Back to Journals » Infection and Drug Resistance » Volume 12
Phage therapy as a renewed therapeutic approach to mycobacterial infections: a comprehensive review
Authors Azimi T ![]() , Mosadegh M
, Mosadegh M ![]() , Nasiri MJ
, Nasiri MJ ![]() , Sabour S, Karimaei S, Nasser A
, Sabour S, Karimaei S, Nasser A
Received 7 June 2019
Accepted for publication 31 August 2019
Published 17 September 2019 Volume 2019:12 Pages 2943—2959
DOI https://doi.org/10.2147/IDR.S218638
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Joachim Wink
Taher Azimi,1,2 Mehrdad Mosadegh,2 Mohammad Javad Nasiri,1 Sahar Sabour,3 Samira Karimaei,2 Ahmad Nasser4
1Department of Microbiology, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 2Department of Pathobiology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran; 3Department of Microbiology, School of Medicine, Ardabil University of Medical Science, Ardabil, Iran; 4Clinical Microbiology Research Center, Ilam University of Medical Science, Ilam, Iran
Correspondence: Taher Azimi
Department of Microbiology, School of Medicine, Shahid Beheshti University of Medical Sciences, PO Box 15468-15514, Tehran, Iran
Tel +98 938 213 0411
Email [email protected]
Abstract: Mycobacterial infections are considered to a serious challenge of medicine, and the emergence of MDR and XDR tuberculosis is a serious public health problem. Tuberculosis can cause high morbidity and mortality around the world, particularly in developing countries. The emergence of drug-resistant Mycobacterium infection following limited therapeutic technologies coupled with the serious worldwide tuberculosis epidemic has adversely affected control programs, thus necessitating the study of the role bacteriophages in the treatment of mycobacterial infection. Bacteriophages are viruses that are isolated from several ecological specimens and do not exert adverse effects on patients. Phage therapy can be considered as a significant alternative to antibiotics for treating MDR and XDR mycobacterial infections. The useful ability of bacteriophages to kill Mycobacterium spp has been explored by numerous research studies that have attempted to investigate the phage therapy as a novel therapeutic/diagnosis approach to mycobacterial infections. However, there are restricted data about phage therapy for treating mycobacterial infections. This review presents comprehensive data about phage therapy in the treatment of mycobacterial infection, specifically tuberculosis disease.
Keywords: mycobacteriophage, tuberculosis, mycobacteria infection, phage therapy
Introduction
Mycobacterium species (spp) can create a variety of infections such as tuberculosis (TB), Searls ulcer, leprosy, and fish tank granuloma.1 TB, caused by Mycobacterium tuberculosis (Mtb), is one of the most serious public health problems that can cause high morbidity and mortality worldwide.2–4 Based on WHO TB report in 2018, TB remained to be a major global health challenge and, in 2017, TB caused an estimated number of 1.3 million deaths among HIV-negative people. In addition, there were an additional number of 300, 000 deaths from TB among HIV-positive people. Moreover, there were an estimated number of 10.0 million new TB cases equal to 133 cases per 100, 000 population, worldwide.5 The rapid spread of infections and the alarming growth of drug resistants, especially the emergence of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) strains, have sounded the alarm to find more new potent drugs; therefore, finding an alternative approach to the controlling and treatment of TB has become extremely vital.6 Phage therapy can be considered as a significant alternative to antibiotics for treating MDR and XDR pathogens.7 The natural and useful capacity of bacteriophages to infect mycobacterial hosts, as well as their ability to kill Mycobacterium spp, has involved numerous research studies for investigaing the phage therapy as a novel therapeutic/diagnosis approach to mycobacterial infections.1,6 However, approapriately relevant data about the efficiency of phage therapy in treating various mycobacterial infections, especially TB, are largely scarce and limited. In this study, a comprehensive review of the literature has been conducted to identify in vitro and in vivo studies associated with the phage therapy in the mycobacterial infection.
Phage therapy
The increasing emergence of infection, coinfection, and drug-resistant pathogens has become a severe challenge for scientists and public health.8,9 In recent years, several novel alternative strategies including therapeutic enzymes, pigments, phytochemicals, antimicrobial polymers, antimicrobial peptides, and silver nanoparticles have led researchers to consider the treatment of infections in the presence of MDR and XDR pathogens.1,10,11 However, since drug-resistant bacteria have become increasingly problematic and challenging, phage therapy is considered to be an important candidate for alternative therapy.12 Bacteriophages are viruses that are isolated from several ecological specimens including sewage, soil, and water.13–15 The isolation of bacteriophage from an environmental sample revealed that bacteriophages did not exert adverse effects on individuals.15–17 It is estimated that there are more than 4200 bacteriophages that exactly infect Mycobacterium spp; it is expected that 1025 phage launches a new infection cycle in every second of the day.18 The application of bacteriophage as a candidate for alternative therapy in non-mycobacterial infection was discovered in the early 20th century, showing that bacteriophage has a high capacity to efficiently eradicate pathogenic bacteria.12,19,20
However, information about the use of phages in the treatment of bacterial infection in humans is little. Phages are considered to be a good candidate in treating and controlling mycobacterial infections; however, these agents have several advantages and limitations for use in humans.21–23 The main advantages and characteristics of bacteriophages are as follows: I) phages cannot infect human cells and replicate only in the target bacterium; II) the selected phage perfectly lyses the pathogen at the site of infection; III) the administration of phages is easier and, after the initial administration, the concentration of phages increases at the site of infection; therefore, very few doses are required;24–26 III) similar to phages TM4 and D29, the selected phage should be highly virulent against Mtb; IV) a non-pathogenic phage should be used in phage therapy; and V) the selected phage should not trigger an immune response and their effects are limited to the site of infection.13,20,27 On the other hand, the use of bacteriophages in humans is subject to several limitations as follows: I) identifying a phage with therapeutic characteristics and demonstrating that a phage is specific to a given bacterial strain is very difficult;21,28 II) the appearance and development of bacterial resistance against phages is theoretically possible, and the production of phage genome without antibiotic-resistant gene, genes encoded bacterial virulence factors, and integrase genes (or without genes for phage-encoded toxins) is complicated;21,29 III) the formulation and stabilization of pharmaceutical preparations of phages is difficult and has several problems;30 IV) it is possible that lysogenic phages integrate their DNA into the bacterial genome and horizontally transfer resistant genes to the bacteria; therefore, new resistant bacteria can develop.31,32 It can be concluded that phages can perhaps set the ground for the emergence and development of antibiotic resistance; and V) it is possible for the immune system to lead to the reduced activity of phages under the in vivo condition.33,34 Results of the previously published studies revealed that phage therapy could be very effective against different pathogenic bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, Staphylococcus aureus, and Escherichia coli.35–38 However, there are limited studies and data about the role of phage therapy in the treatment of mycobacterial infections.
Is mycobacterium smegmatis the ideal delivery system in phage therapy?
Mycobacterium smegmatis (M. smegmatis) is a nonvirulent fast-growing mycobacterium that does not infect people with disease, even immunosuppressed peoples.39 Since bacteriophages do not have the ability to spread throughout the membrane, several strategies are required to deliver the bacteriophage to the intracellular pathogenic bacteria.20,40 The use of nonvirulent mycobacterium, especifically M. smegmatis as a delivery system, is one of the main strategies used for the treatment of mycobacterial infection (Figure 1).41 In the phage therapy process, M. smegmatis plays several roles: I) M. smegmatis acts as a carrier to deliver phage to the intracellular pathogen; II) M. smegmatis can act as a host and lead to the high proliferation of bacteriophage; III) this organism increases bacteriophage infection rates; and IV) M. smegmatis is an appropriate environment for bacteriophage activity within mononuclear cells such as macrophages and monocytes.13,42 Based on the above, it can be concluded that, in phage therapy, the use of nonvirulent M. smegmatis in intracellular infection treatment is a better choice, and this result can be seen as “proof of concept” that opens up new opportunities for further research.
 |
Figure 1 Steps involved in phage mediated Mycobacterium tuberculosis lysis using Mycobacterium smegmatis. |
Phage therapy in tuberculosis
The emergence of MDR and XDR M. tuberculosis and the limitation of finding more potent drugs and alternate therapeutics for the treatment of TB have attracted different research groups to investigate the bacteriophage roles as a suitable alternative to antibiotics in the treatment of TB.6,43 According to the natural capability to infect and kill mycobacteria, bacteriophages can be considered as an appropriate alternative to antibiotics.42 Different bacteriophages were investigated against TB as therapeutic options as follows: 1) Phage DS-6A; 2) Phage TM4; 3) Phage D29; 4) Phage T7; 5) Phage P4; 6) Phage PDRPv; 7) Phage BTCU-1; 8) Phage Bo4; 9) Phage SWU1; 10) Phage GR-21/T; 11) Phage My-327; 12) Phage Ms6; and 13) Phage Bxz2. In this review article, the role of important mycobacteriophages in the treatment of mycobacterial infection, especifically tuberculosis disease, is explained (Table 1).
