Back to Journals » Drug Design, Development and Therapy » Volume 15
Pharmacokinetic and Pharmacodynamic Comparison of Two Formulations of a Fixed-Dose Combination of Gemigliptin/Rosuvastatin 50/20 mg: A Randomized, Open-Label, Single-Dose, Two-Way Crossover Study
Authors Yang E ![]() , Yoo H
, Yoo H ![]() , Jang IJ
, Jang IJ ![]() , Yu KS
, Yu KS ![]() , Lee S
, Lee S ![]()
Received 5 November 2020
Accepted for publication 12 January 2021
Published 17 February 2021 Volume 2021:15 Pages 651—658
DOI https://doi.org/10.2147/DDDT.S288986
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Eunsol Yang, Hyounggyoon Yoo, In-Jin Jang, Kyung-Sang Yu, SeungHwan Lee
Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, Seoul, Republic of Korea
Correspondence: SeungHwan Lee
Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, 101 Daehak-Ro, Jongno-Gu, Seoul, 03080, Republic of Korea
Tel +82-2-2072-2343
Fax +82-2-742-9252
Email [email protected]
Purpose: A fixed-dose combination (FDC) of gemigliptin/rosuvastatin 50/20 mg as a monolayer tablet has been used to treat patients with both type 2 diabetes mellitus and dyslipidemia. To improve the stability of the FDC, a new FDC formulation as a bilayer tablet was developed. This study aimed to compare the pharmacokinetics (PKs) and pharmacodynamics (PDs) of the FDC of gemigliptin/rosuvastatin 50/20 mg between the newly developed bilayer tablet and the approved monolayer tablet in healthy subjects.
Materials and Methods: A randomized, open-label, single-dose, two-treatment, two-way crossover study was conducted. Subjects received a single dose of the FDC of gemigliptin/rosuvastatin 50/20 mg as the bilayer tablet or the monolayer tablet in each period with a 7-day washout. For PK and PD analyses, serial blood samples were collected up to 72 hours after dosing to determine plasma concentrations of gemigliptin, its active metabolite LC15-0636 and rosuvastatin, and plasma dipeptidyl peptidase-4 (DPP-4) activity. PK and PD parameters were calculated using non-compartmental methods and compared between the two formulations.
Results: A total of 48 healthy subjects were randomized, and 45 subjects completed the study. The concentration–time profiles of gemigliptin, LC15-0636 and rosuvastatin were comparable between the two formulations. All geometric mean ratios (90% confidence intervals) of the bilayer tablet to the monolayer tablet for maximum plasma concentration and area under concentration–time curve from 0 to last measurable time point of the three compounds fulfilled the bioequivalence criteria of 0.80– 1.25. Likewise, area under plasma DPP-4 activity inhibition from baseline-time curve from 0 to last measurable time point and maximum inhibition of plasma DPP-4 activity were similar between the two formulations.
Conclusion: The FDC of gemigliptin/rosuvastatin 50/20 mg as the bilayer tablet showed equivalent PK and PD properties with the FDC of gemigliptin/rosuvastatin 50/20 mg as the monolayer tablet in healthy subjects. These results suggest that the newly developed bilayer tablet can become an alternative formulation to the commercially available monolayer tablet.
Keywords: DPP-4 inhibitor, statin, type 2 diabetes, dyslipidemia, bioequivalence
Introduction
Cardiovascular complications are the leading cause of mortality in patients with type 2 diabetes mellitus (T2DM).1,2 Furthermore, about 30–60% of them are also afflicted with dyslipidemia, which has a major role in increasing the risk of cardiovascular complications.3 In patients with T2DM, dipeptidyl peptidase-4 (DPP-4) inhibitors have been widely used as a substitute or an add-on therapy to metformin,4 and several studies reported their potential cardiovascular protective effects.5 Statins are the first-line treatment for the management of dyslipidemia in patients at risk for cardiovascular disease, including those with T2DM.6,7 Therefore, in patients with both T2DM and dyslipidemia, the combination therapy of a DPP-4 inhibitor and a statin is expected to reduce the risk of cardiovascular complications, and ultimately their mortalities.
