Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
pH-sensitive poly(lactide-co-glycolide) nanoparticle composite microcapsules for oral delivery of insulin
Authors Sun S ![]() , Liang N, Yamamoto H, Kawashima Y, Cui F, Yan P
, Liang N, Yamamoto H, Kawashima Y, Cui F, Yan P
Received 28 January 2015
Accepted for publication 20 March 2015
Published 11 May 2015 Volume 2015:10(1) Pages 3489—3498
DOI https://doi.org/10.2147/IJN.S81715
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Shaoping Sun,1 Na Liang,2 Hiromitsu Yamamoto,3 Yoshiaki Kawashima,3 Fude Cui,4 Pengfei Yan1,5
1Key Laboratory of Chemical Engineering Process and Technology for High-efficiency Conversion, College of Heilongjiang Province (Heilongjiang University), School of Chemistry and Material Science, Heilongjiang University, Harbin, People’s Republic of China; 2College of Chemistry and Chemical Engineering, Harbin Normal University, Harbin, People’s Republic of China; 3School of Pharmaceutical Science, Aichi Gakuin University, Nissin, Japan; 4School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, People’s Republic of China; 5Key Laboratory of Functional Inorganic Material Chemistry, Heilongjiang University, Harbin, People’s Republic of China
Abstract: This study proposes a new concept of pH-sensitive poly(lactide-co-glycolide) (PLGA) nanoparticle composite microcapsules for oral delivery of insulin. Firstly, insulin–sodium oleate complex was prepared by the hydrophobic ion pairing method and then encapsulated into PLGA nanoparticles by the emulsion solvent diffusion method. In order to reduce the burst release of insulin from PLGA nanoparticles and deliver insulin to specific gastrointestinal regions, hence to enhance bioavailability of insulin, the PLGA nanoparticles were further encapsulated into Eudragit® FS 30D to prepare PLGA nanoparticle composite microcapsules by organic spray-drying method. The preparation was evaluated in vitro and in vivo, and the absorption mechanism was discussed. The in vitro drug release studies revealed that the drug release was pH dependent, and the in vivo results demonstrated that the formulation of PLGA nanoparticle composite microcapsules was an effective candidate for oral insulin delivery.
Keywords: sodium oleate, complex, PLGA nanoparticles, Eudragit® FS 30D
Introduction
Insulin is a 51 amino acid peptide discovered in 1921 by Banting and Best, together with Macleod and Collip. Excreted by pancreatic β cells, insulin has the ability to control the glucose level in blood by facilitating the uptake of glucose. Despite the significant developments in insulin therapy over the past few decades, subcutaneous injection of insulin remains the preferred approach for the treatment of insulin-requiring diabetic patients. Nevertheless, injection of insulin is painful and inconvenient, resulting in poor patient compliance.1,2 In addition, it was reported that insulin administered via subcutaneous route is delivered to the peripheral circulation, so it cannot mimic the glucose homeostasis observed in normal subjects.3,4 Orally delivered insulin undergoes a hepatic bypass before entering the circulation, so it has the potential to mimic the effects of pancreas-secreted insulin in terms of inhibiting hepatic gluconeogenesis and hepatic glucose output.5–8
Many different strategies have been attempted to develop a biologically active oral insulin formulation. It is generally acknowledged that degradation of protein drug by the harsh conditions of the gastrointestinal (GI) tract, including gastric medium and digestive enzymes, is a major obstacle.9–11 In addition, the intestinal epithelium is another major obstacle to the absorption of hydrophilic macromolecules, as it is difficult for insulin to cross the lipid bilayer cell membrane.12–14
In order to overcome these obstacles mentioned and to enhance the oral bioavailability of insulin, biodegradable polymeric nanoparticles (NPs) such as poly(lactic acid), poly(glycolic acid), and their copolymers poly(lactide-co-glycolide) (PLGA) NPs have been widely studied by our group.15,16
In this study, during encapsulation of hydrophilic insulin into the hydrophobic PLGA copolymer, the hydrophobic ion pairing (HIP) technique was applied to enhance the hydrophobicity of insulin to enhance the entrapment efficiency.16 This technique is simply performed by the interaction between ionic protein and the opposite ionic head group of fatty acid, surface-active agents, or other amphiphilic molecules at suitable pH.17,18 Sodium oleate (S.O) was chosen as the counterpart of insulin to prepare insulin–S.O complex (Ins–S.O-Comp). A serious problem of burst release occurred when Ins–S.O-Comp was encapsulated into PLGA NPs, and this led to the low bioavailability of insulin.16 In order to suppress the burst release of insulin from NPs and control the release of insulin in the GI tract, a novel formulation strategy was adopted where the Ins–S.O-Comp PLGA NPs were encapsulated into the enteric material Eudragit® FS 30D using the organic spray-drying method to prepare Ins–S.O-Comp PLGA NP composite microcapsules. Eudragit® FS 30D is insoluble in acidic media but can be dissolved by salt formation above pH 7.0,19 so the release of insulin from the composite microcapsules can be controlled by both the PLGA NPs and the microcapsules. It was assumed that the resultant composite microcapsule system was intact in the stomach and gradually dissolved in the small intestine and then completely dissolved in the colon. The composite microcapsule system was further studied in diabetic rats following oral administration, and the absorption mechanism was discussed.
