")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 9
Pharmacogenomics of statins: understanding susceptibility to adverse effects
Authors Kitzmiller JP, Mikulik EB, Dauki AM, Mukherjee C, Luzum JA
Received 13 June 2016
Accepted for publication 12 August 2016
Published 3 October 2016 Volume 2016:9 Pages 97—106
DOI https://doi.org/10.2147/PGPM.S86013
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Joseph P Kitzmiller,1 Eduard B Mikulik,1 Anees M Dauki,2 Chandrama Murkherjee,1 Jasmine A Luzum3
1Department of Biological Chemistry and Pharmacology, College of Medicine, 2College of Pharmacy, The Ohio State University, Columbus, OH, 3Department of Clinical Pharmacy, University of Michigan College of Pharmacy, Ann Arbor, MI, USA
Abstract: Statins are a cornerstone of the pharmacologic treatment and prevention of atherosclerotic cardiovascular disease. Atherosclerotic disease is a predominant cause of mortality and morbidity worldwide. Statins are among the most commonly prescribed classes of medications, and their prescribing indications and target patient populations have been significantly expanded in the official guidelines recently published by the American and European expert panels. Adverse effects of statin pharmacotherapy, however, result in significant cost and morbidity and can lead to nonadherence and discontinuation of therapy. Statin-associated muscle symptoms occur in ~10% of patients on statins and constitute the most commonly reported adverse effect associated with statin pharmacotherapy. Substantial clinical and nonclinical research effort has been dedicated to determining whether genetics can provide meaningful insight regarding an individual patient’s risk of statin adverse effects. This contemporary review of the relevant clinical research on polymorphisms in several key genes that affect statin pharmacokinetics (eg, transporters and metabolizing enzymes), statin efficacy (eg, drug targets and pathways), and end-organ toxicity (eg, myopathy pathways) highlights several promising pharmacogenomic candidates. However, SLCO1B1 521C is currently the only clinically relevant pharmacogenetic test regarding statin toxicity, and its relevance is limited to simvastatin myopathy.
Keywords: cholesterol, myopathy, lipids, muscle toxicity, pharmacokinetics, pharmacogenetics
Introduction
Statins are indicated for the prevention of cardiovascular disease and are among the most prescribed classes of medication.1 Their inhibition of HMGCR results in decreased intrahepatic cholesterol synthesis, upregulation of hepatocyte surface low-density lipoprotein cholesterol (LDL-C) receptors, increased LDL-C uptake by hepatocytes, and ultimately decreased systemic concentration of LDL-C. Decreased systemic LDL-C is commonly used as a surrogate measure of statin efficacy, with an estimated reduction in risk of major cardiovascular events of nearly 20% per mmol/L (38 mg/dL) reduction in LDL-C.2 Along with decreased systemic LDL-C, the pleotropic effects (eg, improved endothelial function, increased production of endogenous nitric oxide, enhanced plaque stability, and decreased release of inflammatory interleukins) of statins are beneficial for cardiovascular function and patient health. Contemporary official recommendations from leading expert panels in the US and Europe (eg, American Heart Association, National Lipid Association, European Atherosclerosis Society) emphasize the importance of statin pharmacotherapy in the treatment and prevention of atherosclerotic cardiovascular disease, recently expanding prescribing indications and target populations.3–5
Not all patients, however, respond favorably to statins, and some do not achieve their cholesterol-reduction goals. Furthermore, a considerable number of patients experience adverse effects. Statin myositis and statin-associated muscle symptoms (SAMS) comprise the most commonly reported adverse effect of statins, often leading to poor adherence or discontinuation of statin pharmacotherapy regimens.6 Statin myositis is characterized by inflammation of muscle tissue resulting in muscle pain or weakness and is accompanied by increased blood concentration of CK, a protein biomarker of damaged myocytes. The incidence of statin myopathy, statin myositis with CK levels tenfold greater than the upper limit of normal (ULN), is about one per 1,000 to one per 10,000 person-years.7 SAMS has a broader definition that includes muscle symptoms (subjective or objective) in the absence of elevated CK. As a result, the incidence of SAMS is widely debated. Early pharmaceutical clinical trials in healthy subjects reported incidences <5%.8 This markedly underestimates the true incidence, however, because those studies had stringent criteria (clinical and laboratory) and various potential biases.9 In real-world patient populations, the incidence of SAMS has been significantly higher. The European Atherosclerosis Society Consensus Panel determined the incidences of SAMS to be 7%–29% in registries and observational studies,5 and the Predictions of Muscular Risk in Observational conditions (PRIMO) study, among the largest and more commonly referenced studies, found the incidence of SAMS to be ~10%.10 More often, the symptoms of SAMS occur within the first 6 months of initiating statin therapy and resolve after statin doses are lowered or discontinued.10–13
Another reported adverse effect associated with statins pharmacotherapy is liver toxicity. The incidence of liver toxicity, characterized by elevated blood concentrations of transaminases, is far less than that of SAMS. A meta-analysis of randomized controlled trials of statin use in hyperlipidemic patients found that the proportion of patients with liver toxicity was not significantly different between those receiving statins and those receiving placebo (0.0114 vs 0.0105; 95% confidence interval [CI] for odds ratio [OR] 0.99–1.62).14 Another adverse effect of statins reported to occur with very low incidence is central nervous system (CNS) toxicity. Although recent case reports have received significant attention, they have been highly scrutinized by clinicians and researchers because a substantial amount of published research suggests that statins may in fact improve cognitive function.15,16
There is a paucity of research regarding statin-associated liver and CNS toxicities and reported incidences have been marginal at best. As a result, clinical research investigating statin toxicity has focused primarily on SAMS. Several clinical risk factors for SAMS (Table 1) have been identified and verified in many clinical studies.17–19 Many of these are risk factors because they lead to increased patient exposure to statin and metabolites. They should, therefore, have some relevance also regarding increased risks for statin-associated liver or CNS toxicities. In addition to those risk factors, comorbid conditions affecting liver or CNS function may also be relevant. Risk factors specific to CNS and liver toxicity have not been readily investigated, and similarly, nearly all the focus and literature regarding the pharmacogenomics of statin toxicity has centered on SAMS.
