glimepiride, 50 μL of plasma samples was mixed with 20 μL of clopidogrel as an internal standard (200 ng/mL in 50% methanol). After centrifugation, 100 μL of supernatant was diluted with 100 μL of water, and 10 μL of the resultant solution was injected directly into the column heated at 500°C. The mobile phase was used at a flow rate of 0.25 mL/min. The lower limit of quantification was 1.0 ng/mL. The calibration curve was linear over the concentration range of 1.0–500 ng/mL (correlation coefficient [r2] =0.997). The precision of the assay was <20%, and the accuracy was within the range of 80%–120%.
For rosuvastatin and N-desmethyl rosuvastatin, 50 μL of plasma samples was mixed with 10 μL of valsartan, an internal standard (100 ng/mL in 50% acetonitrile). After the mixture was centrifuged, the supernatant was concentrated and evaporated using a centrifugal vaporizer (CVE-2000; Tokyo RIKAKIKAI Co., Tokyo, Japan) for 20 min (45°C), and the residues were reconstituted with 200 μL of 50% acetonitrile. Then, 10 μL of the resultant solution was injected into the column. The flow rate of the mobile phase was 0.25 mL/min. The lower limit of quantification of rosuvastatin and N-desmethyl rosuvastatin was 0.1 and 0.5 ng/mL, respectively. The calibration curve was linear over the concentration range of 0.1–100 ng/mL (r2=0.997) for rosuvastatin and 0.5–100 ng/mL (r2=0.997) for N-desmethyl rosuvastatin. The precision of the assays was <20%, and the accuracy was within the range of 80%–120%.
PK analysis
The PK parameters of glimepiride, rosuvastatin, and N-desmethyl rosuvastatin were calculated by noncompartmental analysis using the Phoenix 64 WinNonlin 6.3 software (Pharsight, Mountain View, CA, USA). The maximum plasma concentration (Cmax,ss) and the time to reach the Cmax,ss (tmax,ss) at steady state were determined directly from the observed data. The area under the plasma concentration–time curve during the dose interval at steady state (AUCτ,ss) was calculated using the linear trapezoidal rule. The terminal elimination rate constant (λz) was estimated by log-linear regression analysis. The elimination half-life (t1/2) and the apparent plasma clearance (CL/F) were calculated from the equations t1/2= ln(2)/λz and CL/F = dose/AUCinf,ss, respectively.
Safety assessment
Safety was assessed throughout the study. All participants underwent a physical examination, monitoring of vital signs (systolic blood pressure, diastolic blood pressure, pulse rate, and body temperature), 12-lead ECG, and laboratory tests at predefined time points. In addition, adverse events (AEs) were evaluated by self-reporting or monitoring. Any undesirable sign, symptom, or medical condition occurring after the administration of the study drug was recorded, regardless of its suspected relationship to the study medication.
Genetic analysis
Genomic DNA was isolated from a peripheral blood sample using the QIAmp® DNA QIAcube Ht Kit (Qiagen, Hilden, Germany). Single nucleotide polymorphisms (SNPs) in SLCO1B1 521T>C (rs4149056) and CYP2C9*3 (rs1057910) were genotyped using TaqMan® assays (Applied Biosystems, Foster City, CA, USA). The ratio of the genotyping call was >99% and the technical duplicates yielded the same genotype in every case. The real-time PCR reaction was performed in a final volume of 10 μL, including 15 ng of genomic DNA, 5 μL TaqMan® Universal PCR Master Mix, and 0.5 μL of 20× SNP Assay. The thermal cycling conditions were as follows: initial denaturing at 95°C for 10 min, 45 cycles of 95°C for 15 s, and 60°C for 1 min. The HID Real-Time PCR Analysis Software version 1.2 (Thermo Fisher Scientific, Waltham, MA, USA) was used for allelic discrimination. Genotyping was performed on a QuantStudio™ 6 Flex Real-Time PCR System (Applied Biosystems).
Statistical analysis
The PK data were analyzed and compared between each drug monotherapy (glimepiride or rosuvastatin) and the combination therapy. All data are expressed as mean ± standard deviation (SD). The primary PK parameters (Cmax,ss and AUCτ,ss) were log-transformed and analyzed by analysis of variance using a mixed-effects model. To compare the PK parameters, point estimates and 90% confidence intervals (CIs) for the geometric mean ratios (combination therapy/monotherapy) of the log-transformed Cmax,ss and AUCτ,ss were also presented. Demographic characteristics were analyzed using the Wilcoxon rank-sum test for comparison among the two treatment sequences in each study part. All analyses were conducted using the SAS statistical software version 9.2 (SAS Institute, Inc., Cary, NC, USA). All statistical tests were two-sided, and statistical significance was defined as P≤0.05.