Materials and methods
Materials and animals
Porcine insulin (28.8 IU/mg) and streptozotocin (STZ) were purchased from Sigma-Aldrich (St Louis, MO, USA). Polyvinyl alcohol (PVA-403) was kindly supplied by Kuraray Co., Ltd. (Osaka, Japan). S.O and PLGA (75/25, molecular weight of 20 kDa) were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Eudragit® FS 30D was obtained from Rohm Pharma Polymers, Degussa (Darmstadt, Germany). Water was purified and deionized using a Milli-Q system (EMD Millipore, Billerica, MA, USA). All other reagents were of analytical grade.
Male Wistar rats, weighing 200±20 g, 12–13 weeks old, were provided by Aichi Gakuin University. The study protocol was reviewed and approved by the Institutional Animal Care and Use Committee, Aichi Gakuin University, Japan.
Rats were induced into diabetes by injection of STZ (65 mg/kg) in a 10 mM citrate buffer at pH 4.5, as previously described,20 and were considered diabetic when glycemia was higher than 16.67 mmol/dL 7 days after STZ treatment.
Preparation of Ins–S.O-Comp loaded PLGA NPs
Ins–S.O-Comp was prepared by the HIP method.16 Briefly, 1.4 mL of S.O (1 mg/mL) solution was slowly added into 4 mL of 1.0 mg/mL insulin solution in a dropwise manner under magnetic stirring. Then the cloudy mixture was centrifuged at 14,000 rpm for 15 minutes to get the complex. The resultant complex was washed three times with distilled water and lyophilized into powder. Total insulin content in the Ins–S.O-Comp was measured by high-performance liquid chromatography (HPLC). The HPLC mobile phase for insulin determination in this paper was composed of 0.1 mol/L NaH2PO4 aqueous solution, 0.05 mol/L Na2SO4 aqueous solution, and acetonitrile at a ratio of 35:35:30, and the final pH was adjusted to 3.0 with H3PO4 solution. The detector wavelength, column temperature, and flow rate of the mobile phase were set at 214 nm, 35°C and 1 mL/minute, respectively. The injection volume of the test samples was 20 μL. Linear calibration curve (area =18006× concentration −23069) was obtained in the concentration range of 5 and 100 μg/mL with correlation coefficient (r) of 0.9999. All insulin concentrations were calculated by interpolation from the standard curve.
The Ins–S.O-Comp PLGA NPs were prepared using the emulsion solvent diffusion method. More specifically, 100 mg of PLGA polymer and 5 mg of insulin-equivalent Ins–S.O-Comp were dissolved in the organic phase of acetone. The oil phase was then added at a constant flow rate of 2 mL/min to 100 mL of stirred (400 rpm) aqueous solution containing 1% of polyvinyl alcohol. The PLGA NP suspension was centrifuged (20,000 rpm, 4°C), washed three times with distilled water, and lyophilized.
Preparation of Ins–S.O-Comp PLGA NP composite microcapsules
Eudragit® FS 30D is the aqueous dispersion of an anionic copolymer based on methyl acrylate, methyl methacrylate, and methacrylic acid (7:3:1). In order to prepare Ins–S.O-Comp PLGA NP composite microcapsules, Eudragit® FS 30D (30% aqueous dispersion) was freeze dried to yield powder. Afterward, 100 mg of the lyophilized NPs were suspended in 1 mL of water, and the suspension was introduced into a Eudragit® FS 30D (400 mg) ethanol solution (3–5 mL) under magnetic stirring. Unless otherwise noted, all experiments were conducted by varying the amount of ethanol while keeping all of the other process parameters unchanged. The suspension was then spray-dried with a Pulvis mini spray GS 310 apparatus under following conditions: inlet temperature was 50°C; spray pressure was 0.1 MPa; feeding rate of the suspension was 8 mL/min; the powder was collected in the cyclone separator, and the production yield (PY%) and drug recovery (DR%) were calculated using following equations:
PY% = AF/TAEN ×100% | (1) |
DR% = EM/EN ×100% | (2) |
where, AF, TAEN, EM, and EN represent the amount of final product, total amount of Eudragit® FS 30D and NPs added initially, amount of insulin extracted from the composite microcapsules, and amount of insulin extracted from the PLGA NPs, respectively.
Characterization of the PLGA NPs and composite microcapsules
Determination of zeta potential and particle size of the primary NPs
The particle size analysis and zeta potential measurements were performed using photon correlation spectroscopy with a Zetasizer Nano-ZS90 at 25°C. The lyophilized PLGA NP samples were suspended in distilled water before measurement.
Scanning electronic microscopy studies
Morphological characterization of the particles was performed by scanning electron microscopy (SEM) (JSM 5600LV; Jeol Ltd., Tokyo, Japan), which requires ion coating with platinum using a sputter coater (JFC-1300; Jeol Ltd.) for 40 seconds in a vacuum environment at the current intensity of 40 mA after preparing the sample on metallic studs with double-sided conductive tape.