Gemigliptin, a DPP-4 inhibitor, is rapidly absorbed with a time to maximum plasma concentration (Tmax) of 0.5–3.0 hours after dosing and has a terminal half-life (t1/2) of 17.1 ± 1.7 hours.8 It is eliminated via excretion and metabolism with a balanced rate, and ~10% of gemigliptin is metabolized by CYP3A4 into LC15-0636, which is an active metabolite.9,10 Rosuvastatin, a statin, is also rapidly absorbed with a Tmax of 1.0–5.0 hours after dosing, with a terminal t1/2 of 12.3 ± 5.8 hours,11 and is primarily excreted in the feces.12 In previous studies, there was no pharmacokinetic (PK) drug interaction between gemigliptin and rosuvastatin,13 and the fixed-dose combination (FDC) of gemigliptin/rosuvastatin 50/20 mg as a monolayer tablet showed comparable PK and pharmacodynamic (PD) properties with the corresponding loose combination.14 Accordingly, the FDC of gemigliptin/rosuvastatin 50/20 mg as the monolayer tablet (Zemiro® Tab., LG Chem, Ltd., Seoul, Republic of Korea) was approved by the Korea Ministry of Food and Drug Safety (MFDS) in 2017, and has been used to treat patients with both T2DM and dyslipidemia.
Compared to a monolayer formulation, a multilayer formulation can improve overall the chemical stability of a drug product.15 In the case of the marketed monolayer tablet, the two active pharmaceutical ingredients (APIs) of gemigliptin and rosuvastatin are mixed in one layer, and API–API interaction has resulted in generating impurities, which can impact on the stability of the specific condition. Accordingly, to improve the stability of the marketed monolayer tablet, a new FDC formulation as a bilayer tablet was developed. The newly developed bilayer tablet is expected to have an increased stability and extended expiration date for the FDC by separating each API by layers and thus minimizing the interaction between the APIs. Notably, in in vitro accelerated stability tests, the bilayer tablet was more stable than the monolayer tablet and less impurities were formed in the bilayer tablet under the same condition, and therefore these results suggest the improved stability of the bilayer tablet (LG Chem, Ltd., unpublished data, February, 2019; LG Chem, Ltd., unpublished data, April, 2019) (Table 1). Furthermore, in vitro dissolution profiles of the bilayer tablet and monolayer tablet were similar (LG Chem, Ltd., unpublished data, December, 2016; LG Chem, Ltd., unpublished data, October, 2019).
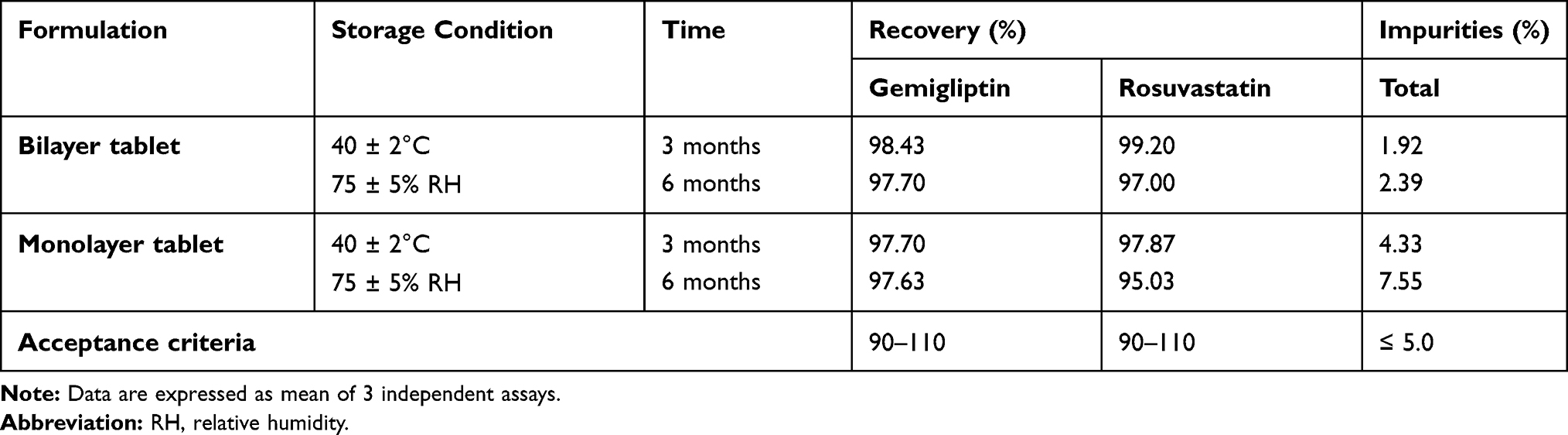 |
Table 1 In Vitro Accelerated Stability Test Results for Fixed-Dose Combination of Gemigliptin/Rosuvastatin 50/20 mg as Bilayer Tablet or Monolayer Tablet |