Results
Study participants
A total of 24 healthy Korean male individuals were enrolled, and 12 participants were randomly assigned to each treatment sequence group in part 1 and 2, respectively. All subjects (n=24) completed the part 1 study. Two participants voluntarily withdrew after study drug administration in part 2, and 22 subjects completed the part 2 study. There were no statistical differences in age, body weight, or body mass index among the sequence groups of each part (P>0.05, Table 1).
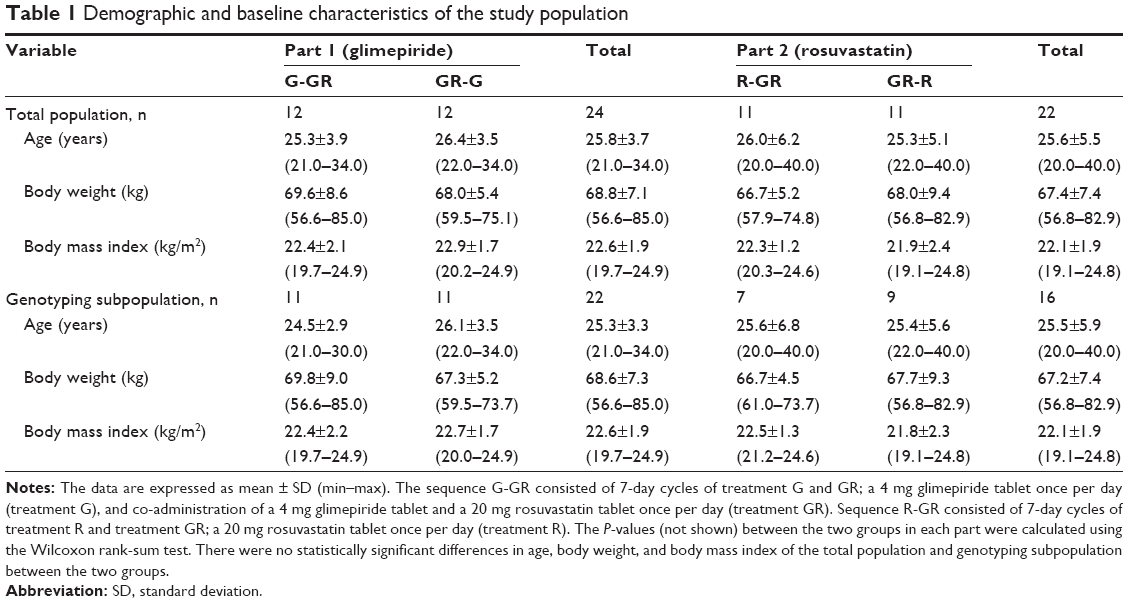 | Table 1 Demographic and baseline characteristics of the study population |
PK parameters
The coadministration of 4 mg glimepiride with 20 mg rosuvastatin once daily for 7 days resulted in a glimepiride mean plasma concentration–time profile at steady state similar to that of glimepiride monotherapy (Figure 1). The calculated PK parameters of glimepiride are shown in Table 2. The PK parameters of glimepiride monotherapy were similar to those of the glimepiride–rosuvastatin combination therapy. The point estimate (with 90% CI) of the geometric mean ratios of the glimepiride Cmax,ss and AUCτ,ss was 1.03 (0.91–1.16) and 1.03 (0.94–1.14), respectively.