 | Table 1 Nongenetic clinical risk factors for statin adverse reactions |
Hundreds of candidate gene studies and several genome-wide association studies (GWASs) have focused on the influence of genetic variants on statin pharmacokinetics (eg, drug and metabolite levels in blood, area under the time–concentration curve [AUC]) and statin pharmacodynamics (lipid lowering, incidence of adverse events, incidence of cardiovascular events). Discussing relevant clinical research, this contemporary review focuses on polymorphisms in several key genes that affect statin pharmacokinetics (eg, transporters and metabolizing enzymes), statin efficacy (eg, drug targets and pathways), and end-organ toxicity (eg, myopathy pathways). As dose–response relationships have repeatedly been demonstrated for both statin efficacy and toxicity,10–14,20 polymorphisms affecting statin pharmacokinetics can directly influence the incidence and severity of statin adverse events. Polymorphisms affecting statin efficacy have the capacity to influence statin toxicity in an indirect manner (eg, prescribers often increase statin doses in patients not achieving substantial lipid reduction). Polymorphisms affecting end-organ toxicity too can directly influence the incidence and severity of statin adverse events.
Pharmacogenomics of statin adverse effects
Genetic polymorphisms affecting statin pharmacokinetics directly affect risk of statin toxicity
Several polymorphisms in SLCO1B1, the gene encoding the SLCO1B1, result in altered transport of statins and their metabolites into the liver. It is within hepatocytes that statins exert their lipid-lowering action, inhibition of the cholesterol-synthesizing enzyme HMGCR. Despite the prominent role of SLCO1B1 in statin transport, variants in SLCO1B1 have been associated with only small effects on statin response. Several studies have demonstrated that SLCO1B1 521C (rs4149056) was associated with statistically significant, albeit marginal (<5%), attenuation of the lipid-lowering effect of simvastatin, atorvastatin, lovastatin, and pravastatin.21,22 SLCO1B1 521C does, however, significantly affect statin pharmacokinetics and risk of statin toxicity. The AUC for simvastatin was approximately double for SLCO1B1 521C carriers compared to wild type (n=41, P<0.01), and in the Genetics of Diabetes Audit and Research in Tayside Scotland database, SLCO1B1 521C homozygous carriers were approximately three times more likely to be statin intolerant (blood concentration of CK > ULN or ALT >1.5ULN) compared to wild type (n=4,340, P<0.01).23 A GWAS of 175 patients receiving simvastatin 80 mg daily determined that SLCO1B1 521C carriers were significantly more likely to develop statin myositis compared to wild type; heterozygote and homozygote carriers were 4.5 and 16.9 times more likely to develop statin myopathy, respectively, compared to wild type, and the ORs for the 40 mg cohorts were 2.6 and 5.2.20 In a case–control study (n=108) reported by Brunham et al, a significant association (P<0.05) with statin myopathy was confirmed for simvastatin (OR: 3.2 per allele) but not atorvastatin.24 Repeatedly demonstrated associations of polymorphisms in SLCO1B1 with decreased simvastatin transport into hepatocytes, increased systemic simvastatin concentrations, and increased risk of myopathy prompted the Clinical Pharmacogenetics Implementation Consortium (CPIC), a partnership between the US National Institutes of Health Pharmacogenomics Research Network and The Pharmacogenomics Knowledge Base (PharmGKB®), to establish formal prescribing recommendations for simvastatin which are based on myopathy risk categories (low, intermediate, or high) defined by SLCO1B1 genotype (Table 2).25,26 Although nearly 200 common variants in SLCO1B1 have been described, SLCO1B1 521C is the most clinically relevant and has the highest level of clinical evidence. In CPIC’s Recommended Dosing of Simvastatin Based on SLCO1B1 Phenotype, all haplotypes conferring increased risk of simvastatin myopathy (*5, *15, and *17) contain the SLCO1B1 521C polymorphism.26 CPIC’s SLCO1B1-based prescribing guidance for simvastatin is a commonly cited example of formal guidance regarding the pharmacogenomics of statin toxicity, and reports from clinician groups utilizing SLCO1B1-based guidance have been encouraging.27–29 Josephine et al reported increased LDL-C reduction and significant improvements in patients’ perception and adherence in 59 patients receiving statin therapy guided by SLCO1B1*5 genotype compared to 58 concurrent controls receiving statin therapy without genotype-based guidance.29