The objective of this study was to compare the PKs and PDs of the FDC of gemigliptin/rosuvastatin 50/20 mg between the newly developed bilayer tablet and the approved monolayer tablet in healthy subjects.
Materials and Methods
The study protocol and informed consent form were approved by the Institutional Review Board of Seoul National University Hospital (No. H-1901-174-1007) and MFDS. Also, this study was registered at ClinicalTrials.gov (NCT03867942). This study was conducted in compliance with Korean Good Clinical Practice guidelines and tenets of the Declaration of Helsinki. Written informed consent was obtained from all subjects before any study procedures were performed.
Study Population
The study population consisted of healthy subjects who were 19–45 years of age, with a body mass index (BMI) of 18.0–27.0 kg/m2 and a fasting plasma glucose level of 70–120 mg/dL. The enrolled subjects presented no clinically significant abnormalities according to their medical histories, clinical laboratory tests, vital signs, physical examination and 12-lead electrocardiogram (ECG) at screening. The major exclusion criteria were the following: aspartate transaminase and alanine transferase > 1.5 × upper limit of normal range; creatine phosphokinase > 2.5 × upper limit of normal range; any hypersensitivity to drugs including gemigliptin and rosuvastatin; and any diseases or histories such as DM, dyslipidemia and drug-induced muscular disorder.
Maximum intra-subject coefficient of variations (CVs) for PK parameters (Cmax and area under concentration–time curve (AUC) from 0 to last measurable time point (AUClast)) were assumed to be 30% for rosuvastatin and 20% for gemigliptin, respectively,13,16 and 30% was conservatively used for calculating the sample size. Considering the drop-out rate as about 20%, a total sample size of 48 subjects was estimated to detect a 20% difference in the PK parameters between the two formulations with an 80% statistical power at a 5% level of significance.
Study Design
This was a randomized, open-label, single-dose, two-treatment, two-period, two-sequence crossover study. The enrolled subjects were randomly assigned to one of two sequences in a ratio of 1:1, in which each treatment consisted of a single oral dose of the FDC of gemigliptin/rosuvastatin 50/20 mg as the bilayer tablet (LG Chem, Ltd.) for test, or the FDC of gemigliptin/rosuvastatin 50/20 mg as the monolayer tablet (Zemiro® Tab., LG Chem, Ltd.) for reference. Between treatment periods, there was a 7-day washout period, which was longer than 5 times the t1/2s of gemigliptin and rosuvastatin previously reported.8,11 According to the subjects’ assigned sequence, each treatment was administered with 150 mL of water after overnight fasting.