 | Table 2 PK parameters of glimepiride after the administration of multiple oral doses of 4 mg glimepiride once per day (treatment G) and coadministration of 4 mg glimepiride and 20 mg rosuvastatin once per day (treatment GR) in healthy volunteers |
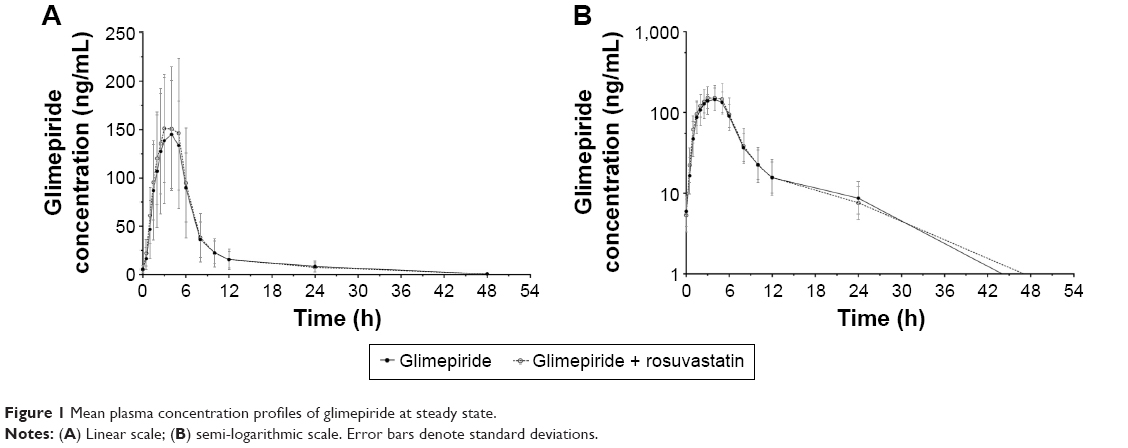 | Figure 1 Mean plasma concentration profiles of glimepiride at steady state. |
The mean plasma concentration–time profiles of rosuvastatin at steady state after 20 mg rosuvastatin with and without 4 mg glimepiride once daily for 7 days are shown in Figure 2. The PK parameters of rosuvastatin were similar to those of the glimepiride–rosuvastatin combination therapy, and the geometric mean ratios (90% CI) of the rosuvastatin Cmax,ss and AUCτ,ss were 1.12 (0.98–1.27) and 0.94 (0.86–1.03), respectively (Table 3). The Cmax,ss of N-desmethyl rosuvastatin in the combination therapy with glimepiride was similar to that of rosuvastatin monotherapy; however, the AUCτ,ss of N-desmethyl rosuvastatin in the combination therapy was decreased compared to that of the rosuvastatin monotherapy (Table 3). The geometric mean ratios (90% CI) of the N-desmethyl rosuvastatin Cmax,ss and AUCτ,s were 0.96 (0.84–1.10) and 0.83 (0.74–0.93), respectively.
 | Table 3 PK parameters of rosuvastatin after administration of multiple oral doses of 20 mg rosuvastatin once per day (treatment R) and coadministration of 4 mg glimepiride and 20 mg rosuvastatin once per day (treatment GR) in healthy volunteers |
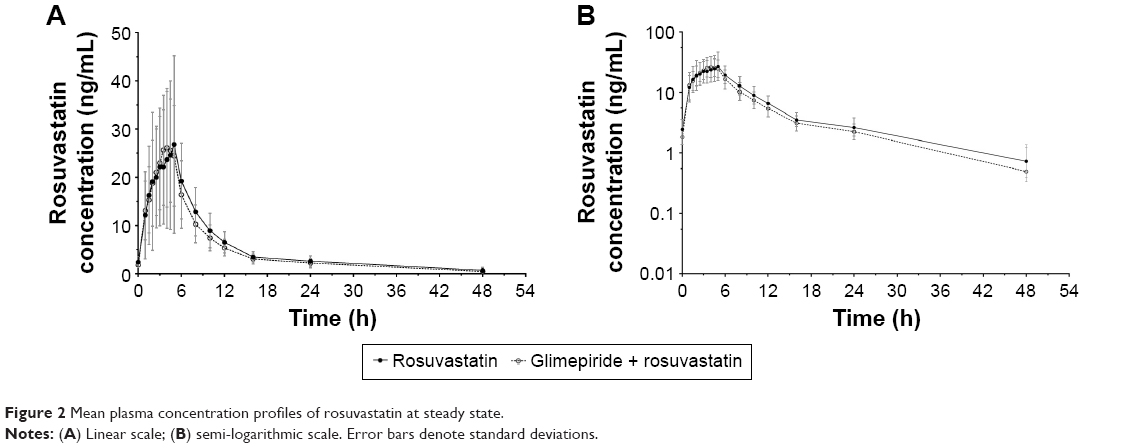 | Figure 2 Mean plasma concentration profiles of rosuvastatin at steady state. |
Genotyping
A total of 38 subjects were categorized according to the SLCO1B1 521T>C and CYP2C9*3 genotype and study groups (part 1 and part 2). The genotype frequency of the SLCO1B1 521T>C and CYP2C9*3 genotype was consistent with that found in other studies of the Korean population.13,14 No variant allele in the CYP2C9*3 genotype was observed in the part 2 study. Age, body weight, and body mass index were not significantly different between the genotype groups in both parts (P>0.05, Table 1).