In vitro release kinetic experiment
The release profiles of insulin from the formulations were investigated in gradual pH changing buffers (pH 1.2, 6.8, and 7.4). This buffer system was selected based on the normal variations of pH and general transit time along the GI tract from the stomach (pH 1.2, 0–2 hours) and small intestine (pH 6.8, 2–6 hours) to the colon region (pH 7.4, 6–24 hours).21 Briefly, test tubes containing samples were placed in a thermostatic-oscillating water bath (100 rpm) and incubated at 37°C. At predetermined time points, aliquots of 100 μL sample were withdrawn and centrifuged for 10 minutes (20,000 rpm, 4°C). The supernatants were used for HPLC analysis. Concentrations were calculated by interpolation from the standard curve. The in vitro release experiments were performed in triplicate. The released amount of insulin was expressed as a percentage of the total insulin encapsulated in PLGA NPs and in composite microcapsules. In order to study the release process of insulin from the composite microcapsules, some of the centrifuged microcapsules at determined time point were dried and observed with SEM.
Confocal imaging studies
Disposition of coumarin-6-loaded formulation in rat GI tract
Ins–S.O-Comp was replaced by the hydrophobic fluorescent marker coumarin-6 (C-6) to facilitate confocal imaging after oral administration of the particulate formulation. C-6 was loaded in a weight ratio of about 1% into PLGA NP composite microcapsules (C-6: PLGA: Eudragit® FS 30D at 1:20:80) using the same preparation procedures as aforementioned. The rats were fasted for 24 hours prior to oral administration of C-6-loaded PLGA NP composite microcapsules (C-6 dose 5 mg/rat). The rats were sacrificed at 4, 6, and 12 hours after the oral administration. The whole GI tract was resected and divided into four segments (stomach, duodenum and ileum, cecum, and colon). The ileum and colon segments were opened longitudinally and imaged by confocal laser scanning microscopy (CLSM) on an LSM 510 System (Carl Zeiss Meditec AG, Jena, Germany) equipped with a laser operating at 488 nm for fluorescence excitation to observe the composite microcapsules’ fate in the GI tract.
CLSM studies for absorption mechanism in ileum
Rats were fasted overnight and gavaged with C-6-loaded PLGA NP composite microcapsules. Four hours after the oral administration, the rats were sacrificed. The ileum was isolated and flushed with normal saline. Then the freshly excised tissues were cryofixed in Tissue-Tek® compound, sectioned (10 μm in thickness) using a cryomicrotome (Leica CM; Wetzlar, Germany), and imaged by the CLSM for fluorescence excitation.
Biological activity of insulin
NIH 3T3 cells (1×103/mL) were suspended in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin solution and seeded into a 24-well tissue culture plate (BD, Franklin Lakes, NJ, USA). A determined amount of free insulin or insulin extracted from Ins–S.O-Comp PLGA NPs or PLGA NP composite microcapsules were added to the cells, and each culture was incubated for 24 hours in a humidified atmosphere of 5% CO2 in air at 37°C. Cell growth was assessed by the [3H]thymidine ([methyl-1′-2′-3′Hthymidine (1.40 TBq/mmol)]; GE Healthcare UK Ltd., Little Chalfont, UK) incorporation assay. Radioactivity was counted with a scintillation counter (MicroBeta TriLux; PerkinElmer Inc., Waltham, MA, USA).
Hypoglycemic effect of the composite microcapsules
STZ-induced Wistar rats were fasted overnight with free access to water. The animals were maintained at constant temperature (22°C) with a fixed 12 hours light/12 hours dark cycle (lights on 7 am to 7 pm). For the experiment, 30 diabetic rats were randomly divided into five groups (six rats per group). The composite microcapsules (20 IU/kg), PLGA NPs (20 IU/kg), insulin solution (20 IU/kg), and saline (control group) were administered intragastrically. Insulin solution (1 IU/kg) was administered subcutaneously to designate its bioavailability as 100%. Blood samples were obtained from retroorbital plexus at different times and centrifuged at 4,000 rpm for 5 minutes to get the plasma. The plasma glucose levels were determined immediately by the Mutarotase-GOD method (Glucose C2; Wako Pure Chemical Industries, Ltd.). According to this method, 3 mL of glucose reagent was added into 20 μL of plasma, and the mixture was incubated at 37°C for 5 minutes. Absorbance values of the standard and plasma samples were measured against the reagent blank at 505 nm using a Hitachi U-3900 UV-visible spectrophotometer (Hitachi Ltd., Tokyo, Japan). The hypoglycemic response to insulin was characterized as follows: the initial plasma glucose was taken as 100% level, and all the following concentrations were given accordingly as the percentage of the initial. Areas above the plasma glucose level-time curves (AAC) were calculated using the trapezoidal rule. The pharmacological relative bioavailability was calculated by the ratio of the respective AAC corrected by the administered oral and subcutaneous doses. The relative bioavailability (RB%) was calculated using the following equation:
RB% = AACOral /AACSC × DoseSC/DoseOral ×100% | (3) |
Results and discussion
Preparation and characterization of Ins–S.O-Comp PLGA NPs
According to our previous study, the portioning coefficient of insulin existing in Ins–S.O-Comp in the form of 1-octanol increased by 3 orders of magnitude compared with free insulin,16 so insulin can be efficiently encapsulated into PLGA NPs. In this study, the encapsulation efficiency of insulin increased to 94.6%, which was about two times as high as free insulin (encapsulation efficiency =42.8%). Furthermore, zeta potential was also slightly increased (−32.6±2.1 mV for Ins–S.O-Comp versus −29.5±1.9 mV for free insulin) due to the introduction of negative S.O. The mean particle size of Ins–S.O-Comp PLGA NPs was about 213 nm. Morphology of the PLGA NPs was studied by SEM. From Figure 1, it can be observed that the particles were monodisperse spherical.