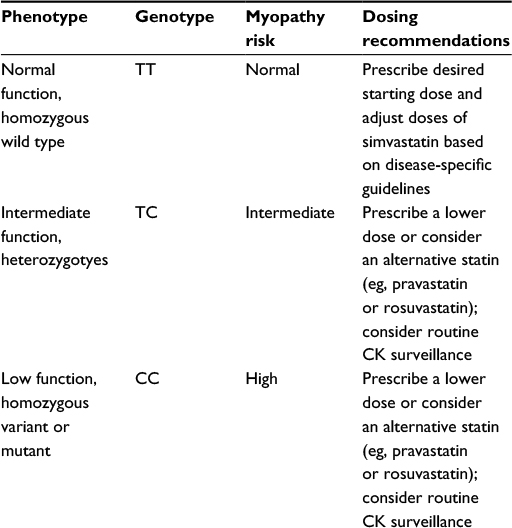 | Table 2 Recommended dosing of simvastatin based on SLCO1B1 phenotype Notes: The minor C allele at rs4149056 is contained within SLCO1B1*5 (rs4149056 alone) as well as the *15 and *17 haplotypes and is associated with lower plasma clearance of simvastatin. The magnitude of this effect is similar for *5, *15, and *17 haplotypes. |
Hepato-biliary and renal–urinary transport of statins and their metabolites occurs largely via ABCB1 transport protein (synonymous with MDR1). Associations with statin pharmacokinetic parameters and with measures of statin toxicity have been demonstrated for three polymorphisms in the ABCB1 gene: 1236T, 2677T, and 3435T (rs1128503, rs2032582, and rs1045642, respectively). In combination with 1236T or 2677T or both, 3435T alters the structure and function of ABCB1 by disrupting proper usage of codons during translation of ABCB1. Those with the TTT haplotype (1236T, 2677T, and 3435T alleles) had an AUC that was nearly 60% greater for simvastatin acid and 55% larger for atorvastatin acid compared to those with the CGT haplotype (P=0.039 and P<0.025, respectively).30 Ferrari et al reported significantly (P=0.013) increased frequencies of the 1236 and 3435 variant alleles in a simvastatin case–control study (23 patients with elevated CK blood concentrations vs 23 controls).31 Hoenig et al reported a significantly higher frequency of the ABCB1 3435T variant in patients with atorvastatin-induced myopathy compared to controls on atorvastatin without myopathy in a 98-patient study (80% vs 62%; P=0.043).32 This finding has not been replicated, and the limited number of patients with myopathy (n=10) included in the analysis markedly increased the scrutiny regarding the report by Hoenig et al. Furthermore, Hermann et al reported no difference in ABCB1 3435T allele frequency in a case–control study of atorvastatin myopathy,33 no association was detected between ABCB1 2677T and atorvastatin blood concentrations in a case–control study reported by DeGorter et al,34 and a reduced frequency of the ABCB1 TTT haplotype in statin myopathy patients compared to controls (20% vs 41%; P=0.03) was reported by Fiegenbaum et al in a prospective trial (n=146) of simvastatin 20 mg daily given for 6 months.35 As the clinical research findings regarding ABCB1 variants have been inconclusive and discordant, routine clinical use of ABCB1 genotyping to predict statin toxicity is not currently recommended. Nonetheless, ABCB1 plays an important role in statin transport, and the future of statin therapy may include multigene guidance that includes ABCB1 variants.
Simvastatin, atorvastatin, and lovastatin are primarily metabolized by cytochrome P450 (CYP) 3A enzymes. Most CYP3A metabolism occurs within hepatocytes, but some also occurs in the small intestine. Significant associations between CYP3A polymorphisms and statin blood concentrations have been reported, and the US Food and Drug Administration-approved product and prescribing label for simvastatin clearly warns clinicians about the marked increase in the risk of simvastatin myotoxicity associated with concomitant use of CYP3A-inhibiting medications (Table 3).36 In addition, other enzymes (CYPs and non-CYPs) are involved in the metabolism of certain statins (Table 4).37 Although less studied than CYP3A, those enzymes too can be significantly altered by the use of certain concomitant medications, resulting potentially in increased risk of statin adverse effects.
 | Table 3 CYP3A-inhibiting medications |
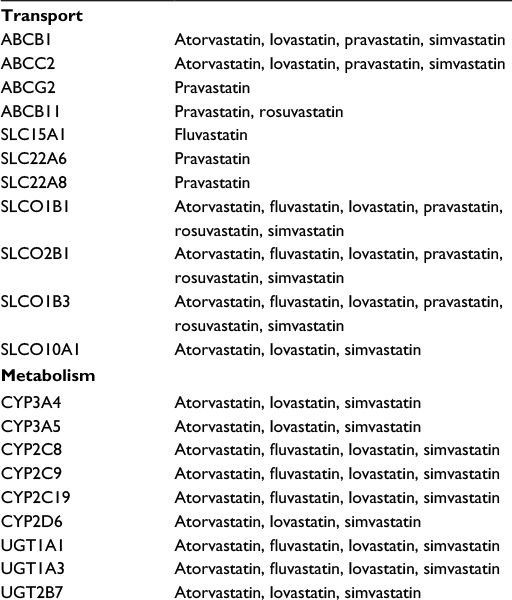 | Table 4 Select transport and metabolism proteins by statin type |
CYP3A4*22 (rs35599367) is a decrease-of-function polymorphism that results in significantly decreased CYP3A4 enzyme level and activity and altered pharmacokinetics and dynamics of simvastatin, atorvastatin, and lovastatin.38 Although the role of CYP3A5 in statin metabolism is less prominent than that of CYP3A4,39 associations with altered statin pharmacokinetics and dynamics have been reported for CYP3A5 polymorphisms. The most frequent and commonly studied CYP3A5 polymorphism is the loss of function CYP3A5*3 (rs776746) allele.40 For simvastatin, which has an annual prescription rate near 80 million in the US alone,1 the following associations have been reported: Tsamandouras et al determined that simvastatin bioavailability was nearly 50% greater in CYP3A4*22 carriers compared to wild type;41 Kim et al determined that simvastatin AUC was 2.3- and 3.3-fold higher in heterozygous and homozygous CYP3A5*3 carriers compared to wild type;42 and Kitzmiller et al determined that 12-hour post-dose concentrations of simvastatin and metabolite in Whites were 20% and 14% higher in *22 carriers compared to wild type and that simvastatin concentration in African Americans was 170% higher in *22 carriers compared to wild type and 33% higher in homozygous CYP3A5*3 carriers compared to those with functional CYP3A5.43