In part 1, there were no statistically significant differences in the Cmax,ss and AUCτ,ss of glimepiride between the SLCO1B1 521TT and TC genotype group within the G or GR group, respectively (P>0.05, Table 4). Comparison of the GR group with the G group showed that the geometric mean ratios (95% CI) of the Cmax,ss and AUCτ,ss of glimepiride within the SLCO1B1 521TT genotype group were similar. However, the Cmax,ss and AUCτ,ss of glimepiride were higher (1.28-fold and 1.26-fold, respectively) in the GR group than those in the G group within the SLCO1B1 521TC genotype group. The CYP2C9 genotyping results showed that the Cmax,ss and AUCτ,ss of glimepiride in the CYP2C9*3 genotype group were higher than those in the CYP2C9*1 genotype group within the G group (P<0.05). In the GR group, the Cmax,ss and AUCτ,ss of glimepiride in the CYP2C9*3 genotype group were higher than those in the CYP2C9*1 group; however, only the difference in the Cmax,ss was statistically significant (P<0.05). There were no significant differences in the Cmax,ss and AUCτ,ss of glimepiride between the G and GR group of the same CYP2C9 genotype group.
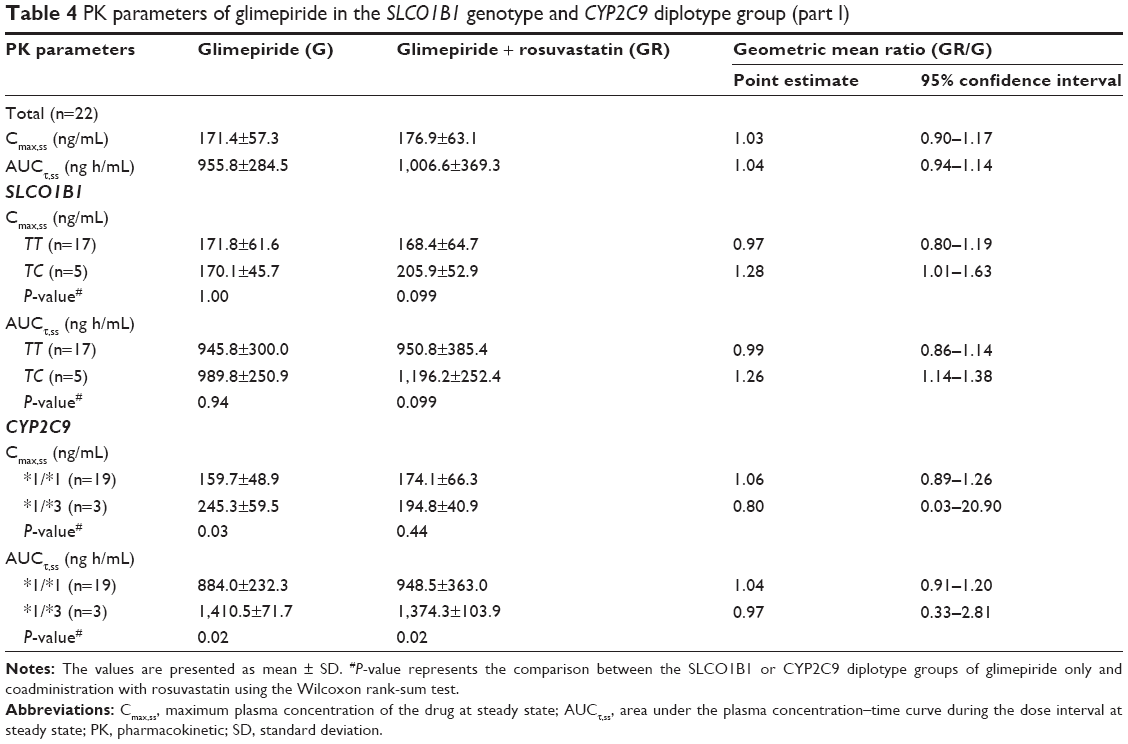 | Table 4 PK parameters of glimepiride in the SLCO1B1 genotype and CYP2C9 diplotype group (part I) |
In part 2, similar Cmax,ss and AUCτ,ss of rosuvastatin were observed between the SLCO1B1 521TT and TC genotype group of the R or GR group, respectively (P>0.05, Table 5). Furthermore, comparison of the GR group with the R group revealed the absence of significant differences in the Cmax,ss and AUCτ,ss of rosuvastatin within the same SLCO1B1 genotype.