 | Figure 1 Scanning electron microscopy showing the morphology of the PLGA NPs. |
Preparation and characterization of Ins–S.O-Comp PLGA NP composite microcapsules
Eudragit® FS 30D is insoluble in acidic media but can be dissolved by salt formation above pH 7.0. Therefore, its dissolution at a higher pH allows the preparation of enteric microcapsules.
From our preliminary work, it was found that the ratio of water to ethanol was a key factor affecting the quality and production yield of composite microcapsules. In the present work, the ratio (water to ethanol) from 1:3 to 1:5 was chosen to study the effect on the formation of composite microcapsules. The effect of the ratio on the characteristics of composite microcapsules is shown in Table 1.
 | Table 1 Effect of the ratio of water to ethanol on the characteristics of composite microcapsules |
Morphology of the PLGA NP composite microcapsules was evaluated by SEM (Figure 2). When the ratio was 1:3, many large pores could be observed on the wall of composite microcapsules (Figure 2A), which caused the burst release of insulin (data is shown below). It can be explained that when ethanol was lower (1:3), the solvent could not dissolve Eudragit® FS 30D completely, which resulted in the low hardness of the wall. As the ratio of water to ethanol decreased to 1:4 and 1:5, the microcapsules displayed a well-defined spherical shape with relatively smooth surface (Figure 2B and C). However, the production yield decreased with decreasing ratio of water to ethanol. Therefore, the ratio of 1:4 was selected for the preparation of composite microcapsules. The spherical microcapsules made via 1:4 ratio determined procedure ranged from 1 μm to 5 μm in size as estimated from Figure 2B. Moreover, the production yield was about 85.4%, with drug recovery of 80.8%.
 | Figure 2 Morphology of the PLGA NP composite microcapsules at varying ratios of water to ethanol of 1:3 (A), 1:4 (B), and 1:5 (C). |
In vitro release kinetic experiment
Based on general transit time of drugs in the GI tract, drug release profiles were investigated in buffers with pH changed gradually (pH 1.2, 6.8, and 7.4). The cumulative release of insulin is shown in Figure 3.
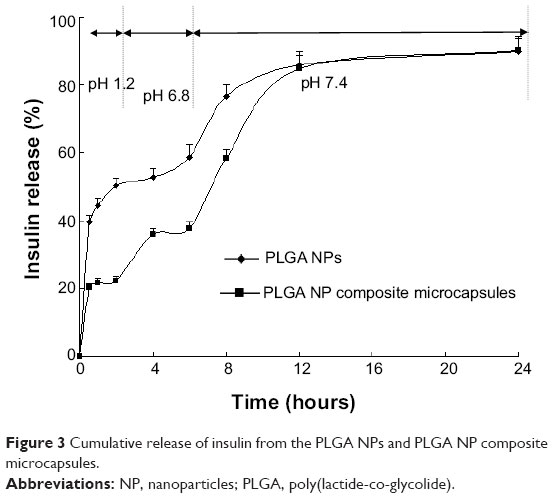 | Figure 3 Cumulative release of insulin from the PLGA NPs and PLGA NP composite microcapsules. |
Figure 3 shows that the cumulative release percentage of insulin from the PLGA NPs and PLGA NP composite microcapsules in pH 1.2 dissolution medium within the first 2 hours was about 50.2%±2.23% and 22.3%±1.24%, respectively. Accordingly, when pH was raised to 6.8 (2–6 hours), the cumulative insulin released from PLGA NPs and the composite microcapsules within the first 6 hours (0–6 hours) was about 58.7%±3.53% and 37.7%±1.91%, respectively. The PLGA NP composite microcapsules allowed rapid and complete drug release when the pH was raised to 7.4. The values within 24 hours (0–24 hours) were 89.9%±4.01% and 90.2%±4.31% for PLGA NPs and PLGA NP composite microcapsules, respectively. These results showed that the release of insulin from the composite microcapsules in pH 1.2 and pH 6.8 solutions was markedly reduced compared with the PLGA NPs.