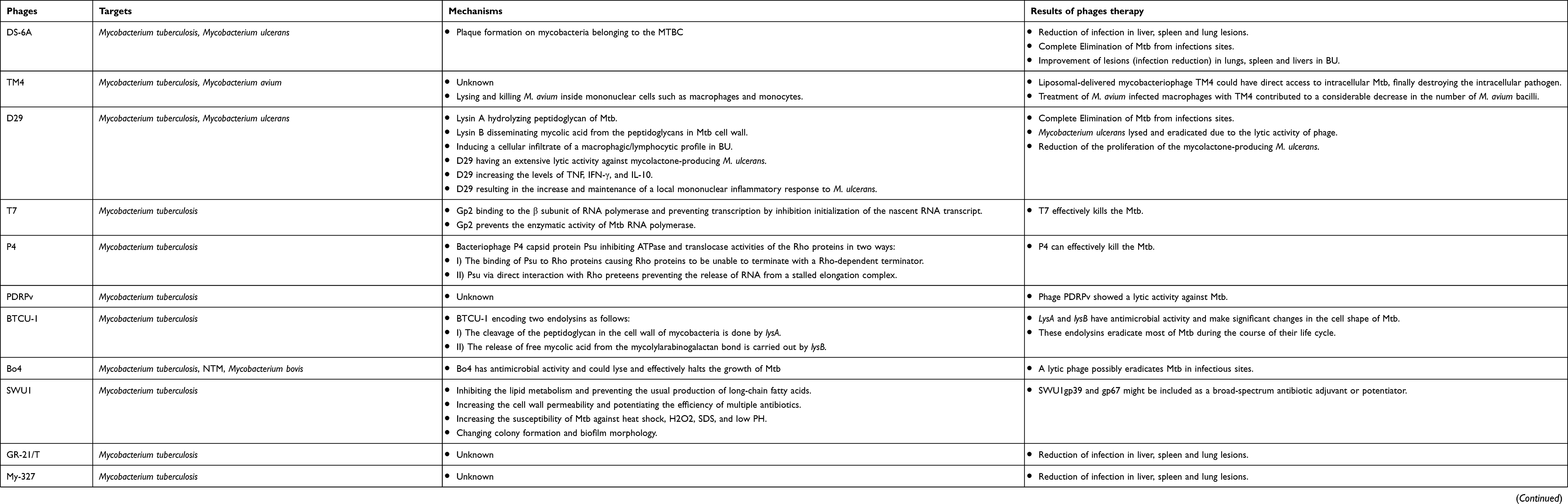 |
Table 1 The list of mycobacteriophages used against Mycobacteria infections: targets, mechanisms, and results of phage therapy |
Phage DS-6A
Mycobacteriophage DS6A has a high specificity to members of the Mycobacterium tuberculosis complex (MTBC), and this unique feature makes this bacteriophage an important and interesting candidate for anti-TB therapy.44 This mycobacteriophage can form plaque only on mycobacteria belonging to the MTBC.45 The results of the previously published studies have shown that the treatment of Mtb infection with mycobacteriophage DS6A has led to a reduction in infection in liver, spleen, and lung lesions; it has shown a high ability to competently eliminate Mtb from infectious sites.44,46 In this scenario, it is presumed that phage-infected Mtb cells may briefly transport mycobacteriophage DS6A to Mtb bacilli within macrophages (Figure 1).13,46 However, more in-vitro and in-vivo studies are required to shed light on the mechanisms that phage uses for Mtb eradication.
Phage TM4
The length of the mycobacteriophage TM4 genome is nearly 52,797 bp, which encodes several proteins with different functions.42 The proteins encoded by TM4 are similar to transcriptional regulators or bear a high similarity to haloperoxidases and glutaredoxins.47 TM4 is a lytic bacteriophage with double-stranded DNA and an extensive host range that could infect both slow-growing and fast-growing strains of mycobacteria.48 Based on the results of studies that evaluated the gene expression and codon usage in different mycobacteriophages, it is revealed that mycobacteriophage TM4 extremely expresses genomes and has probably the strongest capability to eradicate mycobacteria strains in infectious sites.49 In addition to the problem of MDR and XDR Mtb strains, these bacteria infect macrophages and monocytes and grow within them. Finally, in macrophages and monocytes, Mtb begins a dormant or latent phase of infection and produces the restriction effects of used antibiotics against Mtb infections.48 Overall, all mycobacteriophages are not able to penetrate eukaryotic cells, and this limitation against intracellular bacterial infection, such as TB, leads to the introduction and development of a novel vector for phage delivery into intracellular infections.42 Recently, researchers have used the liposomes as a delivery system to transport bacteriophage into pathogen-infected cells.50 Liposomes have significant cell penetration features and, therefore, are quite possible to be an appropriate envelope for phages used against intracellular bacteria, especially Mtb.42,50 Results of a previously published study revealed that liposomes penetrated into infected cells by endocytosis and were found within early endosomes after penetration.51 Mycobacteriophage TM4 has the ability to destroy the intracellular pathogen.52 Since liposomal-delivered mycobacteriophage TM4 could have direct access to intracellular Mtb,42 it is concluded that liposome is a proper vector for mycobacteriophage TM4 therapy of intracellular Mtb infection (Figure 2).
 |
Figure 2 Steps involved in phage TM4 mediated Mycobacterium tuberculosis lysis using liposome. |
Phage D29
The ability of mycobacteriophage D29 to quickly penetrate and eradicate pathogens is the reason why researchers have considered it as a new therapeutic option against MDR and XDR bacteria, especially Mtb.53 This mycobacteriophage is used for several targets, particularly in TB diagnosis and probably in TB treatment.54 In the mycobacteriophage D29 genome, there are three genes that make the lytic cassette. These three genes encode Lysin A, Lysin B, and holin protein.55,56 Lysin A coded by gp10 hydrolyzes peptidoglycan of bacterial cell walls.57 Gp12 encodes Lysin B that leads to the dissemination of mycolic acid from the peptidoglycans in mycobacterial cell wall.53,58 Holin as a membrane pore-forming protein participates in the transfer of lysins from the cytosol to the periplasm.59 Lysin A of mycobacteriophage D29 has three main domains including NTD (lethal to M. smegmatis), LD with catalytic activity in N terminus, and one domain present at the C terminus. Specifically, the C-terminal domain of Lysin A binds to Mtb peptidoglycan and hydrolyzes it.55,57 Therefore, these molecules (LysinA, LysinB, and holin) are potential candidates that develop phage-based therapeutics against Mtb infections (Figure 3).