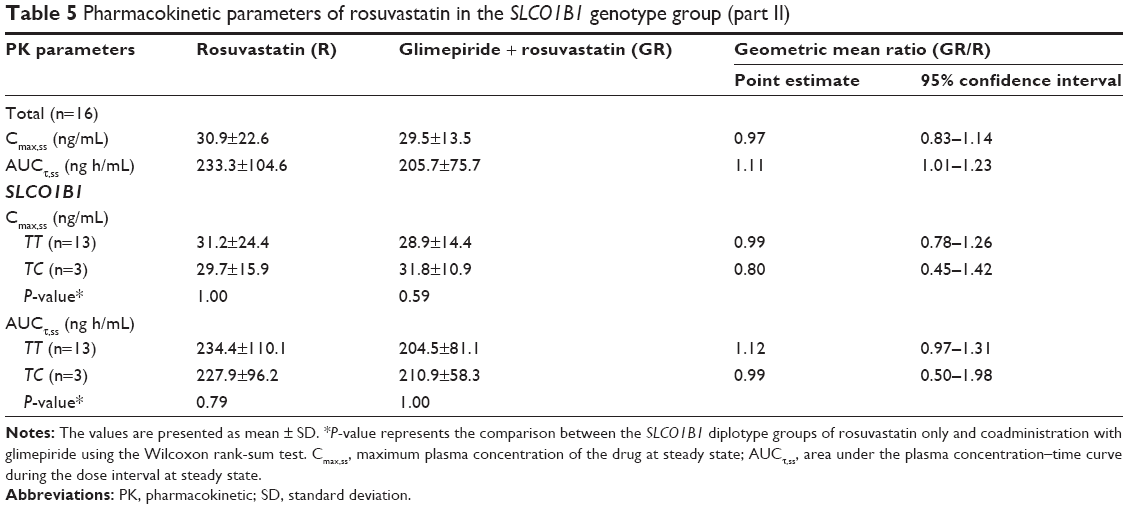 | Table 5 Pharmacokinetic parameters of rosuvastatin in the SLCO1B1 genotype group (part II) |
Safety
There were no serious drug-induced AEs reported in this study. There were 23 AEs in part 1 (14 in the glimepiride monotherapy group and 9 in the combination therapy group) and 26 AEs (5 in the rosuvastatin monotherapy group and 21 in the combination therapy group) in total. Among the AEs, those that were considered to be related to the study drugs are shown in Table 6. All AEs were of mild or moderate severity, and the participants recovered without any complications. In addition, there were no clinically significant changes in the physical examination results, vital signs, clinical laboratory results, or ECG.
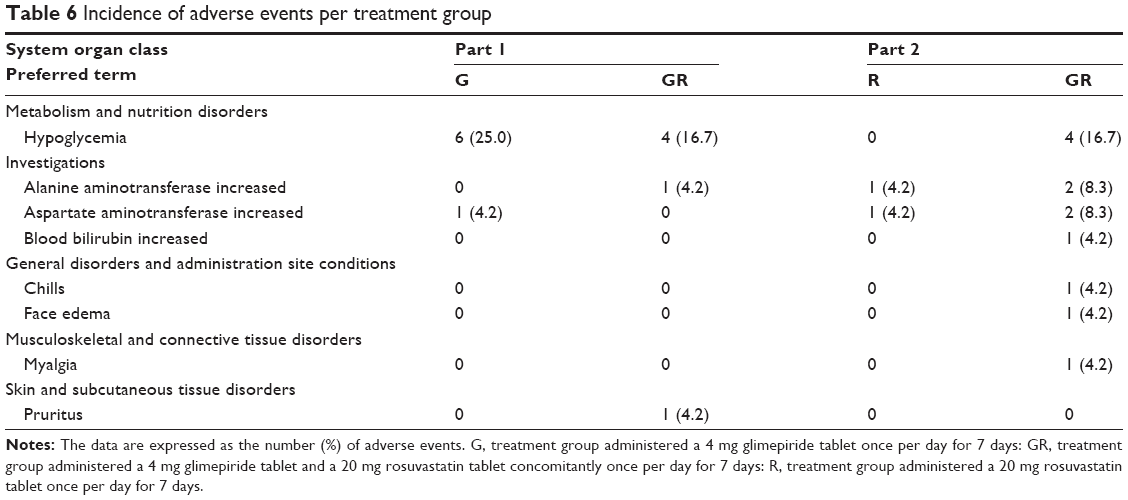 | Table 6 Incidence of adverse events per treatment group |
Discussion
Glimepiride and rosuvastatin are widely used drugs for the treatment of diabetes mellitus and dyslipidemia, respectively, and they are likely to be administered concomitantly. Therefore, this study aimed to evaluate the PK drug–drug interaction between the two drugs in healthy male individuals.