Figure 4A and B clearly shows that the composite microcapsules remained intact when incubated in pH 1.2 dissolution medium for the first 2 hours, and only a small portion of PLGA NPs were released in pH 6.8 phosphate-buffered saline during the next 4 hours (2–6 hours), which can explain the low release of insulin over first 6 hours in the in vitro release profiles. Figure 4C shows that when the pH was changed to 7.4 (pH of the colon region), the enteric polymer was dissolved, and the PLGA NPs were gradually released from the composite microcapsules, then the remaining drug was released from the PLGA NPs in a sustained manner (Figure 4C). It can be concluded that the release of insulin was controlled by both of the microcapsules and PLGA NPs.
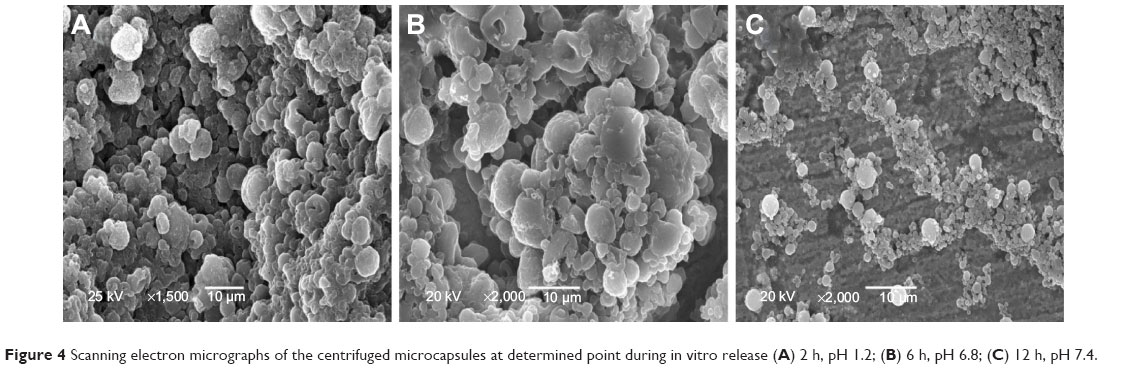 | Figure 4 Scanning electron micrographs of the centrifuged microcapsules at determined point during in vitro release (A) 2 h, pH 1.2; (B) 6 h, pH 6.8; (C) 12 h, pH 7.4. |
There has been increasing interest in targeting peptide and protein drugs to the colon because of the relatively low activity of proteolytic enzymes there.22 Thus, insulin, which is susceptible to acid degradation in the stomach, proteolytic degradation, and deactivation in the upper small intestine, may be more effectively absorbed from the colon. On the other hand, a small portion of PLGA NPs released from the composite microcapsules in the ileum can be directly taken up by Peyer’s patches.23 Thus, this pattern of release profile is ideal for oral administration of insulin.
Disposition of C-6-loaded microcapsules in the GI tract
From the results of in vitro release, it can be deduced that as a pH-sensitive formulation, the composite microcapsules remain intact in the stomach (0–2 hours). When the composite microcapsules arrived at the small intestine, only a small portion of PLGA NPs were released (2–6 hours). The PLGA NPs were then almost completely released in the colon (6–24 hours). This can be proven by the CLSM studies. In Figure 5A, it can be clearly observed that 4 hours after oral administration, a large number of microcapsules arrived at the ileum, and most of them remained intact. However, by 6 hours, very little fluorescence remained on the surface of ileum (Figure 5B), and most of the composite microcapsules got to the colon (Figure 5C). From Figure 5D, it was determined that 12 hours after the formulation administration, most of the PLGA NPs were released from the composite microcapsules, which indicated that the release of insulin could be controlled both by microcapsules and PLGA NPs in the GI tract.