Although many studies have demonstrated the increased risk of statin toxicity with concomitant use of medications that inhibit CYP3A metabolism, there is a paucity of research investigating CYP3A polymorphisms and statin toxicity. Several studies investigating their effects on statin efficacy, however, have been reported. Wang et al determined that CYP3A4*22 carriers required statin doses of atorvastatin, simvastatin, or lovastatin, which were only 20%–60% of those required by wild type in a cohort of 235 dyslipidemic patients receiving statin doses titrated to achieve optimal lipid concentrations (P<0.05).38 Elens et al reported that simvastatin-associated LDL-C reduction in CYP3A4*22 carriers was 7% greater compared to noncarriers (41% vs 48%; P=0.054).44 Kivistö et al reported that the mean percent reduction in total cholesterol was higher (31% vs 17%) in homozygous CYP3A5*3 carriers compared to those with functional CYP3A5 in a study of 69 Caucasians who received lovastatin, simvastatin, or atorvastatin.45 No association, however, between CYP3A5*3 and LDL-C lowering was reported by Ragia et al in a study of 99 patients of European ancestry who received 20 mg simvastatin daily for 6 months,46 no association between CYP3A4*22 and LDL-C lowering was reported by Hu et al in a study of 229 patients of Chinese ancestry who received 40 mg simvastatin daily,47 and no association between CYP3A4*22 and lipid-lowering response was reported by Ragia et al in a study of 209 patients who received 10–40 mg simvastatin daily for 6 months.46 Although findings have been inconsistent and routine clinical use of CYP3A testing is not currently recommended, the importance of CYP3A in the metabolism of atorvastatin, simvastatin, and lovastatin suggests that further investigation may likely uncover clinically relevant effects of CYP3A polymorphisms on the risk of statin adverse effects in certain patient populations.
Genetic variation in the enzymes other than CYP3A which are involved in statin metabolism (Figure 1) also affects statin pharmacokinetic parameters and may alter the risk of statin adverse effects.48 Reports of investigations regarding the effect of altered metabolism in these enzymes (UGT, CYP2D6, and CYP2C) on the risk of statin adverse effects are rare and insufficient to suggest routine genetic testing. Significant biological plausibility exists, but the influence of polymorphisms in these metabolizing enzymes has been considered limited because alternate metabolism pathways can readily compensate or because they have insignificant impact, occur too infrequent, or are yet to be discovered.
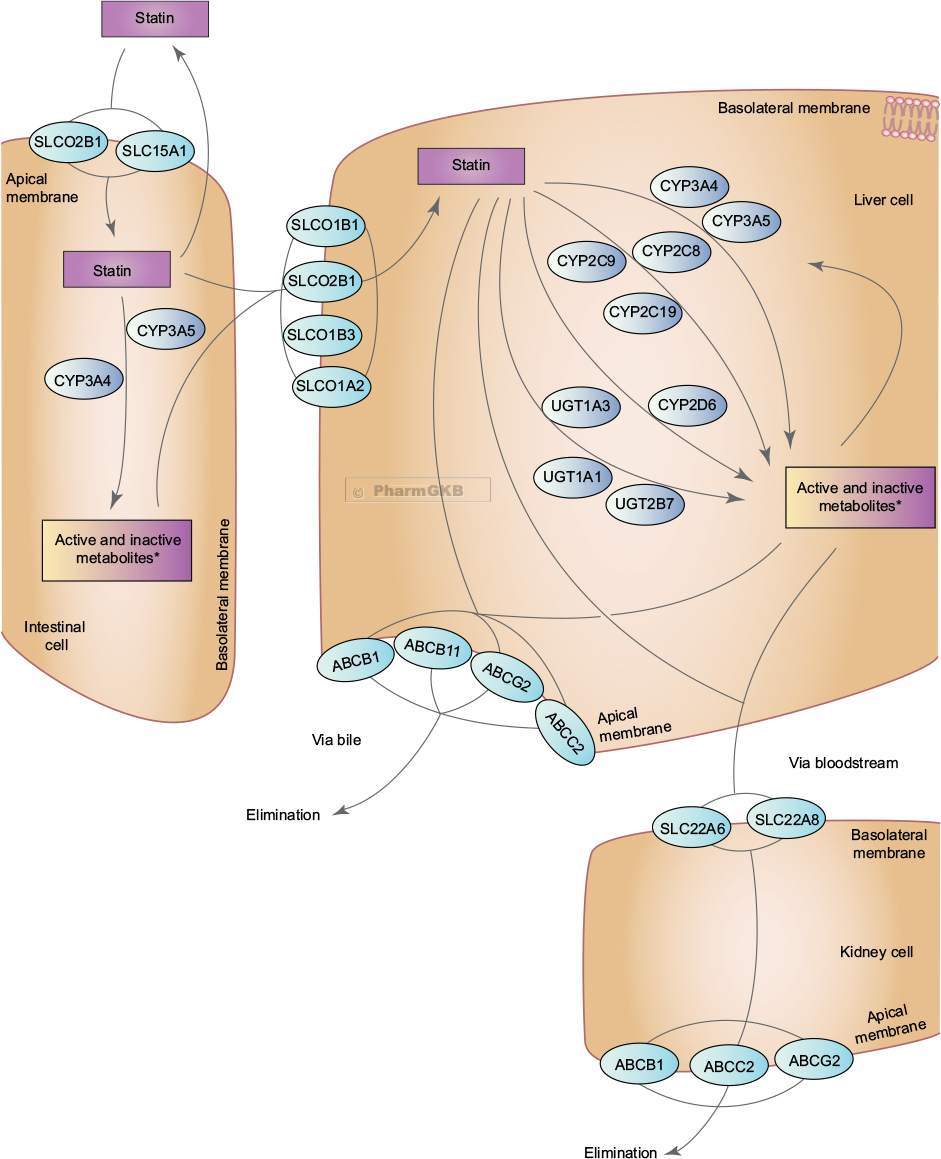 | Figure 1 Representation of the superset of all genes involved in the transport, metabolism and clearance of statin class drugs. ©PharmGKB.37 (Reproduced with permission from the Pharmacogenomics Knowledge Base [PharmGKB] and Stanford University, https://www.pharmgkb.org/pathway/PA145011108)71, Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–417. *Active and inactive metabolites are terms generalized to the medication class and may not be applicable to individual statin types. |
Genetic polymorphisms decreasing statin efficacy indirectly affect risk of statin toxicity
Although many of the new guidelines regarding cardiovascular pharmacotherapies no longer specify lipid goals,3–5 many prescribers continue to utilize lipid-lowering response as a surrogate measure of statin efficacy, titrating statin dose accordingly. Along with various clinical factors (eg, race, sex, ethnicity, and comorbidities) affecting lipid-lowering response to statins, genetic factors too can affect lipid-lowering response, thereby, indirectly affecting the risk of statin toxicity (ie, prescribers increase statin dose in patients not achieving significant lipid reduction, simultaneously increasing the risk of statin toxicity).