Rosuvastatin is efficiently and rapidly taken up from the portal vein into hepatocytes, which is predominantly mediated by OATP1B1, and its hepatic elimination is ~70% of its total elimination.10,11 A previous in vitro study showed that several oral antidiabetic drugs inhibited the OATP1B1-mediated uptake of rosuvastatin.7 Among them, glimepiride, a highly lipophilic drug, showed a higher inhibition of OATP1B1-mediated rosuvastatin uptake.7 In addition, both glimepiride and rosuvastatin are metabolized by CYP2C9.6,11 The metabolism of glimepiride was decreased when coadministered with rosuvastatin, and rosuvastatin competitively inhibited the CYP2C9 metabolism of glimepiride in vitro.4 On the basis of these data, it is likely that these two drugs have PK interactions with each other.
In this study, it was found that both glimepiride and rosuvastatin did not interact with each other, even though some data suggested the possibility of an interaction between the two drugs. When the PK parameters of glimepiride monotherapy were compared with those of the glimepiride–rosuvastatin combination therapy, the point estimates for the Cmax,ss and AUCτ,ss of glimepiride were both 1.03, and the 90% CI for those PK parameters was within the commonly accepted criteria of 0.8 to 1.25.15 For rosuvastatin, the point estimates (90% CI) of the Cmax,ss and AUCτ,ss were 1.12 (0.98–1.27) and 0.94 (0.86–1.03), respectively. The 90% CI of the AUCτ,ss met the abovementioned criteria; however, the 90% CI of the Cmax,ss was not between 0.8 and 1.25. Although the 90% CI of the Cmax,ss did not meet the abovementioned criteria, the 12% increase in the Cmax,ss of rosuvastatin in the glimepiride–rosuvastatin combination therapy group was lower than the intrasubject variability in the Cmax,ss of rosuvastatin (25.0%). Accordingly, the change in the Cmax,ss of rosuvastatin in combination with glimepiride is not considered clinically significant.
These results were similar to those found in an in vivo study in rats, which showed that there were no significant interactions between glimepiride and rosuvastatin.4 This discrepancy between the data from in vitro and in vivo studies could be attributed to the plasma protein binding of drugs. The concentration of the unbound drugs is generally below the IC50 values observed in in vitro studies because of the high plasma protein binding of oral antidiabetics (>98%).7 Glimepiride is also a drug showing high plasma protein binding (>99%),16 and the plasma concentration of glimepiride may be too low to inhibit OATP1B1-mediated uptake of rosuvastatin. Similar results were observed in an interaction study between pitavastatin and glyburide.17 Although glyburide inhibited OATP1B1-mediated pitavastatin uptake in vitro, it is unlikely that interactions between these two drugs occur in vivo because of the high plasma protein binding of glyburide.
Drug uptake from the blood into hepatocytes is a prerequisite for intracellular drug action or subsequent drug metabolism before biliary excretion.18 Therefore, uptake transporters located in the basolateral hepatocyte membrane play an important role herein.
Therefore, in this study, we explored whether the interaction between glimepiride and rosuvastatin might be related to the SLCO1B1 polymorphism. There were no statistical differences in the glimepiride PK between the SLCO1B1 521TT and TC genotype group when administering glimepiride as monotherapy or as glimepiride–rosuvastatin combination therapy. However, compared with the glimepiride PK within the SLCO1B1 521T>C polymorphism group, the Cmax,ss and AUCτ,ss of glimepiride in the SLCO1B1 521TC group were significantly higher for the glimepiride–rosuvastatin combination therapy than for glimepiride monotherapy. All subjects in the SLCO1B1 521TC group had the CYP2C9*1/*1 genotype. This result suggests that the hepatocyte uptake of glimepiride is mediated by OATP1B1, even though, to the best of our knowledge, there are no published studies that have shown this. Furthermore, given that the affinity of rosuvastatin for OATP1B1 in the liver is significantly higher than that for other drugs,19 this result also suggests that rosuvastatin can inhibit the OATP1B1-mediated uptake of glimepiride in subjects with the SLCO1B1 521T>C polymorphism. However, we cannot exclude the possibility that this result may be a chance observation because this study is limited by the relatively small number of subjects studied. In addition, we could not conclude that the subjects with the SLCO1B1 521TC polymorphism would experience hypoglycemia more frequently after administration of rosuvastatin and glimepiride because we studied only healthy individuals. A previous study assessing the relationship between the SLCO1B1 polymorphism and lipid response revealed that this polymorphism did not affect the lipid-lowering effect of rosuvastatin, even though the SLCO1B1 521T>C polymorphism was associated with an increased plasma concentration of rosuvastatin.20 Therefore, large-scale polymorphism studies correlating the PK and glycemic parameters in patients are warranted to confirm this finding.