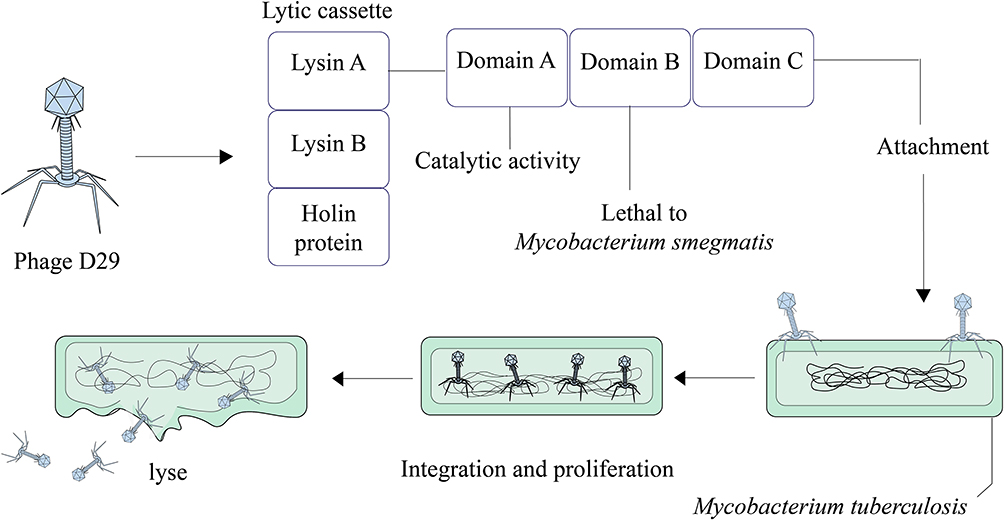 |
Figure 3 Overview of phage D29 mediated Mycobacterium tuberculosis lysis. |
Phage T7
Bacteriophage T7, an obligate lytic phage, was defined in 1945 as one of the several different bacteriophages that infects and replicates in Escherichia coli.60 Bacteriophage T7 enjoys several advantages that make it a suitable model system for genome strategy and engineering. These advantages are as follows: 1) T7 is relatively independent of complex host physiology, and 2) most of the T7 genomes are transported into a newly infected cell through RNA polymerase.61,62 Six important proteins formed the main T7 phage particle including I) gp10A as a primary capsid protein; II) gp10B as a secondary capsid protein; III) gp8 as a connector; IV) gp17 as a tail fiber; and V) gp11 and gp12 as tail proteins.60,63 A new method for treating infectious diseases, such as TB, is used to define how bacteriophages can be effective in killing bacterial cells. Bacteriophages could produce small encoded proteins that bind to RNA polymerase.64 RNA polymerase is responsible for transcription in microorganism, and the binding of the small phage-encoded proteins to RNA polymerase leads to the suppression of bacterial gene transcription. T7 phage protein, Gp2, encoded by the rpoC gene, in Escherichia coli, binds to the section including amino acids 1145–1198 in the beta-prime subunit of RNAP, thus inhibiting the productive engagement of RNA polymerase with the promoter.65–68 Gp2 prevents the enzymatic activity of bacterial RNA polymerase by several mechanisms in the following fashion: I) Gp2 inhibits functionally necessary alterations in RNA polymerase; II) Gp2 inhibits the interaction between catalytic site of RNA polymerase and DNA; III) in the DNA binding channel, Gp2 inhibits the binding of DNA.64 One of the main antibiotics used in TB treatment is rifampicin. Results of an in silico study revealed that, in the case of Mtb infection, Gp2, similar to rifampicin, binds to the β subunit of RNA polymerase, encoded by the rpoB gene; in addition, it prevents transcription by inhibiting the nascent RNA transcript, yet to a lesser degree than that in Escherichia coli (Figure 4).64,69
 |
Figure 4 Steps involved in phage T7 mediated Mycobacterium tuberculosis lysis. |
Phage P4
Rho is a homohexameric transcription terminator in various pathogens including Gram-negative and Gram-positive bacteria that regulates gene expression and many physiological processes and is a potential antibiotic target.70–74 Accordingly, Rho is involved in many physiological processes; therefore, the inhibition of this transcription terminator could be beneficial as a synergistic antimicrobial treatment strategy.75 In different bacteria such as Escherichia coli, Psu is a bacteriophage P4 capsid protein that acts as an unconventional capsid organizing protein and inhibits ATPase and translocases activities of these Rho proteins.76 In recent years, the results of a study revealed that the Rho-dependent termination had the main role in the pathogenicity of Mtb.77 Similar to the same bacteria, in Mtb, Psu binds to Rho protein and antagonizes Rho in trans by forming a mechanical interference to Rho translocation (Figure 5).78 In total, the expression of Psu can kill various bacteria such as Mtb in two ways: I) the binding of Psu to Rho proteins makes Rho proteins unable to terminate with a Rho-dependent terminator; II) Psu through direct interaction with Rho proteins prevents the release of RNA from a stalled elongation complex.76 Hence, Psu could be useful as a synergistic antibiotic treatment against Mycobacterium.
 |
Figure 5 Overview of phage P4 mediated Mycobacterium tuberculosis lysis. |
Phage PDRPv
So far, twenty-seven clusters of mycobacteriophages have been identified in the Actinobacteriophage database (Phagesdb.org). Mycobacteriophages (PDRPv) belong to Siphoviridae family and B1 sub-cluster. The length of the mycobacteriophage PDRPv genome is approximately 69,110 bp with a G+C content of ~66%, containing 106 open reading frames (ORFs).12,79 The results of another study revealed that phage PDRPv had a lytic activity against Mtb. However, the exact anti-tuberculosis mechanism of this phage was not determined.12
Phage BTCU-1
Mycobacteriophage BTCU-1 belongs to Siphoviridae family and has been isolated from soil specimen, obtained from eastern Taiwan.80 The length of the mycobacteriophage BTCU-1 genome is approximately 46 kb, and this bacteriophage has a linear double-stranded DNA with an icosahedral head and a very long tail.81 The genome of BTCU-1 encodes several proteins with predefined functions. One of these proteins is identified as a putative phosphoribosyl transferase (PRT) and is particularly found in mycobacteriophages that infect Mycobacterium.80,82 These proteins eradicate most bacteria during the course of their life cycle.80 Moreover, different mycobacteriophages encode lytic endolysins that have antimicrobial activity and can be effective against MDR and XDR MTB strains.83–85 BTCU-1_ORF7 (lysA) and BTCU-1_ORF8 (lysB) are two presumed lytic genes in the genome of mycobacteriophage BTCU-1 that encode two endolysins with antimycobacterial activities.81 These endolysins possess two separate basic functions. The cleavage of the peptidoglycan in the cell wall of mycobacteria is done by lysA. On the other hand, it is presumed that the release of free mycolic acid from the mycolylarabinogalactan bond is carried out by lysB.81,86 Finally, it can be concluded that lysA and lysB can make significant changes in the cell shape of mycobacterium, and these findings recommend that these endolysins are good candidates for treating and controlling mycobacterial infections (Figure 6).
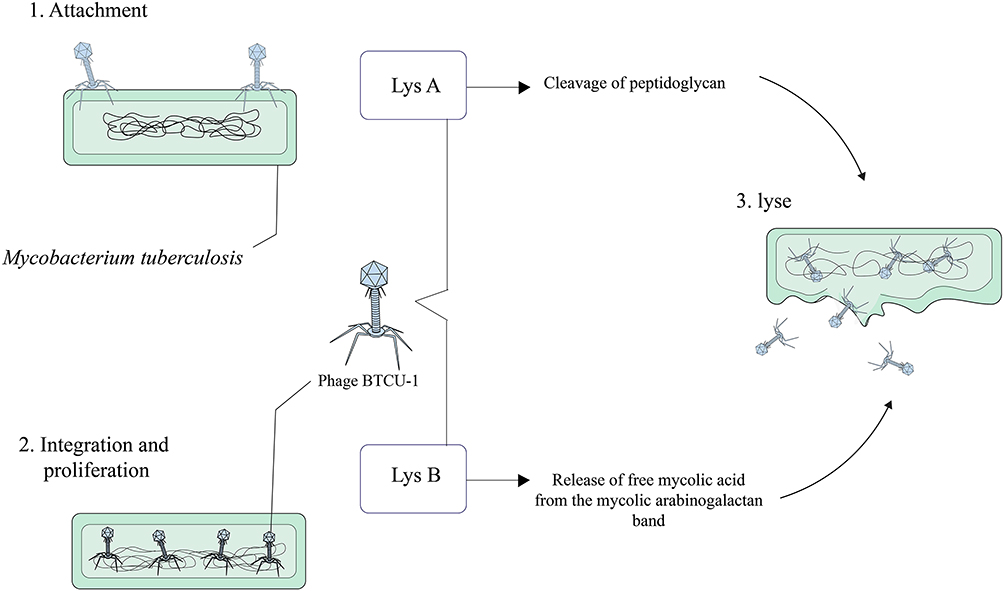 |
Figure 6 Overview of phage BTCU-1 mediated Mycobacterium tuberculosis lysis. |
Phage Bo4
The length of the mycobacteriophage Bo4 genome is approximately 39,318 bp, and this bacteriophage has a dsDNA genome with a G+C content of 66.76%. It is identified that genome of mycobacteriophage Bo4 contains 58 ORFs.87 This mycobacteriophage has a long noncontractile tail with isometric and icosahedral heads.87,88 These features revealed that mycobacteriophage Bo4 could be considered as a lytic phage that infected and possibly eradicated pathogenic mycobacteria in the infected sites. Moreover, this phage can be a valuable tool for phage typing of Mtb.89 Different mycobacterial species including non-tuberculosis mycobacterium (NTM), mycobacterium bovis, and Mtb can be infected by this mycobacteriophages.87 However, mycobacteriophage Bo4 has the capability to infect MDR and XDR Mtb. In vivo conditions, in blood, and in lysosomal macrophages, Bo4 could lyse and effectively halt the growth of Mtb, showing that mycobacteriophage Bo4 has antimicrobial activity.87 Finally, these features make it an ideal candidate and a potentially useful tool for diagnosing and developing phage-based anti-TB therapies (Figure 1).