Several candidate genes have been investigated, but the majority of reported findings regarding statin efficacy focus on variants in HMGCR and in CETP. The pharmacologic target of statins, HMGCR, is polymorphic, and genetic variation can significantly affect statin efficacy. Although not a pharmacologic target of statins, CETP plays an important role in cholesterol metabolism, bringing cholesterol esters into the liver and transferring triglycerides from LDL to high-density lipoprotein.49 Polymorphisms in CETP have been associated with cholesterol levels, clinical outcomes (eg, myocardial infarction or stroke), and response to statins. Patients carrying the CETP Taq 1B polymorphism (rs708272) had lower concentrations of CETP, higher concentrations of high-density lipoproteins, and less atherosclerotic progression compared to wild type.49–51 The 10-year mortality rate for male statin-treated patients was higher for carriers of the Taq 1B variant compared to wild type.52 Together, these findings suggest that although untreated patients with the Taq 1B variant have less risk of atherosclerotic progression compared to wild type, statins may be more efficacious in wild type compared to those with the Taq 1B variant. In a meta-analysis (n=13,677) reported by Boekholdt et al, associations among the Taq 1B variant and concentrations of high-density lipoprotein and risk of atherosclerosis were confirmed; however, the Taq 1B association with statin treatment was not substantiated.53 In vitro studies have not yet been successful in elucidating the mechanisms by which the Taq 1B variant may affect cholesterol levels, cardiovascular risk, or response to statin pharmacotherapies. More recently, Papp et al reported that cholesterol concentrations and sex-dependent cardiovascular risk were significantly associated with transcription-altering polymorphisms in the promoter and enhancer regions of CETP and with a polymorphism in exon 9 of CETP that leads to formation of a nonfunctional or dominant-negative splice isoform of CETP.54 Further clinical association studies of these polymorphisms may help to better determine whether polymorphisms in CETP effect cardiovascular risk and response to statin pharmacotherapy.
Within hepatocytes, statins and metabolites inhibit HMGCR, the rate-limiting step of cholesterol synthesis. Decreased intrahepatic cholesterol synthesis leads to upregulation of hepatic surface LDL-C receptors and increased LDL-C uptake by hepatocytes. Systemic LDL-C is decreased as hepatic LDL-C uptake is increased. Ultimately, lower blood concentrations of LDL-C reduce the progression of atherosclerotic cardiovascular disease. Polymorphisms in HMGCR, however, can result in significantly diminished response to statin pharmacotherapy. Comprising three polymorphisms (rs17244841, rs3846662, and rs17238540), H7 haplotype of HMGCR appears to result in attenuated lipid-lowering response to statin pharmacotherapy.55 LDL-C reduction was ~20% less in a combined analysis of the Cholesterol and Pharmacogenomics (CAP) study, the Genetics of Diabetes Audit and Research in Tayside Scotland, and the Pravastatin Inflammation/CRP Evaluation (PRINCE) trial.55–57 The findings from a combined analysis of additional patient cohorts (Treatment to New Targets [TNT], Atorvastatin Comparative Cholesterol Efficacy and Safety Study [ACCESS], Assessment of Lescol in Renal Transplantation [ALERT], and Prospective Study of Pravastatin in the Elderly at Risk [PROPSER]) did not replicate an association of the H7 haplotype with diminished LDL-C response.58–61 Like CETP, HMGCR plays an important role in statin efficacy. The research to date, however, does not support CETP or HMGCR testing for guiding statin therapy. In fact, no efficacy-based pharmacogenomic biomarkers are currently recommended for guiding statin pharmacotherapy.
Genetic polymorphisms affecting end-organ toxicity pathways directly affect risk of statin toxicity
The decrease-of-function SLCO1B1 521C polymorphism results in decreased statin transport into liver cells. SLCO1B1 521C, therefore, should theoretically confer a decreased risk of statin-associated liver toxicity due to attenuated hepatic statin exposure. This, however, has not been studied. More importantly, SLCO1B1 521C does result in increased systemic statin exposure and increased risk of statin myopathy.20,23,25,26,31,34 Likewise, polymorphisms affecting transport across the blood–brain barrier would have the capacity to affect the risk of CNS toxicity, but this too has not been studied. No studies have been reported regarding the investigation of polymorphisms in CNS or liver as related to risk of statin-associated liver or CNS toxicity. Reports of polymorphisms in muscle, however, have been reported to influence risk of statin myopathy.