Regarding the rosuvastatin PK, some studies have shown that the SLCO1B1 521TC polymorphism resulted in a higher rosuvastatin exposure compared to that observed for the SLCO1B1 521TT polymorphism.13,21 In this rosuvastatin PK study, no statistically significant differences were observed within the SLCO1B1 521T>C polymorphism groups. However, because of the small number of subjects included in this study, it is unlikely that SLCO1B1 is not related to the rosuvastatin PK. In this study, the SLCO1B1 521TC polymorphism was found in only three subjects, and none of the subjects carried the SLCO1B1 521CC polymorphism. Thus, studies with a larger sample size are needed to assess the effect of the SLCO1B1 polymorphisms on the PK parameters of and interaction between glimepiride and rosuvastatin.
Glimepiride is mainly metabolized by CYP2C9, and rosuvastatin, although to a lesser extent, is also metabolized by CYP2C9.6,11 In this study, we explored if the interactions between these two drugs were related to the CYP2C9 genotype. Previous studies have shown that the AUC in subjects with the CYP2C9*1/*3 polymorphism was >100% higher, and the oral clearance ~75% lower than those parameters in subjects with the CYP2C9*1/*1 polymorphism.22,23 The present study showed similar results: the Cmax,ss and AUCτ,ss of glimepiride were higher in the subjects in the CYP2C9*1/*3 polymorphism group than in the CYP*1/*1 group. However, the CYP2C9 polymorphism did not significantly affect the influence of rosuvastatin on glimepiride exposure, which was consistent with the results of another study showing that the PK of rosuvastatin was not associated with the CYP2C9 polymorphism.21 This result is most likely due to the PK characteristics of rosuvastatin because rosuvastatin is only partly metabolized by CYP2C9 and is predominantly eliminated unchanged.10 In the present rosuvastatin PK study, none of the subjects carried the CYP2C9*3 allele. Thus, a potential effect of glimepiride on rosuvastatin exposure due to the CYP2C9 polymorphism could not be determined.
Throughout the study, the administration of 4 mg glimepiride and 20 mg rosuvastatin, either as monotherapy or combination therapy, was well tolerated by all the participants, and there were no serious AEs. The most common treatment-emergent AE was hypoglycemia. All subjects with hypoglycemia consumed a 3 g glucose candy and recovered without any complications. There was no difference in the total AEs between glimepiride and rosuvastatin monotherapy or combination therapy, except for the higher frequency of hypoglycemia in the glimepiride monotherapy group.
Conclusion
In healthy males, there were no significant PK interactions between glimepiride and rosuvastatin at 4 mg glimepiride and 20 mg rosuvastatin dosage regimens when these drugs were administered as combination therapy. Furthermore, both drug therapies showed tolerable safety profiles. Although the sample size in the genotyping study was relatively small, our study suggests that the exposure to glimepiride could be affected by rosuvastatin in the presence of the SLCO1B1 521T>C polymorphism. Nevertheless, further studies are needed to confirm the effect of the SLCO1B1 521T>C polymorphism on the drug–drug interactions between glimepiride and rosuvastatin. In addition, studies including diabetic patients with dyslipidemia are recommended to investigate the clinical significance of these PK interactions between these two drugs.