Serial blood samples for PK and PD analyses were collected at 0 (before dosing), 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, 48 and 72 hours after dosing. For PK evaluation, 8 mL of blood was taken in a heparinized tube for each sampling point and subsequently centrifuged at 688 g for 8 minutes at 4°C. For gemigliptin, 0.5 mL of supernatants were transferred to tubes containing 0.5 mL of 5% formic acid solution and then mixed well, and for rosuvastatin, 1.0 mL of supernatants were transferred to tubes.9 For PD evaluation, 3 mL of blood was taken in an ethylenediaminetetraacetic acid tube for each sampling point and centrifuged, and 0.5 mL of supernatants were transferred to tubes. Samples for PK and PD evaluations were stored at −70°C until the sample analysis.
Determination of Plasma Gemigliptin, LC15-0636 and Rosuvastatin Concentrations
Plasma concentrations of gemigliptin, its active metabolite LC15-0636 and rosuvastatin were determined by a validated liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS; API 5500, SCIEX for gemigliptin and LC15-0636; Shimadzu UFLC, SHIMADZU and API5000(1), SCIEX for rosuvastatin).17,18 The samples were separated under gradient conditions in the LC system, and positive electrospray ionization mode and multiple reaction monitoring mode were used in the MS/MS system.
For quality control samples, the accuracy ranges were 97.9–102.3% for gemigliptin, 99.1–102.3% for LC15-0636 and 99.8–105.5% for rosuvastatin, and the CVs were ≤ 4.9% for gemigliptin, ≤ 6.5% for LC15-0636 and ≤ 4.0% for rosuvastatin. The lower limits of quantification were 0.5 μg/L for gemigliptin, 0.25 μg/L for LC15-0636 and 0.1 μg/L for rosuvastatin.
Determination of Plasma DPP-4 Activity
Plasma DPP-4 activity was determined by a continuous spectrophotometric assay as previously described.19
PK and PD Analyses
PK and PD analyses were performed in subjects who had completed the study without major deviation affecting PK and PD results. The following PK and PD parameters were calculated by non-compartmental methods using WinNonlin® software version 8.0 (Certara USA Inc., Princeton, NJ, USA). Maximum plasma concentration (Cmax) and Tmax were determined directly from observed plasma concentration–time profiles, and AUClast was calculated using linear-up and log-down trapezoidal rule. AUC from 0 to infinity (AUCinf) was calculated with the following formula: AUCinf = AUClast + Clast/λz, in which Clast is the last measurable concentration, and λz is the terminal elimination rate constant. Apparent clearance was calculated as a single dose divided by AUCinf, and terminal elimination t1/2 was calculated as 0.693/λz. Area under plasma DPP-4 activity inhibition from baseline-time curve from 0 to last measurable time point (AUEClast) was calculated using linear trapezoidal rule, and maximum inhibition of plasma DPP-4 activity (Imax) was obtained from the observed value.
Safety Assessment
Safety was evaluated in subjects who had administered the treatment at least once, based on adverse events (AEs), clinical laboratory tests, vital signs, physical examination and 12-lead ECG. The clinical significance and the relationship with the treatment of all findings from the safety parameters were determined by investigators.
Statistical Analysis
Statistical analysis was performed using SAS® software version 9.4 (SAS Institute, Cary, NC, USA). PK (Cmax, AUClast) and PD (AUEClast, Imax) parameters were log-transformed, and the geometric mean ratios (GMRs) of the bilayer tablet to the monolayer tablet and its confidence intervals (CIs) were estimated from the linear mixed-effect model, including sequence, period, group and treatment as fixed effects, and subject nested within sequence as a random effect. If the GMRs and its 90% CIs for these PK/PD parameters were contained within the conventional bioequivalence criterion of 0.80–1.25, the two formulations were judged to be bioequivalent. The incidences of AEs and adverse drug reactions (ADRs) were compared between the two formulations using Fisher’s exact test.