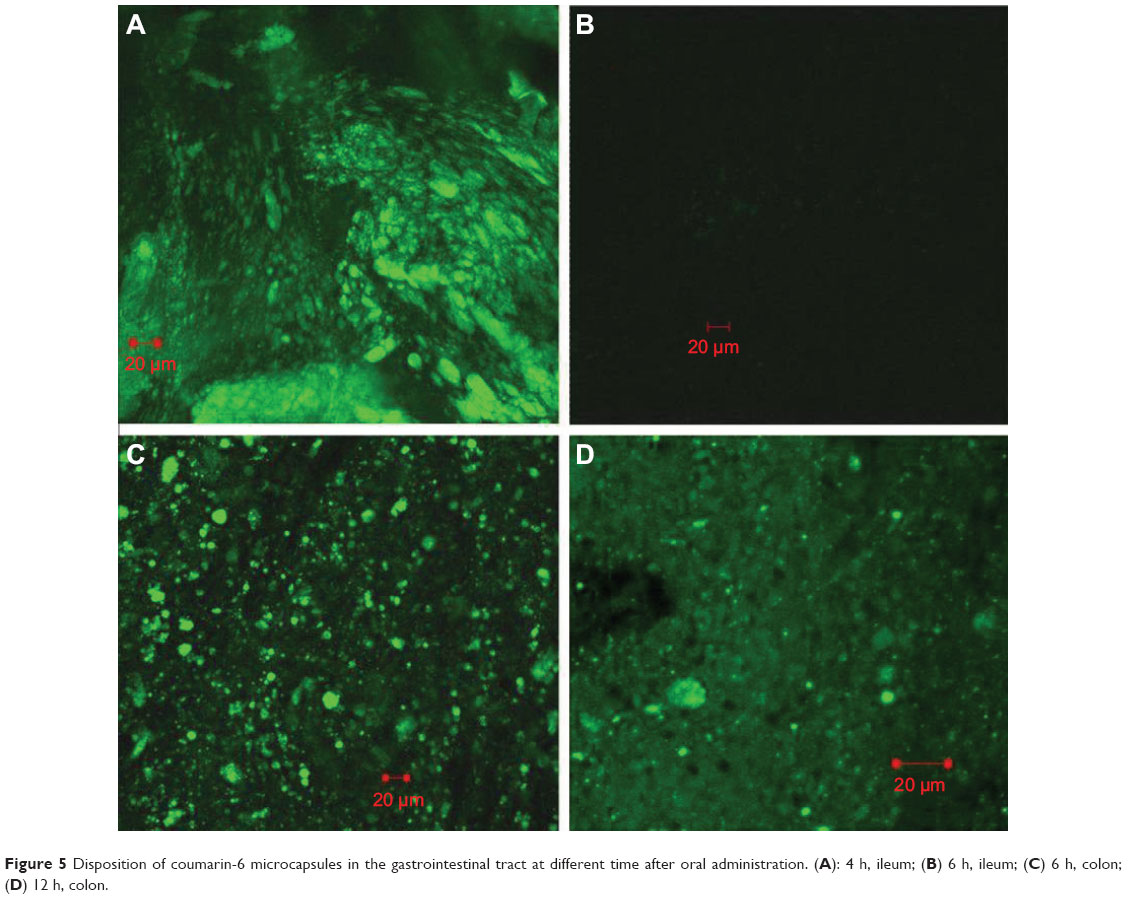 | Figure 5 Disposition of coumarin-6 microcapsules in the gastrointestinal tract at different time after oral administration. (A): 4 h, ileum; (B) 6 h, ileum; (C) 6 h, colon; (D) 12 h, colon. |
Biological activity of insulin
As insulin is readily destroyed, it is important to determine whether insulin maintains its biological activity during the preparation process. As we all know, insulin acts as a growth factor. NIH 3T3 mouse fibroblasts have few endogenous insulin receptors and are insensitive to insulin but express insulin-like growth factor (IGF)-receptors and are responsive to IGF-1.24 Insulin and IGF regulate certain cellular functions via both overlapping receptor and postreceptor signaling pathways.25 Therefore, we used these cells to test the biological activity of insulin extracted from Ins–S.O-Comp PLGA NPs and the composite microcapsules compared with that of normal insulin. In control cells, [3H]thymidine uptake was 9,472.7±1,189.9 dpm (Figure 6). The radioactivity of cells treated with extracted insulin has similar potency to that of normal insulin. This showed insulin retained its bioactivity during the preparation process.
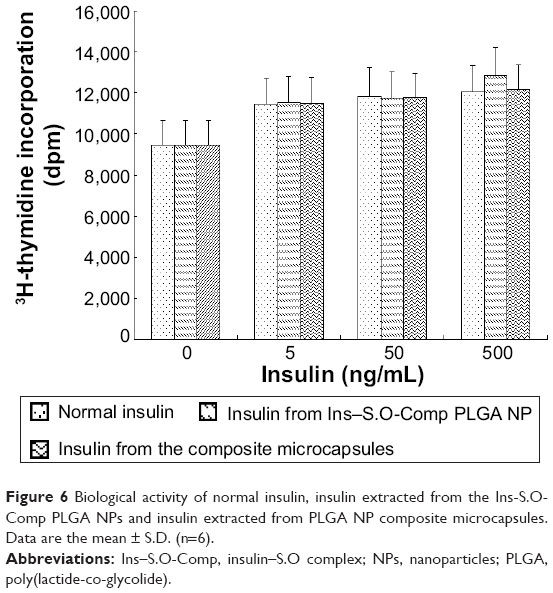 | Figure 6 Biological activity of normal insulin, insulin extracted from the Ins-S.O-Comp PLGA NPs and insulin extracted from PLGA NP composite microcapsules. Data are the mean ± S.D. (n=6). |
CLSM studies for absorption mechanism in the ileum
In order to further study the potential absorption mechanism of insulin in the ileum, CLSM studies were carried out with C-6-loaded PLGA NP composite microcapsules. PLGA NPs released from the composite microcapsules were capable of passing through the internal wall of the ileum via Peyer’s patches (Figure 7).
 | Figure 7 Confocal laser scanning microscopy image of Peyer’s patches cross-sections prepared 4 h after oral administration of coumarin-6-loaded PLGA NP composite microcapsules. |
Furthermore, many composite microcapsules reached the colon, where they released PLGA NPs. The colon is the ideal site for insulin absorption because of fewer digestive enzymes, longer residence time, and lower fluid volume compared to other GI regions.