Phage SWU1
In recent years, a novel mycobacteriophage SWU1 has been isolated from environmental samples, especially from a soil sample in China using M. smegmatis mc2155 as the host microorganism.90 The length of the mycobacteriophage SWU1 genome is approximately 52,474 bp with a G+C content of 62.4%, containing 94 and 3 candidate protein-coding and tRNA genes, respectively.91 SWU1gp39 is a new gene from mycobacteriophage SWU1, which is absent in other mycobacteriophages and the exact function of this gene has not yet been determined.41 Overall, the degree of antibiotic resistance in Mtb is closely related to cell wall permeability. SWU1gp39 can inhibit the lipid metabolism of Mycobacterium and prevent the usual production of long-chain fatty acids.41,92 Therefore, SWU1gp39 could increase the cell wall permeability in Mtb and potentiate the efficiency of multiple antibiotics such as rifampicin, isoniazid, vancomycin, ofloxacin, ciprofloxacin, norfloxacin, ampicillin, and erythromycin.41 Moreover, SWU1gp39 increases the susceptibility of Mtb to various stresses including heat shock, H2O2, SDS, and low PH.41,93 On the other hand, a putative GTPase-activating protein (GAP) is encoded with mycobacteriophage SWU1 A321_gp67.94 The GAP superfamily comprises 6 subfamilies including Ras, Rho, Ran, Rab, Rheb, and ARF.95 These proteins can be involved in several processes including signal transduction, cell differentiation, cell cycle, and protein synthesis through regulating the activity of GTPase.96 Gp67 is a mycobacteriophage SWU1 late-stage gene that could change colony formation and biofilm morphology and may play a role in the reproduction and release of the phage progeny.97 Gp67 can downregulate the transcription of various genes such as MSMEG_0235, MSMEG_6092, MSMEG_1876, and mmpL4b.94 These genes have multiple roles in biofilm formation, cell wall integrity, and development of colony morphology.98 Moreover, gp67 can increase the susceptibility of Mtb against different antibiotics such as streptomycin and capreomycin via several procedures including (a) making changes in cell wall integrity and cell wall structure and (b) preventing and disrupting biofilm formation.94 In total, it can be concluded that SWU1gp39 and gp67 might be utilized as a broad-spectrum antibiotic adjuvant or potentiator and be included into the existing antibiotic regimen for better control and greater efficiency of anti-tuberculosis drugs (including isoniazid and rifampicin) in bacterial killing (Figure 7).
 |
Figure 7 Overview of phage SWU1 mediated Mycobacterium tuberculosis lysis. |
Phage Ms6
Mycobacteriophage Ms6 is a temperate phage with a linear double-stranded DNA that has a lytic cassette composed of five genes.99,100 Similar to the phage D29, in Ms6, Lysin A (a 384 amino acid polypeptide) has a central peptidoglycan recognition protein (PGRP) in a super-family conserved domain and hydrolyzes peptidoglycan of bacterial cell walls.101 Ms6 Gp1 is highly similar to a chaperone-like protein and participates in the transport of LysA to the extracytoplasmic setting.43 In the lysis cassette, Ms6 LysB is localized between lysA and hol genes. The length of Ms6 LysB is 996 bp that encodes a protein with lipolytic activity and possesses a capability to hydrolyze a wide spectrum of fatty acid esters.99–102 Results of several studies revealed that, in M. smegmatis, Ms6 LysB targets the outer membrane, leading to the cleavage of ester bond among arabinogalactan and mycolic acids in the mycolyl-arabinogalactan-peptidoglycan (mAGP) complex.58,101,102 Moreover, in Mycobacterium tuberculosis H37Ra and Mycobacterium bovis BCG, Ms6 LysB leads to the cleavage of the ester bond between trehalose and mycolic acids in trehalose 6,6′-dimycolate (TDM).101 In addition to LysA (gp2) and LysB (gp1 and gp3), in Ms6 lytic cassette, holin is encoded by gp4 (hol).99 In Ms6, holin protein has several roles including controlling the activation of the endolysin and controlling the access of endolysin to murein.43,103 Finally, according to the above-mentioned statements, it can be concluded that mycobacteriophage Ms6 with different endolysins is a worthy candidate in Mtb infection therapy (Figure 8).
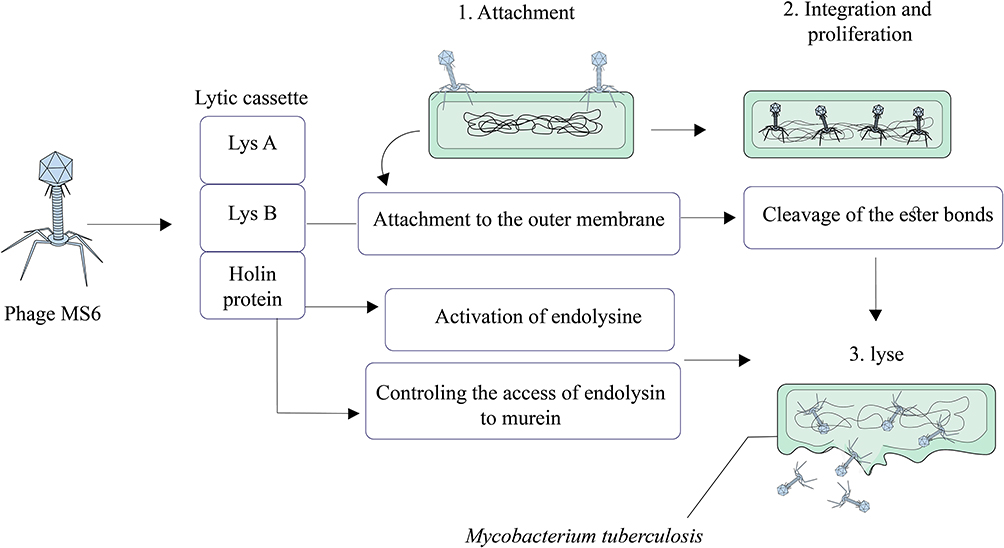 |
Figure 8 Overview of phage MS6 mediated Mycobacterium tuberculosis lysis. |
Phage therapy in mycobacterium avium infections
Mycobacterium avium (M. avium) is the slowest growing intracellular pathogen that replicates and persists within the mononuclear phagocytes.104,105 This pathogen causes the dissemination of infection in immunocompromised patients. The patients with acquired immune deficiency syndrome (AIDS), especially patients with <50 CD4+ T cells/mm3, are susceptible to disseminated infection caused by this organism.106 However, disseminated infections caused by M. avium have been reported to be highly frequency in non-AIDS individuals.48 The application of protease inhibitors in the treatment of human immunodeficiency virus (HIV-1) infection has a substantial effect and contributes to a significant reduction in the occurrence of M. avium bacteremia.107 Nevertheless, when the anti-HIV treatment stops, the incidence of M. avium bacteremia increases.108 M. avium shows resistance to a wide range of antituberculosis antibiotics, and merely a few antibiotics including clarithromycin, azithromycin, and roxithromycin (Macrolides) have shown activities against M. avium in vitro and in vivo.48 On the other hand, the other problem is that M. avium is able to infect and replicate within mononuclear cells including macrophages and monocytes.109 The intracellular growth of organism within macrophages and monocytes justifies the latent phase of infection in the host.48 Therefore, the antimicrobial agents that require a microbial target in active replication are not able to eradicate these infections. Of note, it is important for the new alternative therapies to be evaluated from the viewpoint of the following two facts: 1) clarithromycin, azithromycin, and roxithromycin are used in the prophylactic form for the M. avium infection, and the emergence of resistance in one antibiotic will be equal to resistance to all macrolides and 2) intracellular growth of organism.110 The use of bacteriophage against M. avium infection is a significant alternative and is useful as an antimycobacterial regimen for treating drug-resistant bacteria.109 TM4 is a lytic mycobacteriophage that infects M. avium and does not form persistent lysogens.42,111 TM4 can be delivered by a nontuberculous mycobacterium (Mycobacterium smegmatis) and has the ability to lyse and kill M. avium inside mononuclear cells such as macrophages and monocytes.48 It has been described previously that the treatment of M. avium infected macrophages with TM4 infected M. smegmatis and contributed to a considerable decrease in the number of M. avium bacilli.109 Moreover, the treatment of M. avium infected macrophages with TM4 infected M. smegmatis led to the fusion of vacuole concealing M. smegmatis infected by TM4 with the M. avium vacuole in macrophages.48 However, accordingly, M. avium naturally inhibits phagosome-lysosome fusion in macrophages, and it is predictable that the mycobacterial vacuole still has the capacity to be fused with endosomal.112 Results of another study revealed that the coinfection of macrophage with M. avium and Coxiella burnetti led to the fusion of the two vacuoles and, finally, created new vacuoles that are acidic and contain Coxiella and M. avium.113 Moreover, it is possible for TM4 to reach the M. avium vacuole in different ways, and that when vacuoles are lysed, the bacteria-containing vacuole becomes acidic.48,109 It can be concluded that these findings proposed a new concept to kill intracellular mycobacteria and warrant the upcoming progression. However, it appears that in order to understand the exact role of mycobacteriophage in M. avium infection treatment, further studies are required.