Results
Study Population
A total of 48 healthy Korean subjects were enrolled and randomized, and 45 subjects completed the study. Two subjects withdrew their consent before the first and the second administration, respectively, and the other subject dropped out after the first administration due to an AE not related to the treatment. The mean ± standard deviation values for age, height, weight and BMI of the enrolled subjects were 32.1 ± 6.1 years, 168.6 ± 8.8 cm, 65.4 ± 9.2 kg and 23.0 ± 2.3 kg/m2, respectively. Thirty-one (64.6%) subjects were male and 17 (35.4%) subjects were female. PK and PD characteristics were analyzed in 45 subjects who had completed the study without major deviation affecting PK and PD results, and safety was assessed in 47 subjects who had taken the treatment at least once.
Pharmacokinetics
After a single administration of the FDC of gemigliptin/rosuvastatin 50/20 mg as the bilayer tablet or the monolayer tablet, the mean plasma concentration–time profiles and PK characteristics of gemigliptin, LC15-0636 and rosuvastatin were similar between the two formulations (Figure 1, Table 2). The GMRs (90% CIs) of the bilayer tablet to the monolayer tablet for Cmax and AUClast were 0.9798 (0.8998–1.0669) and 0.9714 (0.9491–0.9941) for gemigliptin, 1.0269 (0.9593–1.0992) and 0.9998 (0.9844–1.0154) for LC15-0636, and 1.0233 (0.9370–1.1175) and 0.9931 (0.9471–1.0413) for rosuvastatin, respectively (Table 2). All of the GMRs and the 90% CIs for Cmax and AUClast of the three compounds fulfilled the bioequivalence criterion of 0.80–1.25.
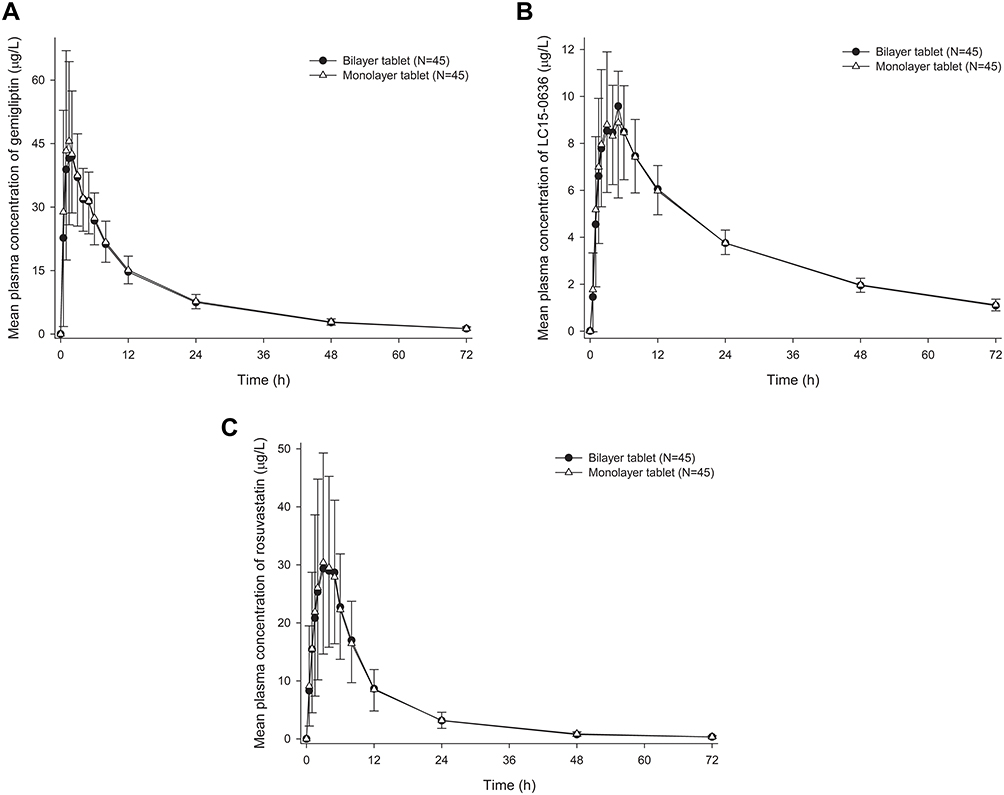 |
Figure 1 Mean plasma concentration–time profiles of (A) gemigliptin, (B) LC15-0636 and (C) rosuvastatin after a single administration of fixed-dose combination of gemigliptin/rosuvastatin 50/20 mg as bilayer tablet or monolayer tablet. Note: Error bars represent standard deviation. |
 |
Table 2 Pharmacokinetic Parameters of Gemigliptin, LC15-0636 and Rosuvastatin After a Single Administration of Fixed-Dose Combination of Gemigliptin/Rosuvastatin 50/20 mg as Bilayer Tablet or Monolayer Tablet |
Pharmacodynamics
The bilayer tablet and the monolayer tablet exhibited similar mean plasma DPP-4 activity-time profiles (Figure 2). Likewise, AUEClast and Imax, which represent the degree of inhibition of plasma DPP-4 activity from baseline, were comparable between the two formulations (Table 3). The GMRs (90% CIs) of the bilayer tablet to the monolayer tablet for AUEClast and Imax were 0.9915 (0.9807–1.0025) and 0.9970 (0.9871–1.0069), respectively (Table 3).