Associations between genetic variation in COQ2 and statin myopathy have been reported. Puccetti et al determined that rs4693075, a polymorphism in the COQ2, was associated with statin muscle intolerance in their analysis of 76 cases (46 and 30 cases treated with atorvastatin and rosuvastatin, respectively) and matched controls.62 Allele frequencies of rs4693075 were 0.11 and 0.56 in rosuvastatin-tolerant and rosuvastatin-intolerant patients, respectively, and 0.12 and 0.24 in atorvastatin-tolerant and atorvastatin-intolerant patients, respectively. In rosuvastatin-treated patients, the 95% CI of the OR was 1.7–4.4 (P<0.001). In the atorvastatin-treated patients, the 95% CI of the OR was 1.9–6.4 (P<0.001).62 Ruaño et al reported an association between another polymorphism in COQ2, rs4693570, and statin-induced myalgia in a case–control study (n=377 cases vs 416 controls) of patients receiving various statins (P<0.01).63 Although some patient reports have described benefits from COQ2 supplementation in cases of statin myopathy,64 neither COQ2 supplementation nor COQ2 testing is currently recommended for routine use in patients receiving statin pharmacotherapy.
GATM is the rate-limiting enzyme in the creatine biosynthesis pathway. Providing an important source of cellular energy, creatine is predominantly synthesized in the liver and kidneys and subsequently transported to skeletal muscle. A GATM polymorphism, rs9806699, was associated with decreased risk of statin myopathy (95% CI for meta-analysis OR was 0.45–0.81) in an analysis of 172 cases of statin myopathy.65 Mangravite et al suggested that the protective effect of the GATM polymorphism may likely have been the result of the attenuation of cellular processes (via diminished myocellular capacity for phosphocreatine energy storage as a result of decreased creatine availability) necessary for development of statin myopathy.65 The detected effect reported by Mangravite et al was not substantiated in three separate cohorts: analyses reported by Luzum et al, Carr et al, and Floyd et al found no associations between rs9806699 and statin myopathy (n=609, 150, and 175 cases, respectively).66–68 Furthermore, the mechanism (regarding the apparent protective effect of GATM rs9806699) proposed by Mangravite et al is not consistent with the reported findings that suggest GATM deficiency as a contributing factor in cases of myopathy.69,70 The clinical significance of GATM rs9806699 is therefore uncertain, and additional research is necessary to determine whether the variant truly confers a protective effect against statin myopathy in certain patient populations.
Future considerations and directions
With proven efficacy and relatively few adverse effects, statins remain among the most commonly prescribed medication classes. Most patients benefit; however, some experience atherosclerotic events despite statin therapy, and some experience adverse effects. Despite the completion of hundreds of candidate gene studies and numerous GWASs, the only clinically relevant pharmacogenetic test regarding statin toxicity is SLCO1B1 521C and its relevance is limited to simvastatin myopathy. The research findings regarding polymorphisms in other key genes (eg, ABCB1, CYP3A, HMGCR, CETP, GATM, and COQ2) suggest that additional research is warranted and that clinically meaningful genetic testing for risk of statin toxicity may ultimately be summative (eg, multigene risk score modeling). Future research must also incorporate nongenetic risk factors and consider interactions (eg, gene–gene, gene–environment). Flexibility to accommodate risk factors specific to statin type and the ability to adjust for race and ethnicity are also essential. Ultimately, a pharmacogenomic test, in the form of a multi-polymorphism and multigene array, could provide opportunity for prescriber and patient to better assess the expected benefit and potential risks of statin pharmacotherapy, allowing for more informed strategies for selecting statin dose and type.
Disclosure
The authors report no conflicts of interest in this work.
References
IMS Institute for Healthcare Informatics. Medicines use and spending shifts: a review of the use of medicines in the U.S. in 2014. Parsippany, NJ: IMS Institute for Healthcare Informatics; 2015. | ||
Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomisedtrials of statins. Lancet. 2005;366:1267–1278. | ||
Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;63(25):2889–2934. | ||
Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia. Part 1 – full report. J Clin Lipidol. 2015;9(2):129–169. | ||
Stroes ES, Thompson PD, Corsini A, et al. Statin-associated muscle symptoms: impact on statin therapy–European Atherosclerosis Society consensus panel statement on assessment, aetiology and management. Eur Heart J. 2015;36(17):1012–1022. | ||
Leusink M, Onland-Moret NC, de Bakker PI, de Boer A, Maitland-van der Zee AH. Seventeen years of statin pharmacogenomics: a systematic review. Pharmacogenomics. 2016;17(2):163–180. | ||
Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97(8A):52C–60C. | ||
Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA. 2003;289(13):1681–1690. | ||
Cohen JD, Brinton EA, Ito MK, Jacobson TA. Understanding Statin Use in America and Gaps in Patient Education (USAGE): an internet-based survey of 10,138 current and former statin users. J Clin Lipidol. 2012;6(3):208–215. | ||
Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc Drugs Ther. 2005;19(6):403–414. | ||
Simpson RJ, Mendys P. The effects of adherence and persistence on clinical outcomes in patients treated with statins: a systematic review. J Clin Lipidol. 2010;4(6):462–471. | ||
The West of Scotland Coronary Prevention Study Group. Compliance and adverse event withdrawal: their impact on the West of Scotland Coronary Prevention Study. Eur Heart J. 1997;18(11):1718–1724. | ||
McGinnis BD, Olson KL, Delate TM, Stolcpart RS. Statin adherence and mortality in patients enrolled in a secondary prevention program. Am J Manag Care. 2009;15(10):689–695. | ||
de Denus S, Spinler SA, Miller K, Peterson AM. Statins and liver toxicity: a meta-analysis. Pharmacotherapy. 2004;24(5):584–591. | ||
Suraweera C, de Silva V, Hanwella R. Simvastatin-induced cognitive dysfunction: two case reports. J Med Case Rep. 2016;10(1):83. | ||
Sallustio F, Studer V. Targeting new pharmacological approaches for Alzheimer’s disease: potential for statins and phosphodiesterase inhibitors. CNS Neurol Disord Drug Targets. 2016;15(6):647–659. | ||
Stulc T, Ceška R, Gotto AM. Statin intolerance: the clinicians’ perspective. Curr Atheroscler Rep. 2015;17(12):69. | ||
Niemi M, Pasanen MK, Neuvone PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63(1):157–181. | ||
Feng Q, Wilke RA, Baye TM. Individualized risk for statin-induced myopathy: current knowledge, emerging challenges and potential solutions. Pharmacogenomics. 2012;13(5):579–594. | ||
Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359(8):789–799. | ||
Peters BJ, Rodin AS, Klungel OH, et al. Pharmacogenetic interactions between ABCB1 and SLCO1B1 tagging SNPs and the effectiveness of statins in the prevention of myocardial infarction. Pharmacogenomics. 2010;11(8):1065–1076. | ||
Yang GP, Yuan H, Tang B, et al. Lack of effect of genetic polymorphisms of SLCO1B1 on the lipid-lowering response to pitavastatin in Chinese patients. Acta Pharmacol Sin. 2010;31(3):382–386. | ||
Donnelly LA, Doney AS, Tavendale R, et al. Common nonsynonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: a go-DARTS study. Clin Pharmacol Ther. 2011;89(2):210–216. | ||
Brunham LR, Lansberg PJ, Zhang L, et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharmacogenomics J. 2012;12(3):233–237. | ||
Wilke RA, Ramsey LB, Johnson SG, et al. The Clinical Pharmacogenomics Implementation Consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther. 2012;92(1):112–117. | ||
Ramsey LB, Johnson SG, Caudle KE, et al. The Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin Pharmacol Ther. 2014;96(4):423–428. | ||
Maningat P, Gordon BR, Breslow JL. How do we improve patient compliance and adherence to long-term statin therapy? Curr Atheroscle Rep. 2013;15(1):291. | ||
Haga SB, Lapointe NM. The potential impact of pharmacogenetic testing on medication adherence. Pharmacogenomics J. 2013;13(6):481–483. | ||
Josephine LH, Joy SV, Haga SB, et al. Genetically guided statin therapy on statin perceptions, adherence, and cholesterol lowering: a pilot implementation study in primary care patients. J Pers Med. 2014;4(2):147–162. | ||
Keskitalo J, Kurkinen K, Neuvonen P, Niemi M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin Pharmacol Ther. 2008;84(4):457–461. | ||
Ferrari M, Guasti L, Maresca A, et al. Association between statin-induced creatine kinase elevation and genetic polymorphisms in SLCO1B1, ABCB1 and ABCG2. Eur J Clin Pharmacol. 2014;70(5):539–547. | ||
Hoenig MR, Walker PJ, Gurnsey C, Beadle K, Johnson L. The C3435T polymorphism in ABCB1 influences atorvastatin efficacy and muscle symptoms in a high-risk vascular cohort. J Clin Lipidol. 2011;5(2):91–96. | ||
Hermann M, Bogsrud MP, Molden E, et al. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin Pharmacol Ther. 2006;79(6):532–539. | ||
DeGorter MK, Tirona RG, Schwarz UI, et al. Clinical and pharmacogenetic predictors of circulating atorvastatin and rosuvastatin concentrations in routine clinical care. Circ Cardiovasc Genet. 2013;6(4):400–408. | ||
Fiegenbaum M, da Silveira FR, van der Sand CR, et al. The role of common variants of ABCB1, CYP3A4, and CYP3A5 genes in lipid-lowering efficacy and safety of simvastatin treatment. Clin Pharmacol Ther. 2005;78(5):551–558. | ||
United States Federal Drug Administration. FDA drug safety communication: new restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. US FDA Safety Announcement 06-08-2011. Washington, DC: FDA; 2011. | ||
Whirl-Carrillo M, McDonagh EM, Herbert JM, et al. Pharmacgenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–417. | ||
Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 2011;11(4):274–286. | ||
Park JE, Kim KB, Bae SK, Moon BS, Liu KH, Shin JG. Contribution of cytochrome P450 3A4 and 3A5 to the metabolism of atorvastatin. Xenobiotica. 2008;38(9):1240–1251. | ||
Westlind-Johnsson A, Malmebo S, Johansson A, et al. Comparative analysis of CYP3A expression in human liver suggests only a minor role for CYP3A5 in drug metabolism. Drug Metab Dispos. 2003;31(6):755–761. | ||
Tsamandouras N, Dickinson G, Guo Y, et al. Identification of the effect of multiple polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid using a population-modeling approach. Clin Pharmacol Ther. 2014;96(1):90–100. | ||
Kim KA, Park PW, Lee OJ, Kang DK, Park JY. Effect of polymorphic CYP3A5 genotype on the single-dose simvastatin pharmacokinetics in healthy subjects. J Clin Pharmacol. 2007;47(1):87–93. | ||