Phage therapy in mycobacterium ulcerans infection
Buruli Ulcer (BU) is the third most common mycobacterial infection in immunocompromised individuals, especially in HIV positive patients.52 This infectious disease is caused by Mycobacterium ulcerans (M. ulcerans) and is a serious and chronic necrotizing skin-infection disease, which is reported to be active in more than 30 countries worldwide with high frequency in West Africa.114,115 Although M. ulcerans is proposed as an extracellular pathogen, evidence shows that this microorganism can be found in macrophages throughout the initial phase of infection.116 M. ulcerans secretes a lipidic exotoxin diagnosed as mycolactone. This lipid toxin was shown to be cytopathic to cultured L929 murine fibroblasts and induced apoptosis in mammalian cells.117,118 Moreover, this exotoxin was characterized by immunosuppressive properties and, finally, led to the typically clinical sign of ulcerative BU skin lesions.115,119 The clinical manifestation of BU is characterized by various forms including preulcerative nodule, papules, plaque, and oedematous lesions; these lesions can tend towards characteristic necrotic ulcerative forms with undermined edges.114,116 According to the data on BU disease, the design and preparation of controlling programs for prevention purposes can be very difficult. Moreover, no vaccine against BU is available so far; however, evidence has shown that vaccination with Bacille Calmette-Guérin (BCG) could provide temporary protection against BU.120 To date, BU is conventionally treated by surgical resection of affected skin followed by grafting, if required.121 However, as of 2004, the World Health Organization (WHO) recommended the combination of antituberculosis drugs rifampicin and streptomycin as standard therapy for the treatment of BU patients.122 Although the application of antituberculotic drugs decreases the relapse rates, this treatment is susceptible to numerous drawbacks: 1) this treatment does not resolve widespread lesions and patients are often left with scars and lifelong disabilities;123 2) muscular injection of streptomycin for a long time requires skilled personnel; 3) the mutations related to rifampicin resistance have already identified an in-vivo experimental condition after monotherapy; 4) the application of these antituberculosis drugs is related to several adverse side effects; 5) the consumption of these drugs may contribute to the deterioriation of the lesion with paradoxical reactions or the emergence of new lesions.124–128 The use of bacteriophages as a diagnostic and treatment method for BU provides several benefits for patients: 1) extracellular microorganisms that prevail in progressive lesions are lysed and eradicated due to the lytic activity of phages; 2) phages can be administered topically in necrotic infection sites for the treatment of ulcerative lesions; and 3) M. ulcerans is naturally found as an extracellular pathogen and, finally, this pathogen might be nearly available by lytic phages.114,129 Among different bacteriophages used for the treatment of bacterial infections, plaques of phage D29 are comparatively large and adsorption of phage particles seems to be efficient, which might be the best choice for the treatment of BU.52 Moreover, the use of phage DS-6A in animal models with disseminated tuberculosis leads to the reduction of lesions in lungs, spleen, and livers.114 Mycobacteriophage D29 is a lytic phage, and the results of experimentally infected animal models demonstrated that a single subcutaneous inoculation of this phage reduced the proliferation of the mycolactone-producing M. ulcerans 1615.52,114 Notably, it is revealed that lytic activity of mycobacteriophage D29 may not be restricted to M. ulcerans 1615, and this phage also shows the lytic activity against numerous other M. ulcerans isolates in vitro.130 One of the main characteristics of mycobacteriophage D29 is that this phage can be detected in several organs including blood and spleen (in post-injection 2 h) after subcutaneous injection.114 Moreover, mycobacteriophage D29 could be found in the draining lymph nodes for longer periods of time (at least 15 days).114,130 Results of a previously published study revealed that the application of mycobacteriophage D29 to the treatment of BU in vivo led to the pathologic reduction and the prevention of ulceration.114 In total, it can be concluded that mycobacteriophage D29 reduces the number of M. ulcerans through several mechanisms: 1) mycobacteriophage D29 induces a cellular infiltrate of a macrophagic/lymphocytic profile; 2) mycobacteriophage D29 has an extensive lytic activity against mycolactone-producing M. ulcerans isolates, especially M. ulcerans 1615; 3) mycobacteriophage D29 increases the levels of TNF, IFN-γ, and IL-10 in vivo; and 4) treatment with mycobacteriophage D29 leads to the increase and maintenance of a local mononuclear inflammatory response to M. ulcerans.114–116,131
Discussion and conclusion
The rapid prevalence of mycobacterial infections and drug-resistance bacteria, especially the emergence of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB), has prompted researchers to find a novel alternative approach to controlling and treating TB. Mycobacteriophages are considered as natural antibacterial agents and are parasites on bacteria. Moreover, mycobacteriophages are extremely specific to their host. Therefore, phage therapy can be considered as a novel candidate for treating and controlling mycobacterial infections. Although phage therapy is a novel therapeutic approach against bacterial infection, especially MDR and XDR bacteria, the clinical use of this approach is susceptible to several limitations as follows: I) the administration of a large dose of phages in patients probably leads to the onset of the immunological response; therefore, this limitation restricts the use of specific phage more than once; II) the rate of clearance of phages in the body is very high; III) typically, the intracellular pathogens do not have access to the phage, and the transport of phages inside intracellular pathogens requires a delivery system such as non-virulent mycobacteria (M. smegmatis) or liposomes. Finally, it is suggested dedicating a greater body of in vivo and in vitro research to demonstrate the exact role and efficiency of phage therapy in the treatment of mycobacterial infection, particularly TB.
Acknowledgment
We greatly appreciate the input from Dr. Melika Khanzadeh Tehrani (from Department of Pathobiology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran) for her collaboration with us in figures design for revising of the manuscript and his helpful comments. Moreover, we would like to thank “School of Public Health, Tehran University of Medical Sciences, Tehran, Iran” for their kind cooperation. The authors received no specific funding for this work.
Author contributions
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data. They played an active role in drafting the article or revising it critically to achieve important intellectual content, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Disclosure
All of the authors declare that there are no commercial, personal, political, and any other potential conflicting interests related to the published manuscript.
References
1. Gondil V, Chhibber S. Exploring potential of phage therapy for tuberculosis using model organism. Biomed Biotechnol Res J. 2018;2(1):9–15. doi:10.4103/bbrj.bbrj_93_17
2. Azimi T, Shariati A, Fallah F, et al. Mycobacterium tuberculosis genotyping using MIRU-VNTR typing. J Mazandaran Univ Med Sci. 2017;27(149):40–48.
3. Azimi T, Nasiri MJ, Zamani S, et al. High genetic diversity among Mycobacterium tuberculosis strains in Tehran, Iran. J Clin Tuberc Other Mycobact Dis. 2018;11:1–6. doi:10.1016/j.jctube.2018.01.001
4. Taghipour A, Azimi T, Javanmard E, et al. Immunocompromised patients with pulmonary tuberculosis; a susceptible group to intestinal parasites. Gastroenterol Hepatol Bed Bench. 2018;11(Suppl 1):S134–S139.
5. WHO. Global Tuberculosis Report 2018. World Health Organization; 2018.
6. Hatfull GF. Mycobacteriophages: windows into tuberculosis. PLoS Pathog. 2014;10(3):e1003953. doi:10.1371/journal.ppat.1003953
7. Wei L, Wu J, Liu H, et al. A mycobacteriophage-derived trehalose-6, 6′-dimycolate-binding peptide containing both antimycobacterial and anti-inflammatory abilities. Faseb J. 2013;27(8):3067–3077. doi:10.1096/fj.13-227454
8. Sharahi JY, Azimi T, Shariati A, Safari H, Tehrani MK, Hashemi A. Advanced strategies for combating bacterial biofilms. J Cell Physiol. 2019;234(9):14689–14708. doi:10.1002/jcp.v234.9
9. Lu TK, Collins JJ. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc National Acad Sci. 2009;106(12):4629–4634. doi:10.1073/pnas.0800442106
10. Ergene C, Yasuhara K, Palermo EF. Biomimetic antimicrobial polymers: recent advances in molecular design. Polym Chem. 2018;9(18):2407–2427. doi:10.1039/C8PY00012C
11. Li W, Tailhades J, O’Brien-Simpson NM, et al. Proline-rich antimicrobial peptides: potential therapeutics against antibiotic-resistant bacteria. Amino Acids. 2014;46(10):2287–2294. doi:10.1007/s00726-014-1820-1
12. Bajpai U, Mehta AK, Eniyan K, et al. Isolation and characterization of bacteriophages from India, with lytic activity against Mycobacterium tuberculosis. Can J Microbiol. 2018;64(7):483–491. doi:10.1139/cjm-2017-0387
13. Guo S, Ao Z. Phage in the diagnosis and treatment of tuberculosis. Front Biosci. 2012;17:2691–2697. doi:10.2741/4080
14. Nasser A, Azizian R, Tabasi M, et al. Specification of bacteriophage isolated against clinical methicillin-resistant staphylococcus aureus. Osong Public Health Res Perspect. 2019;10(1):20–24. doi:10.24171/j.phrp.2019.10.1.05
15. Azizian R, Nasser A, Askari H, et al. Sewage as a rich source of phage study against Pseudomonas aeruginosa PAO. Biologicals. 2015;43(4):238–241. doi:10.1016/j.biologicals.2015.05.004
16. Reza A, Farid AJ, Zamberi S, et al. Dynamics of bacteriophages as a promising antibiofilm agents. J Pure Appl Microbiol. 2014;8(2):1015–1019.
17. Nasser A, azizi J, azizian R, askari H, pakzad I. The use of mrsa phage as a super specialized antibiotic against lethal methicillin-resistant staphylococcus aureus. J Ilam Univ Med Sci. 2014;22:171–179.