Acknowledgments
This study was supported by a grant of the Korean Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C1062), and funded by Ildong Pharmaceutical Corp., Seoul, Korea. We thank the staff at the Severance Hospital Clinical Trials Center for their generous cooperation. We also thank the Won Choi for overall review and general support. The studies were not registered at ClinicalTrials.gov and, hence, no identifier numbers are available at this website. This study was sponsored by Ildong Pharmaceutical Corp., Seoul, Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103(2):137–149. | ||
Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: a comparative risk assessment. Lancet Diabetes Endocrinol. 2014;2(8):634–647. | ||
Dake AW, Sora ND. Diabetic dyslipidemia review: an update on current concepts and management guidelines of diabetic dyslipidemia. Am J Med Sci. 2016;351(4):361–365. | ||
Galani V, Vyas M. In vivo and in vitro drug interactions study of glimepiride with atorvastatin and rosuvastatin. J Young Pharm. 2010;2(2):196–200. | ||
International Diabetes Federation Clinical Guidelines Task Force [webpage on the Internet]. Global Guidelines for Type 2 Diabetes. 2012. Available from: http://www.idf.org/guideline-type-2-diabetes. Accessed January 17, 2017. | ||
Langtry HD, Balfour JA. Glimepiride. A review of its use in the management of type 2 diabetes mellitus. Drugs. 1998;55(4):563–584. | ||
van de Steeg E, Greupink R, Schreurs M, et al. Drug-drug interactions between rosuvastatin and oral antidiabetic drugs occurring at the level of OATP1B1. Drug Metab Dispos. 2013;41(3):592–601. | ||
Snow V, Aronson MD, Hornbake ER, Mottur Pilson C, Weiss KB; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Lipid control in the management of type 2 diabetes mellitus: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2004;140(8):644–649. | ||
Lai SW, Lin CL, Liao KF. Rosuvastatin and risk of acute pancreatitis in a population-based case-control study. Int J Cardiol. 2015;187:417–420. | ||
Martin PD, Warwick MJ, Dane AL, Brindley C, Short T. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin Ther. 2003;25(10):2553–2563. | ||
Elsby R, Hilgendorf C, Fenner K. Understanding the critical disposition pathways of statins to assess drug-drug interaction risk during drug development: it’s not just about OATP1B1. Clin Pharmacol Ther. 2012;92(5):584–598. | ||
Zachařová A, Siller M, Spičáková A, et al. Rosuvastatin suppresses the liver microsomal CYP2C11 and CYP2C6 expression in male Wistar rats. Xenobiotica. 2012;42(8):731–736. | ||
Birmingham BK, Bujac SR, Elsby R, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in Caucasian and Asian subjects residing in the United States. Eur J Clin Pharmacol. 2015;71(3):329–340. | ||
Lee Y, Byeon J, Kim Y, et al. Effects of CYP2C9*1/*3 genotype on the pharmacokinetics of flurbiprofen in Korean subjects. Arch Pharm Res. 2015;38(6):1232–1237. | ||
US Department of Health and Human Services Food and Drug Administration CfDEaRF, CDER. Guidance for Industry: Drug Interaction Studies-Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed January 30, 2015. | ||
Rosenkranz B, Malerczyk V, Lehr K, Profozic V, Mrzljak V, Skrabalo Z. Pharmacokinetics of Glimepiride in Kidney-Disease. New York, NY: Nature Publishing Group; 1991:170. | ||
Hirano M, Maeda K, Shitara Y, Sugiyama Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos. 2006;34(7):1229–1236. | ||
Klatt S, Fromm MF, König J. The influence of oral antidiabetic drugs on cellular drug uptake mediated by hepatic OATP family members. Basic Clin Pharmacol Toxicol. 2013;112(4):244–250. | ||
Brown C, Windass A, Bleasby K, Lauffart B. Rosuvastatin is a high affinity substrate of hepatic organic anion transporter OATP-C. Atheroscler Suppl. 2001;2(2):90. | ||
Lee H, Hu M, Lui SS, Ho C, Wong C, Tomlinson B. Effects of polymorphisms in ABCG2, SLCO1B1, SLC10A1 and CYP2C9/19 on plasma concentrations of rosuvastatin and lipid response in Chinese patients. Pharmacogenomics. 2013;14(11):1283–1294. | ||
DeGorter MK, Tirona RG, Schwarz UI, et al. Clinical and pharmacogenetic predictors of circulating atorvastatin and rosuvastatin concentrations in routine clinical care. Circ Cardiovasc Genet. 2013;6(4):400–408. | ||
Niemi M, Cascorbi I, Timm R, Kroemer HK, Neuvonen PJ, Kivistö KT. Glyburide and glimepiride pharmacokinetics in subjects with different CYP2C9 genotypes. Clin Pharmacol Ther. 2002;72(3):326–332. | ||
Wang R, Chen K, Wen S, Li J, Wang S. Pharmacokinetics of glimepiride and cytochrome P450 2C9 genetic polymorphisms. Clin Pharmacol Ther. 2005;78(1):90–92. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.