 |
Figure 2 Mean plasma dipeptidyl peptidase-4 activity-time profiles after a single administration of fixed-dose combination of gemigliptin/rosuvastatin 50/20 mg as bilayer tablet or monolayer tablet. Note: Error bars represent standard deviation. |
 |
Table 3 Plasma Dipeptidyl Peptidase-4 (DPP-4) Activity Inhibition from Baseline After a Single Administration of Fixed-Dose Combination of Gemigliptin/Rosuvastatin 50/20 mg as Bilayer Tablet or Monolayer Tablet |
Safety
There were no clinically meaningful changes in the clinical laboratory tests, vital signs, physical examination and 12-lead ECG before and after the administration of the treatments. Throughout the study, a total of 14 AEs were observed in nine subjects; 10 AEs were observed in six subjects who received the bilayer tablet; and four AEs were observed in three subjects who received the monolayer tablet. Of the 14 AEs, 10 AEs in seven subjects were assessed as related to the bilayer tablet (eight AEs in six subjects) or the monolayer tablet (two AEs in one subject), which were ADRs. One subject was withdrawn from the study due to an AE (rhabdomyolysis), but this AE occurred before the first administration and was assessed as not drug-related. All AEs and ADRs were mild in intensity, and no serious AE occurred. Moreover, there was no statistically significant difference in the incidence rates of the AEs (p-value = 0.4850) and ADRs (p-value = 0.1106) between the bilayer tablet and the monolayer tablet.
Discussion
This study aimed to compare the PK and PD profiles of the FDC of gemigliptin/rosuvastatin 50/20 mg as the bilayer tablet and the monolayer tablet in healthy subjects. The two formulations showed similar PK and PD characteristics. Furthermore, the GMRs for AUEClast and Imax as well as Cmax and AUClast of gemigliptin, its active metabolite LC15-0636 and rosuvastatin were close to 1, and the corresponding 90% CIs were included in the conventional bioequivalence range of 0.80–1.25. These results indicate that the bilayer tablet is pharmacokinetically and pharmacodynamically equivalent to the monolayer tablet, thus supporting the substitutability of the bilayer tablet with the monolayer tablet.
LC15-0636, a major active metabolite of gemigliptin, is primarily formed by CYP3A4 through systemic metabolism and has about a 2-fold higher in vitro DPP-4 inhibitory potency compared to gemigliptin.20 Considering that LC15-0636 would largely contribute to the antidiabetic effect of gemigliptin, this study analyzed not only the parent drug but also its active metabolite, LC15-0636. Because the plasma concentration–time profiles of gemigliptin were superimposable between the two formulations, the PK profiles of its metabolite would not be different depending on the formulation. Accordingly, LC15-0636 showed similar PK profiles and almost the same values for the metabolic ratio in both formulations. Though it is recommended by regulatory agencies that the bioequivalence assessment is applied to the parent drug,21 Cmax and AUClast of LC15-0636 also met the bioequivalence criterion. The results for LC15-0636 can act as supportive evidence for the comparable efficacy between the two formulations.