18. Suttle CA. Marine viruses—major players in the global ecosystem. Nat Rev Microbiol. 2007;5(10):801–812. doi:10.1038/nrmicro1750
19. Summers WC. The strange history of phage therapy. Bacteriophage. 2012;2(2):130–133. doi:10.4161/bact.20757
20. Sulakvelidze A, Alavidze Z, Morris JG. Bacteriophage therapy. Antimicrob Agents Chemother. 2001;45(3):649–659. doi:10.1128/AAC.45.3.649-659.2001
21. Principi N, Silvestri E, Esposito S. Advantages and limitations of bacteriophages for the treatment of bacterial infections. Front Pharmacol. 2019;10:513. doi:10.3389/fphar.2019.00848
22. Dublanchet A, Patey O, Mazure H, Liddle M, Smithyman AM. Indications and Limitations of Phage Therapy in Human Medicine: Personal Experience and Literature Review. Reprints. 2018.
23. Torres-Barceló C. Phage therapy faces evolutionary challenges. Viruses. 2018;10(6):323–330. doi:10.3390/v10060323
24. Kakasis A, Panitsa G. Bacteriophage therapy as an alternative treatment for human infections. A comprehensive review. Int J Antimicrob Agents. 2018;53(1):16–21. doi:10.1016/j.ijantimicag.2018.09.004
25. Bogovazova G, Voroshilova N, Bondarenko V. The efficacy of Klebsiella pneumoniae bacteriophage in the therapy of experimental Klebsiella infection. Zh Mikrobiol Epidemiol Immunobiol. 1991;68(4):5–8.
26. Del Pozo J, Alonso M, Arciola C, et al. Biotechnological war against biofilms. Could phages mean the end of device-related infections? Int J Artif Organs. 2007;30(9):805–812.
27. Vitiello CL, Merril CR, Adhya S. An amino acid substitution in a capsid protein enhances phage survival in mouse circulatory system more than a 1000-fold. Virus Res. 2005;114(1–2):101–103. doi:10.1016/j.virusres.2005.05.014
28. Mattila S, Ruotsalainen P, Jalasvuori M. On-demand isolation of bacteriophages against drug-resistant bacteria for personalized phage therapy. Front Microbiol. 2015;6:1271. doi:10.3389/fmicb.2015.01271
29. Seed KD. Battling phages: how bacteria defend against viral attack. PLoS Pathog. 2015;11(6):e1004847. doi:10.1371/journal.ppat.1004847
30. Vandenheuvel D, Lavigne R, Brüssow H. Bacteriophage therapy: advances in formulation strategies and human clinical trials. Annu Rev Virol. 2015;2:599–618. doi:10.1146/annurev-virology-100114-054915
31. Brabban A, Hite E, Callaway T. Evolution of foodborne pathogens via temperate bacteriophage-mediated gene transfer. Foodborne Pathog Dis. 2005;2(4):287–303. doi:10.1089/fpd.2005.2.287
32. Maiques E, Úbeda C, Tormo MÁ, et al. Role of staphylococcal phage and SaPI integrase in intra-and interspecies SaPI transfer. J Bacteriol. 2007;189(15):5608–5616. doi:10.1128/JB.00619-07
33. Kaźmierczak Z, Piotrowicz A, Owczarek B, et al. Molecular imaging of T4 phage in mammalian tissues and cells. Bacteriophage. 2014;4(2):e28364. doi:10.4161/bact.28364
34. Dąbrowska K, Miernikiewicz P, Piotrowicz A, et al. Immunogenicity studies of proteins forming the T4 phage head surface. J Virol. 2014;88(21):12551–12557. doi:10.1128/JVI.02043-14
35. Rhoads D, Wolcott R, Kuskowski M, Wolcott B, Ward L, Sulakvelidze A. Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J Wound Care. 2009;18(6):237–243. doi:10.12968/jowc.2009.18.6.42801
36. Biswas B, Adhya S, Washart P, et al. Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect Immun. 2002;70(1):204–210. doi:10.1128/iai.70.1.204-210.2002
37. Kaur S, Harjai K, Chhibber S. Bacteriophage-aided intracellular killing of engulfed methicillin-resistant Staphylococcus aureus (MRSA) by murine macrophages. Appl Microbiol Biotechnol. 2014;98(10):4653–4661. doi:10.1007/s00253-014-5643-5
38. Gondil VS, Chhibber S. Evading antibody mediated inactivation of bacteriophages using delivery systems. J Virol Curr Res. 2017;1:555–574.
39. Grover N, Paskaleva EE, Mehta KK, Dordick JS, Kane RS. Growth inhibition of Mycobacterium smegmatis by mycobacteriophage-derived enzymes. Enzyme Microb Technol. 2014;63:1–6. doi:10.1016/j.enzmictec.2014.04.018
40. Morello E, Saussereau E, Maura D, Huerre M, Touqui L, Debarbieux L. Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: first steps towards treatment and prevention. PLoS One. 2011;6(2):e16963. doi:10.1371/journal.pone.0016963
41. Li Q, Zhou M, Fan X, Yan J, Li W, Xie J. Mycobacteriophage SWU1 gp39 can potentiate multiple antibiotics against Mycobacterium via altering the cell wall permeability. Sci Rep. 2016;6:28701. doi:10.1038/srep28701
42. Nieth A, Verseux C, Barnert S, Süss R, Römer W. A first step toward liposome-mediated intracellular bacteriophage therapy. Expert Opin Drug Deliv. 2015;12(9):1411–1424. doi:10.1517/17425247.2015.1043125
43. Catalão MJ, Gil F, Moniz‐Pereira J, Pimentel M. The mycobacteriophage Ms6 encodes a chaperone‐like protein involved in the endolysin delivery to the peptidoglycan. Mol Microbiol. 2010;77(3):672–686. doi:10.1111/j.1365-2958.2010.07239.x
44. McNerney R, Traore H. Mycobacteriophage and their application to disease control. J Appl Microbiol. 2005;99(2):223–233. doi:10.1111/jam.2005.99.issue-2
45. Mayer O, Jain P, Weisbrod TR, et al. Fluorescent reporter DS6A mycobacteriophages reveal unique variations in infectibility and phage production in mycobacteria. J Bacteriol. 2016;198(23):3220–3232. doi:10.1128/JB.00592-16
46. Sula L, Sulova J, Stolcpartova M. Therapy of experimental tuberculosis in guinea pigs with mycobacterial phages DS-6A, GR-21 T, My-327. Czech Med. 1981;4(4):209–214.
47. Ford M, Stenstrom C, Hendrix R, Hatfull G. Mycobacteriophage TM4: genome structure and gene expression. Tuber Lung Dis. 1998;79(2):63–73. doi:10.1054/tuld.1998.0007
48. Broxmeyer L, Sosnowska D, Miltner E, et al. Killing of Mycobacterium avium and Mycobacterium tuberculosis by a mycobacteriophage delivered by a nonvirulent mycobacterium: a model for phage therapy of intracellular bacterial pathogens. J Infect Dis. 2002;186(8):1155–1160. doi:10.1086/343812
49. Ranjan A, Vidyarthi AS, Poddar R. Evaluation of codon bias perspectives in phage therapy of Mycobacterium tuberculosis by multivariate analysis. In Silico Biol. 2007;7(4,5):423–431.
50. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi:10.1016/j.addr.2012.09.037
51. Un K, Sakai-Kato K, Oshima Y, Kawanishi T, Okuda H. Intracellular trafficking mechanism, from intracellular uptake to extracellular efflux, for phospholipid/cholesterol liposomes. Biomaterials. 2012;33(32):8131–8141. doi:10.1016/j.biomaterials.2012.07.030
52. Rybniker J, Kramme S, Small PL. Host range of 14 mycobacteriophages in Mycobacterium ulcerans and seven other mycobacteria including Mycobacterium tuberculosis–application for identification and susceptibility testing. J Med Microbiol. 2006;55(1):37–42. doi:10.1099/jmm.0.46238-0
53. Joshi H, Seniya SP, Suryanarayanan V, Patidar ND, Singh SK, Jain V. Dissecting the structure–function relationship in lysozyme domain of mycobacteriophage D29‐encoded peptidoglycan hydrolase. FEBS Lett. 2017;591(20):3276–3287. doi:10.1002/1873-3468.12848
54. Peng L, Luo Y, Chen B, et al. Therapeutic effect of bacteriophage D29 in the treatment for guinea pigs infected with sensitive strain of Mycobacterium tuberculosis. Chin J Zoonoses. 2009;25(8):733–736.