Hypoglycemic effect of the composite microcapsules
From Figure 8, it was observed that the oral administered free insulin (20 IU/kg) did not significantly lower the blood glucose levels of animals. This can be illustrated as follows: when insulin is administered orally, the insulin molecules will be degraded in the GI tract by digestive enzymes and acidity, but the absorption of a small fraction of insulin prior to its degradation should not be disregarded. It was reported that when introduced in the lumen of the rat duodenum and colon, insulin could be rapidly internalized by the epithelial cells and transferred through a transcytotic pathway via the Golgi apparatus to the interstitial space from which it reached the blood circulation.26 After oral administration of Ins–S.O-Comp PLGA NPs, the hypoglycemic effect was observed to occur in a biphasic way. Two hours after oral administration of Ins–S.O-Comp PLGA NPs, the blood glucose level decreased to 74.88%±7.87%, then the level recovered to 85.07%±6.89% at 6 hours. It was interesting that this level reached 77.32%±6.84% again at 8 hours and maintained a sustained reduction for 12 hours. This may be related to the typical biphasic drug release pattern, with an initial burst release and prolonged release from polymer NPs.27–29 Initial burst release of insulin from PLGA NPs in the intestinal lumen was responsible for the first physiological effect. Then, after arrival to the ileum and colon, polymer NPs could be taken up and absorbed, resulting in a prolonged hypoglycemic effect compared with oral administration of insulin solution.30
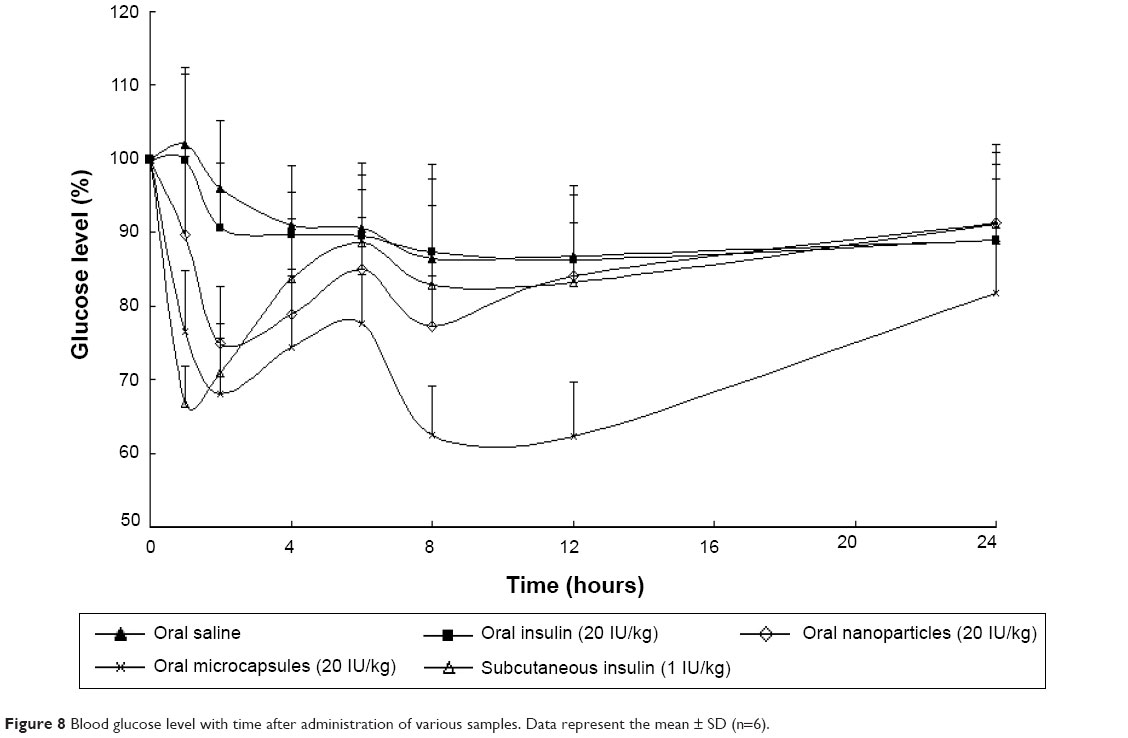 | Figure 8 Blood glucose level with time after administration of various samples. Data represent the mean ± SD (n=6). |
The hypoglycemic effect also occurred in a biphasic manner after oral administration of the PLGA NP composite microcapsules. The blood glucose level was reduced to 67.97%±7.69% at 2 hours, and then it recovered to 77.58%±6.76% at 6 hours. At the time point of 8 hours, the level reached 62.53%±6.58% again and maintained a sustained reduction for 24 hours. There was a significant difference between the PLGA NPs and composite microcapsules (P<0.05) at each time point except 2, 4, and 6 hours. The duration of reduction in blood glucose level was less in the case of PLGA NPs (12 hours) than PLGA NP composite microcapsules (24 hours). This can be explained that in the acidic environment of the stomach, a large amount of insulin was quickly released from PLGA NPs, while Eudragit® FS 30D can protect the insulin in the composite microcapsules. When the composite microcapsules came in contact with the alkaline conditions at the intestine (pH 6.8) and colon (pH 7.4), the enteric polymer Eudragit® FS 30D was gradually dissolved, and then the PLGA NPs and insulin were released.