Because the degree of inhibition of DPP-4 activity is a direct PD biomarker of DPP-4 inhibitors,14,22,23 this study measured the plasma DPP-4 activity to evaluate the PDs of gemigliptin in the two formulations, and similar values for AUEClast and Imax were observed regardless of the formulation. Recently, several studies showed that the degree of inhibition of DPP-4 activity after administration of DPP-4 inhibitors was comparable in normoglycemic and diabetic subjects.24 Referring to these points, it is expected that the bilayer tablet will exhibit comparable anti-glycemic effects also in patients with T2DM compared to the monolayer tablet, similar to the results of this study in healthy subjects.
Conclusion
This study demonstrates that the FDC of gemigliptin/rosuvastatin 50/20 mg as the bilayer tablet has equivalent PK and PD properties with the FDC of gemigliptin/rosuvastatin 50/20 mg as the monolayer tablet in healthy subjects. Therefore, the newly developed bilayer tablet can become an alternative formulation to the commercially available monolayer tablet.
Data Sharing Statements
The individual de-identified participant data supporting published results are available with approval from the corresponding author on reasonable request, at any time after publication.
Acknowledgments
This study was sponsored by LG Chem, Ltd., Seoul, Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Taskinen MR, Borén J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis. 2015;239(2):483–495. doi:10.1016/j.atherosclerosis.2015.01.039
2. Mooradian AD. Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5(3):150–159. doi:10.1038/ncpendmet1066
3. Feingold KR Dyslipidemia in diabetes. In: Feingold KR, Anawalt B, Boyce A, et al., eds. Endotext. South Dartmouth (MA):MDText.com, Inc. Copyright © 2000–2020, MDText.com, Inc.; 2000.
4. Kim MK, Ko SH, Kim BY, et al. Clinical practice guidelines for type 2 diabetes mellitus in Korea. Diabetes Metab J. 2019;43(4):398–406. doi:10.4093/dmj.2019.0137
5. Xie W, Song X, Liu Z. Impact of dipeptidyl-peptidase 4 inhibitors on cardiovascular diseases. Vascul Pharmacol. 2018;109:17–26. doi:10.1016/j.vph.2018.05.010
6. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the american association of clinical endocrinologists and american college of endocrinology on the comprehensive type 2 diabetes management algorithm - 2018 executive summary. Endocr Pract. 2018;24(1):91–120. doi:10.4158/CS-2017-0153
7. American Diabetes Association. 10. Cardiovascular disease and risk management: standards of medical care in diabetes-2019. Diabetes Care. 2019;42(Suppl1):S103–s123. doi:10.2337/dc19-S010
8. Lim KS, Kim JR, Choi YJ, et al. Pharmacokinetics, pharmacodynamics, and tolerability of the dipeptidyl peptidase iv inhibitor lc15-0444 in healthy korean men: a dose-block-randomized, double-blind, placebo-controlled, ascending single-dose, Phase I study. Clin Ther. 2008;30(10):1817–1830. doi:10.1016/j.clinthera.2008.10.013
9. Kim N, Patrick L, Mair S, et al. Absorption, metabolism and excretion of [14c]gemigliptin, a novel dipeptidyl peptidase 4 inhibitor, in humans. Xenobiotica. 2014;44(6):522–530. doi:10.3109/00498254.2013.865856
10. Kim SH, Yoo JH, Lee WJ, Park CY. Gemigliptin: an update of its clinical use in the management of type 2 diabetes mellitus. Diabetes Metab J. 2016;40(5):339–353. doi:10.4093/dmj.2016.40.5.339
11. Kang WY, Seong SJ, Ohk B, et al. Pharmacokinetic and bioequivalence study comparing a fimasartan/rosuvastatin fixed-dose combination with the concomitant administration of fimasartan and rosuvastatin in healthy subjects. Drug Des Devel Ther. 2018;12:3607–3615. doi:10.2147/DDDT.S161917
12. Highlights of prescribing information. crestor (rosuvastatin calcium) tablets AstraZeneca pharmaceuticals LP. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021366s040s041lbl.pdf. Published 2020.