Kitzmiller JP, Luzum JA, Baldassarre D, Krauss RM, Medina MW. CYP3A4*22 and CYP3A5*3 are associated with increased levels of plasma simvastatin concentrations in the cholesterol and pharmacogenetics study cohort. Pharmacogenet Genomics. 2014;24(10):486–491. | ||
Elens L, Becker ML, Haufroid V, et al. Novel CYP3A4 intron 6 single nucleotide polymorphism is associated with simvastatin-mediated cholesterol reduction in the Rotterdam Study. Pharmacogenet Genomics. 2011;21(12):861–866. | ||
Kivistö KT, Niemi M, Schaeffeler E, et al. Lipid-lowering response to statins is affected by CYP3A5 polymorphism. Pharmacogenetics. 2004;14(8):523–525. | ||
Ragia G, Kolovou V, Tavridou A, et al. No effect of CYP3A4 intron 6 C>T polymorphism (CYP3A4*22) on lipid-lowering response to statins in Greek patients with primary hypercholesterolemia. Drug Metab Pers Ther. 2015;30(10):43–48. | ||
Hu M, Mak VW, Xiao Y, Tomlinson B. Associations between the genotypes and phenotype of CYP3A and the lipid response to simvastatin in Chinese patients with hypercholesterolemia. Pharmacogenomics. 2013;14(1):25–34. | ||
Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–417. | ||
Freeman DJ, Griffin BA, Holmes AP, et al. Regulation of plasma: associations between the TaqI B RFLP in the CETP gene and smoking and obesity. Arterioscler Thromb. 1994;14(3):336–344. | ||
Kuivenhoven JA, Jukema JW, Zwinderman AH, et al. The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The Regression Growth Evaluation Statin Study Group. N Engl J Med. 1998;338(2):86–93. | ||
Willer 2008. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40(2):161–169. | ||
Regieli JJ, Jukema JW, Grobbee DE, et al. CETP genotype predicts increased mortality in statin-treated men with proven cardiovascular disease: an adverse pharmacogenetic interaction. Eur Heart J. 2008;29(22):2792–2799. | ||
Boekholdt SM, Sacks FM, Jukema JW, et al. Cholesteryl ester transfer protein TaqIB variant, high-density lipoprotein cholesterol levels, cardiovascular risk, and efficacy of pravastatin treatment: individual patient meta-analysis of 13,677 subjects. Circulation. 2005;111(3):278–287. | ||
Papp AC, Pinsonneault JK, Wang D, et al. Cholesteryl ester transfer protein (CETP) polymorphisms affect mRNA splicing, HDL levels, and sex-dependent cardiovascular risk. PLoS One. 2012;7(3):e31930. | ||
Krauss RM, Mangravite LM, Smith JD, et Aal. Variation in the 3-hydroxyl-3-methylglutaryl coenzyme a reductase gene is associated with racial differences in low-density lipoprotein cholesterol response to simvastatin treatment. Circulation. 2008;117(12):1537–1544. | ||
Chasman DI, Posada D, Subrahmanyan L, Cook NR, Stanton VP Jr, Ridker PM. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA. 2004;291(23):2821–2827. | ||
Singer JB, Holdaas H, Jardine AG, et al. Genetic analysis of fluvastatin response and dyslipidemia in renal transplant recipients. J Lipid Res. 2007;48(9):2072–2078. | ||
Donnelly LA, Doney AS, Dannfald J, et al. A paucimorphic variant in the HMG-CoA reductase gene is associated with lipid-lowering response to statin treatment in diabetes: a GoDARTS study. Pharmacogenet Genomics. 2008;18(12):1021–1026. | ||
Thompson J, Man M, Johnson K, et al. An association study of 43 SNPs in 16 candidate genes with atorvastatin response. Pharmacogenomics J. 2005;5(6):352–358. | ||
Polisecki E, Muallem H, Maeda N, et al. Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) Investigators. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis. 2008;200(1):109–114. | ||
Thompson JF, Hyde CL, Wood LS, et al. Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the treating to new targets (TNT) cohort. Circ Cardiovasc Gen. 2009;2(2):173–181. | ||
Puccetti L, Ciani F, Auteri A. Genetic involvement in statins induced myopathy. Preliminary data from an observational case–control study. Atherosclerosis. 2010;211(1):28–29. | ||
Ruaño G, Windemuth A, Wu AHB, et al. Mechanisms of statin induced myalgia assessed by physiogenomic associations. Atherosclerosis. 2011;218(2):451–456. | ||
Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol. 2007;49(23):2231–2237. | ||
Mangravite LM, Engelhardt BE, Medina MW, et al. A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature. 2013;502(7471):377–380. | ||
Luzum JA, Kitzmiller JP, Isackson PJ, et al. GATM polymorphism associated with the risk for statin-induced myopathy does not replicate in case-control analysis of 715 dyslipidemic individuals. Cell Metab. 2015;21(4):622–627. | ||
Carr DF, Alfirevic A, Johnson R, Chinoy H, van Staa T, Pirmohamed M. GATM gene variants and statin myopathy risk. Nature. 2014;513(7518):E1. | ||
Floyd JS, Bis JC, Brody JA, Heckbert SR, Rice K, Psaty BM. GATM locus does not replicate in rhabdomyolysis study. Nature. 2014;513(7518):E1–E3. | ||
Edvardson S, Korman SH, Livne A, et al. L-arginine:glycine amidinotransferase (AGAT) deficiency: clinical presentation and response to treatment in two patients with a novel mutation. Mol Genet Metab. 2010;101(2–3):228–232. | ||
Nouioua S, Cheillan D, Zaouidi S, et al. Creatine deficiency syndrome. A treatable myopathy due to arginine-glycine amidinotransferase (AGAT) deficiency. Neuromuscul Disord. 2013;23(8):670–674. | ||
Pharmacogenomics Knowledge Base (PharmGKB) and Stanford University. Available from: https://www.pharmgkb.org/pathway/PA145011108. Accessed September 20, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.