55. Pohane AA, Joshi H, Jain V. Molecular dissection of phage endolysin: an interdomain interaction confers host specificity in Lysin A of Mycobacterium phage D29. J Biol Chem. 2014;289(17):12085–12095. doi:10.1074/jbc.M113.529594
56. Hassan S, Dusthackeer A, Subramanyam B, Ponnuraja C, Sivaramakrishnan GN, Kumar V. Lytic efficiency of mycobacteriophages. Open Syst Biol J. 2010;3(1):21–28. doi:10.2174/1876392801003010021
57. Kamilla S, Jain V. Mycobacteriophage D29 holin C‐terminal region functionally assists in holin aggregation and bacterial cell death. Febs J. 2016;283(1):173–190. doi:10.1111/febs.13565
58. Payne K, Sun Q, Sacchettini J, Hatfull GF. Mycobacteriophage Lysin B is a novel mycolylarabinogalactan esterase. Mol Microbiol. 2009;73(3):367–381. doi:10.1111/j.1365-2958.2009.06775.x
59. Catalao MJ, Gil F, Moniz-Pereira J, Sao-Jose C, Pimentel M. Diversity in bacterial lysis systems: bacteriophages show the way. FEMS Microbiol Rev. 2013;37(4):554–571. doi:10.1111/1574-6976.12006
60. Deng X, Wang L, You X, Dai P, Zeng Y. Advances in the T7 phage display system (Review). Mol Med Rep. 2018;17(1):714–720. doi:10.3892/mmr.2017.7994
61. Wisuthiphaet N, Yang X, Young GM, Nitin N. Rapid detection of Escherichia coli in beverages using genetically engineered bacteriophage T7. AMB Express. 2019;9(1):55. doi:10.1186/s13568-019-0776-7
62. Chan LY, Kosuri S, Endy D. Refactoring bacteriophage T7. Mol Syst Biol. 2005;1:
63. Sipley J, Stassi D, Dunn J, Goldman E. Analysis of bacteriophage T7 gene 10A and frameshifted 10B proteins. Gene Expression J Liver Res. 1991;1(2):127–136.
64. Du Plessis J, Cloete R, Burchell L, et al. Exploring the potential of T7 bacteriophage protein Gp2 as a novel inhibitor of mycobacterial RNA polymerase. Tuberculosis. 2017;106:82–90. doi:10.1016/j.tube.2017.07.004
65. Sheppard C, Cámara B, Shadrin A, et al. Inhibition of Escherichia coli RNAp by T7 Gp2 protein: role of negatively charged strip of amino acid residues in Gp2. J Mol Biol. 2011;407(5):623–632. doi:10.1016/j.jmb.2011.02.013
66. Mekler V, Minakhin L, Sheppard C, Wigneshweraraj S, Severinov K. Molecular mechanism of transcription inhibition by phage T7 gp2 protein. J Mol Biol. 2011;413(5):1016–1027. doi:10.1016/j.jmb.2011.09.029
67. Shadrin A, Sheppard C, Severinov K, Matthews S, Wigneshweraraj S. Substitutions in the Escherichia coli RNA polymerase inhibitor T7 Gp2 that allow inhibition of transcription when the primary interaction interface between Gp2 and RNA polymerase becomes compromised. Microbiology. 2012;158(Pt 11):2753–2764. doi:10.1099/mic.0.062547-0
68. Shadrin A, Sheppard C, Savalia D, Severinov K, Wigneshweraraj S. Overexpression of Escherichia coli udk mimics the absence of T7 Gp2 function and thereby abrogates successful infection by T7 phage. Microbiology. 2013;159(Pt 2):269–274. doi:10.1099/mic.0.064527-0
69. McClure WR, Cech CL. On the mechanism of rifampicin inhibition of RNA synthesis. J Bio Chem. 1978;253(24):8949–8956.
70. Boudvillain M, Nollmann M, Margeat E. Keeping up to speed with the transcription termination factor Rho motor. Transcription. 2010;1(2):70–75. doi:10.4161/trns.1.2.12232
71. Banerjee S, Chalissery J, Bandey I, Sen R. Rho-dependent transcription termination: more questions than answers. J Microbiol. 2006;44(1):11–22.
72. Peters JM, Vangeloff AD, Landick R. Bacterial transcription terminators: the RNA 3′-end chronicles. J Mol Biol. 2011;412(5):793–813. doi:10.1016/j.jmb.2011.03.036
73. Ray-Soni A, Bellecourt MJ, Landick R. Mechanisms of bacterial transcription termination: all good things must end. Annu Rev Biochem. 2016;85:319–347. doi:10.1146/annurev-biochem-060815-014844
74. Kalyani BS, Muteeb G, Qayyum MZ, Sen R. Interaction with the nascent RNA is a prerequisite for the recruitment of Rho to the transcription elongation complex in vitro. J Mol Biol. 2011;413(3):548–560. doi:10.1016/j.jmb.2011.08.053
75. Grylak-Mielnicka A, Bidnenko V, Bardowski JK, Bidnenko E. Transcription termination factor Rho: a hub linking diverse physiological processes in bacteria. Microbiology. 2016;162(3):433–447. doi:10.1099/mic.0.000244
76. Ghosh G, Reddy J, Sambhare S, Sen R. A bacteriophage capsid protein is an inhibitor of a conserved transcription terminator of various bacterial pathogens. J Bacteriol. 2018;200(1):e00380–e00317. doi:10.1128/JB.00380-17
77. Botella L, Vaubourgeix J, Livny J, Schnappinger D. Depleting Mycobacterium tuberculosis of the transcription termination factor Rho causes pervasive transcription and rapid death. Nat Commun. 2017;8:14731. doi:10.1038/ncomms14731
78. Pani B, Banerjee S, Chalissery J, et al. Mechanism of inhibition of Rho-dependent transcription termination by bacteriophage P4 protein Psu. J Biol Chem. 2006;281(36):26491–26500. doi:10.1074/jbc.M603982200
79. Smith KC, Castro-Nallar E, Fisher JN, Breakwell DP, Grose JH, Burnett SH. Phage cluster relationships identified through single gene analysis. BMC Genomics. 2013;14(1):410. doi:10.1186/1471-2164-14-181
80. Li L, Lee E, Shaw N. Expression, purification and crystallization of phosphoribosyl transferase from a mycobacteriophage. Acta Crystallogr F Struct Biol Commun. 2018;74(3):161–165.
81. Lai M-J, Liu -C-C, Jiang S-J, et al. Antimycobacterial activities of endolysins derived from a mycobacteriophage, BTCU-1. Molecules. 2015;20(10):19277–19290. doi:10.3390/molecules201019277
82. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi:10.1093/nar/28.1.235
83. Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2010;54(4):1603–1612. doi:10.1128/AAC.01625-09
84. Hermoso JA, García JL, García P. Taking aim on bacterial pathogens: from phage therapy to enzybiotics. Curr Opin Microbiol. 2007;10(5):461–472. doi:10.1016/j.mib.2007.08.002
85. Borysowski J, Weber-Dąbrowska B, Górski A. Bacteriophage endolysins as a novel class of antibacterial agents. Exp Biol Med. 2006;231(4):366–377. doi:10.1177/153537020623100402
86. Payne KM, Hatfull GF. Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS One. 2012;7(3):e34052.
87. Gan Y, Wu T, Liu P, Guo S. Characterization and classification of Bo4 as a cluster G mycobacteriophage that can infect and lyse M. tuberculosis. Arch Microbiol. 2014;196(3):209–218. doi:10.1007/s00203-014-0954-6
88. W T. Preliminary Research about Mycobacteriophage Cocktail Therapy against Drug-resistance Tuberculosis. Chongqing, China: Chongqing Medical University; 2012.
89. Jr WD J, Good RC, Thompson NJ, Kelly GD. Bacteriophage types of Mycobacterium tuberculosis in the United States. Am Rev Respir Dis. 1982;125(6):740–745. doi:10.1164/arrd.1982.125.6.740
90. Fan X, Teng T, Wang H, Xie J. Biology of a novel mycobacteriophage, SWU1, isolated from Chinese soil as revealed by genomic characteristics. Am Soc Microbiol. 2012;86(18):10230–10231.
91. Hatfull GF, Jacobs-Sera D, Lawrence JG, et al. Comparative genomic analysis of 60 mycobacteriophage genomes: genome clustering, gene acquisition, and gene size. J Mol Biol. 2010;397(1):119–143. doi:10.1016/j.jmb.2010.01.011
92. Molshanski-Mor S, Yosef I, Kiro R, et al. Revealing bacterial targets of growth inhibitors encoded by bacteriophage T7. Proc National Acad Sci. 2014;111(52):18715–18720. doi:10.1073/pnas.1413271112
93. Banerjee A, Dubnau E, Quemard A, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263(5144):227–230. doi:10.1126/science.8284673
94. Yan S, Xu M, Duan X, et al. Mycobacteriophage putative GTPase-activating protein can potentiate antibiotics. Appl Microbiol Biotechnol. 2016;100(18):8169–8177. doi:10.1007/s00253-016-7681-7
95. Johnson DS, Chen YH. Ras family of small GTPases in immunity and inflammation. Curr Opin Pharmacol. 2012;12(4):458–463.
96. Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. nature. 1991;349(6305):117–127. doi:10.1038/349117a0
97. Merril CR, Scholl D, Adhya SL. The prospect for bacteriophage therapy in Western medicine. Nat Rev Drug Discov. 2003;2(6):489–497. doi:10.1038/nrd1111
98. Pang JM, Layre E, Sweet L, et al. The polyketide Pks1 contributes to biofilm formation in Mycobacterium tuberculosis. J Bacteriol. 2012;194(3):715–721. doi:10.1128/JB.06304-11
99. Catalão MJ, Milho C, Gil F, Moniz-Pereira J, Pimentel M. A second endolysin gene is fully embedded in-frame with the lysA gene of mycobacteriophage Ms6. PLoS One. 2011;6(6):e20515. doi:10.1371/journal.pone.0020515
100. Catalão MJ, Gil F, Moniz-Pereira J, Pimentel M. Functional analysis of the holin-like proteins of mycobacteriophage Ms6. J Bacteriol. 2011;193(11):2793–2803. doi:10.1128/JB.01519-10
101. Gil F, Grzegorzewicz AE, Catalao MJ, Vital J, McNeil MR, Pimentel M. Mycobacteriophage Ms6 LysB specifically targets the outer membrane of Mycobacterium smegmatis. Microbiology. 2010;156(Pt 5):1497–1504. doi:10.1099/mic.0.032821-0
102. Gil F, Catalao MJ, Moniz-Pereira J, Leandro P, McNeil M, Pimentel M. The lytic cassette of mycobacteriophage Ms6 encodes an enzyme with lipolytic activity. Microbiology. 2008;154(5):1364–1371. doi:10.1099/mic.0.2007/014621-0
103. Waldor MK, Friedman DI, Adhya SL. Phages: their role in bacterial pathogenesis and biotechnology. ASM Press. 2005;59(9):1003–1004.
104. Timms VJ, Gehringer MM, Mitchell HM, Daskalopoulos G, Neilan BA. How accurately can we detect Mycobacterium avium subsp. paratuberculosis infection? J Microbiol Methods. 2011;85(1):1–8. doi:10.1016/j.mimet.2011.01.026
105. Basra S, Anany H, Brovko L, Kropinski AM, Griffiths MW. Isolation and characterization of a novel bacteriophage against Mycobacteriumavium subspecies paratuberculosis. Arch Virol. 2014;159(10):2659–2674. doi:10.1007/s00705-014-2122-3
106. Busatto C, Vianna JS, Da Silva LVJ, Ramis IB, Da Silva PEA. Mycobacterium avium: an overview. Tuberculosis (Edinb). 2019;114:127–134. doi:10.1016/j.tube.2018.12.004
107. Jr FJ P, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N Engl J Med. 1998;338(13):853–860. doi:10.1056/NEJM199803263381301
108. Kaplan JE, Hanson D, Dworkin MS, et al. Epidemiology of human immunodeficiency virus-associated opportunistic infections in the United States in the era of highly active antiretroviral therapy. Clin Infect Dis. 2000;30(Supplement_1):S5–S14. doi:10.1086/313843
109. Danelishvili L, Young LS, Bermudez LE. In vivo efficacy of phage therapy for Mycobacterium avium infection as delivered by a nonvirulent mycobacterium. Microb Drug Resist. 2006;12(1):1–6. doi:10.1089/mdr.2006.12.1
110. van der Paardt A-F, Wilffert B, Akkerman OW, et al. Evaluation of macrolides for possible use against multidrug-resistant Mycobacterium tuberculosis. Eur Respir J. 2015;46(2):444–455. doi:10.1183/09031936.00147014
111. Foley-Thomas EM, Whipple DL, Bermudez LE, Barletta RG. Phage infection, transfection and transformation of Mycobacterium avium complex and Mycobacterium paratuberculosis. Microbiology. 1995;141(5):1173–1181. doi:10.1099/13500872-141-5-1173
112. Pradhan G, Shrivastva R, Mukhopadhyay S. Mycobacterial PknG Targets the Rab7l1 signaling pathway to inhibit phagosome–lysosome fusion. J Immunol. 2018;201(5):1421–1433. doi:10.4049/jimmunol.1800530
113. Gomes MS, Paul S, Moreira AL, Appelberg R, Rabinovitch M, Kaplan G. Survival of Mycobacterium avium andMycobacterium tuberculosis in acidified vacuoles of murine macrophages. Infect Immun. 1999;67(7):3199–3206.
114. Trigo G, Martins TG, Fraga AG, et al. Phage therapy is effective against infection by Mycobacterium ulcerans in a murine footpad model. PLoS Negl Trop Dis. 2013;7(4):e2183. doi:10.1371/journal.pntd.0002183
115. Sarfo FS, Phillips R, Wansbrough‐Jones M, Simmonds RE. Recent advances: role of mycolactone in the pathogenesis and monitoring of Mycobacterium ulcerans infection/Buruli ulcer disease. Cell Microbiol. 2016;18(1):17–29. doi:10.1111/cmi.12547
116. Bolz M, Kerber S, Zimmer G, Pluschke G. Use of recombinant virus replicon particles for vaccination against Mycobacterium ulcerans disease. PLoS Negl Trop Dis. 2015;9(8):e0004011. doi:10.1371/journal.pntd.0004011
117. Torrado E, Fraga AG, Logarinho E, et al. IFN-γ–dependent activation of macrophages during experimental infections by Mycobacterium ulcerans is impaired by the toxin mycolactone. J Immunol. 2010;184(2):947–955. doi:10.4049/jimmunol.0902717
118. Torrado E, Adusumilli S, Fraga AG, Small PL, Castro AG, Pedrosa J. Mycolactone-mediated inhibition of tumor necrosis factor production by macrophages infected with Mycobacterium ulcerans has implications for the control of infection. Infect Immun. 2007;75(8):3979–3988. doi:10.1128/IAI.00290-07
119. George KM, Chatterjee D, Gunawardana G, et al. Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science. 1999;283(5403):854–857. doi:10.1126/science.283.5403.854
120. Einarsdottir T, Huygen K. Buruli ulcer. Hum Vaccin. 2011;7(11):1198–1203. doi:10.4161/hv.7.11.17751
121. Yeboah‐Manu D, Peduzzi E, Mensah‐Quainoo E, et al. Systemic suppression of interferon‐γ responses in Buruli ulcer patients resolves after surgical excision of the lesions caused by the extracellular pathogen Mycobacterium ulcerans. J Leukoc Biol. 2006;79(6):1150–1156. doi:10.1189/jlb.1005581
122. WHO, Initiative GBU. Provisional Guidance on the Role of Specific Antibiotics in the Management of Mycobacterium Ulcerans Disease (buruli Ulcer). Geneva: World Health Organization; 2004.
123. Kibadi K, Boelaert M, Fraga AG, et al. Response to treatment in a prospective cohort of patients with large ulcerated lesions suspected to be Buruli Ulcer (Mycobacterium ulcerans disease). PLoS Negl Trop Dis. 2010;4(7):e736. doi:10.1371/journal.pntd.0000736
124. Nienhuis WA, Stienstra Y, Thompson WA, et al. Antimicrobial treatment for early, limited Mycobacterium ulcerans infection: a randomised controlled trial. Lancet. 2010;375(9715):664–672. doi:10.1016/S0140-6736(09)61962-0
125. Sarfo FS, Phillips R, Asiedu K, et al. Clinical efficacy of combination of rifampin and streptomycin for treatment of Mycobacterium ulcerans disease. Antimicrob Agents Chemother. 2010;54(9):3678–3685. doi:10.1128/AAC.00299-10
126. Ruf M-T, Chauty A, Adeye A, et al. Secondary Buruli ulcer skin lesions emerging several months after completion of chemotherapy: paradoxical reaction or evidence for immune protection? PLoS Negl Trop Dis. 2011;5(8):e1252. doi:10.1371/journal.pntd.0001370
127. Sopoh GE, Dossou AD, Brun LV, et al. Severe multifocal form of Buruli ulcer after streptomycin and rifampin treatment: comments on possible dissemination mechanisms. Am J Trop Med Hyg. 2010;83(2):307–313. doi:10.4269/ajtmh.2010.09-0617
128. Gordon CL, Buntine JA, Hayman JA, et al. All-oral antibiotic treatment for buruli ulcer: a report of four patients. PLoS Negl Trop Dis. 2010;4(11):e770. doi:10.1371/journal.pntd.0000770
129. Jikia D, Chkhaidze N, Imedashvili E, et al. The use of a novel biodegradable preparation capable of the sustained release of bacteriophages and ciprofloxacin, in the complex treatment of multidrug‐resistant Staphylococcus aureus‐infected local radiation injuries caused by exposure to Sr90. Clin Exp Dermatol. 2005;30(1):23–26. doi:10.1111/j.1365-2230.2004.01600.x
130. Gorski A. Bacteriophage penetration in vertebrates. J Appl Microbiol. 2005;98(1):7–13. doi:10.1111/j.1365-2672.2004.02422.x
131. Mve-Obiang A, Lee RE, Portaels F, Small P. Heterogeneity of mycolactones produced by clinical isolates of Mycobacterium ulcerans: implications for virulence. Infect Immun. 2003;71(2):774–783. doi:10.1128/iai.71.2.774-783.2003
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.