13. Choi HY, Lim HS, Kim YH, et al. Evaluation of the pharmacokinetics of the DPP-4 inhibitor gemigliptin when coadministered with rosuvastatin or irbesartan to healthy subjects. Curr Med Res Opin. 2015;31(2):229–241. doi:10.1185/03007995.2014.980886
14. Kim E, Park KR, Jang IJ, Yu KS, Lee S. A fixed-dose combination of gemigliptin and rosuvastatin exhibits similar pharmacokinetics, pharmacodynamics, and safety compared to that of a loose combination in healthy subjects. Drug Des Devel Ther. 2019;13:3879–3885. doi:10.2147/DDDT.S197054
15. Desai D, Wang J, Wen H, Li X, Timmins P. Formulation design, challenges, and development considerations for fixed dose combination (FDC) of oral solid dosage forms. Pharm Dev Technol. 2013;18(6):1265–1276. doi:10.3109/10837450.2012.660699
16. Thota S, Tippabhotla SK, Khan S, Nakkawar M. Two-way crossover bioequivalence study of rosuvastatin tablets 5 mg in healthy, adult, asian-indian male volunteers under fasting condition. Int J Pharm Pharm Sci. 2013;5(3):289–293.
17. Han S. Analysis of Gemigliptin and LC15-0636 in Human Plasma by LC-MS/MS (Study No.: AN1-18-116). International Scientific Standards, Inc. 2019
18. Park J. Sample Analytical Report (Quantitative analysis of Rosuvastatin in human plasma by LC/MS/MS), (Clinical Study Title: a randomized, open-label, single dose, two-way crossover clinical trial to investigate the pharmacokinetics/pharmacodynamics, safety, and tolerability of the monolayer combination of gemigliptin/rosuvastatin 50/20 mg in comparison to the bilayer combination of gemigliptin/rosuvastatin 50/20 mg administered in healthy volunteers, Study No.: DDS19-643AN_R (Ver.01). BioCore Co., Ltd. 2019
19. Shin D, Cho YM, Lee S, et al. Pharmacokinetic and pharmacodynamic interaction between gemigliptin and metformin in healthy subjects. Clin Drug Investig. 2014;34(6):383–393. doi:10.1007/s40261-014-0184-3
20. Noh YH, Lim HS, Jin SJ, et al. Effects of ketoconazole and rifampicin on the pharmacokinetics of gemigliptin, a dipeptidyl peptidase-iv inhibitor: a crossover drug-drug interaction study in healthy male korean volunteers. Clin Ther. 2012;34(5):1182–1194. doi:10.1016/j.clinthera.2012.04.001
21. Guidance for Industry. Bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an anda. U.S. Food and Drug Administration. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioequivalence-studies-pharmacokinetic-endpoints-drugs-submitted-under-abbreviated-new-drug. Published 2013.
22. Park SI, Lee H, Oh J, et al. A fixed-dose combination tablet of gemigliptin and metformin sustained release has comparable pharmacodynamic, pharmacokinetic, and tolerability profiles to separate tablets in healthy subjects. Drug Des Devel Ther. 2015;9:729–736. doi:10.2147/DDDT.S75980
23. Jin X, Kim E, Huh KY, et al. Pharmacokinetics and pharmacodynamics of a fixed-dose combination of gemigliptin/metformin sustained release 25/500 mg compared to the loose combination in healthy male subjects. Transl Clin Pharmacol. 2020;28(1):43–54. doi:10.12793/tcp.2020.28.e2
24. Pathak R, Bridgeman MB. Dipeptidyl peptidase-4 (DPP-4) inhibitors in the management of diabetes. P T. 2010;35(9):509–513.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.