More specifically, a little portion of PLGA NPs were released from the composite microcapsules in the intestine, and the PLGA NPs could be taken up as a whole by Peyer’s patches, then insulin can be absorbed.31 While in the colon, most of the composite microcapsules can be dissolved, and insulin will be gradually released from PLGA NPs and absorbed.32 The enteric nature of the polymer particles thus helped in improving the in vivo response for 24 hours from one single application, and the relative bioavailability was about 15.6% for the composite microcapsules while for free insulin it was less than 0.5%.
Conclusion
The present investigation explored the possibility of formulating an oral insulin delivery system by loading PLGA NPs into microcapsules, combining HIP method, emulsion solvent diffusion method, and organic spray-drying method. From in vitro results, it can be concluded that the PLGA NP composite microcapsules were intact in the stomach and gradually dissolved in small intestine and then almost completely dissolved in colon. The in vivo study demonstrated that PLGA NP composite microcapsules can increase the oral bioavailability of insulin, and they appear to be a promising candidate for oral insulin delivery.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (No 51403057), the Heilongjiang Returned Overseas Fund (No LC2013C24), Heilongjiang Postdoctoral Foundation, and the Doctoral Scientific Research Startup Foundation of Harbin Normal University (XKB201304).
Disclosure
The authors report no conflicts of interest in this work.
References
Song L, Zhi ZL, Pickup JC. Nanolayer encapsulation of insulin-chitosan complexes improves efficiency of oral insulin delivery. Int J Nanomedicine. 2014;9:2127–2136. | ||
Sonia TA, Sharma CP. An overview of natural polymers for oral insulin delivery. Drug Discov Today. 2012;17(13–14):784–792. | ||
Arbit E, Kidron M. Oral insulin: the rationale for this approach and current developments. J Diabetes Sci Technol. 2009;3(3):562–567. | ||
Arbit E. The physiological rationale for oral insulin administration. Diabetes Technol Ther. 2004;6(4):510–517. | ||
Sonaje K, Lin KJ, Wey SP, et al. Biodistribution, pharmacodynamics and pharmacokinetics of insulin analogues in a rat model: Oral delivery using pH-responsive nanoparticles vs subcutaneous injection. Biomaterials. 2010;31(26):6849–6858. | ||
Lewis GF, Zinman B, Groenewoud Y, Vranic M, Giacca A. Hepatic glucose production is regulated both by direct hepatic and extrahepatic effects of insulin in humans. Diabetes. 1996;45(4):454–462. | ||
Damgé C, Maincent P, Ubrich N. Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J Control Release. 2007;117(2):163–170. | ||
Chaturvedi K, Ganguly K, Nadagouda MN, Aminabhavi TM. Polymeric hydrogels for oral insulin delivery. J Control Release. 2013;165(2):129–138. | ||
Sullivan CO, Birkinshaw C. In vitro degradation of insulin-loaded poly(n-butylcyanoacrylate) nanoparticles. Biomaterials. 2004;25(18):4375–4382. | ||
Yun Y, Cho YW, Park K. Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev. 2013;65(6):822–832. | ||
Maroni A, Zema L, Del Curto MD, Foppoli A, Gazzaniga A. Oral colon delivery of insulin with the aid of functional adjuvants. Adv Drug Deliv Rev. 2012;64(6):540–556. | ||
Li X, Guo S, Zhu C, et al. Intestinal mucosa permeability following oral insulin delivery using core shell corona nanolipoparticles. Biomaterials. 2013;34(37):9678–9687. | ||
Soares S, Costa A, Sarmento B. Novel non-invasive methods of insulin delivery. Expert Opin Drug Deliv. 2012;9(12):1539–1558. | ||
Mukhopadhyay P, Mishra R, Rana D, Kundu PP. Strategies for effective oral insulin delivery with modified chitosan nanoparticles: a review. Prog Polym Sci. 2012;37(11):1457–1475. | ||
Sun S, Liang N, Kawashima Y, Xia D, Cui F. Hydrophobic ion pairing of an insulin-sodium deoxycholate complex for oral delivery of insulin. Int J Nanomedicine. 2011;6:3049–3056. | ||
Sun S, Liang N, Piao H, Yamamoto H, Kawashima Y, Cui F. Insulin-S.O (sodium oleate) complex-loaded PLGA nanoparticles: formulation, characterization and in vivo evaluation. J Microencapsul. 2010;27(6):471–478. | ||
Quintanar-Guerrero D, Allémann E, Fessi H, Doelker E. Applications of the ion-pair concept to hydrophilic substances with special emphasis on peptides. Pharm Res. 1997;14(2):119–127. | ||
Meyer JD, Manning MC. Hydrophobic ion pairing: altering the solubility properties of biomolecules. Pharm Res. 1998;15(2):188–193. | ||
Kshirsagar SJ, Bhalekar MR, Umap RR. In vitro in vivo comparison of two pH sensitive Eudragit polymers for colon specific drug delivery. J Pharm Sci Res. 2009;1(4):61–70. | ||
Damgé C, Michel C, Aprahamian M, Couvreur P. New approach for oral administration of insulin with polyalkylcyanoacrylate nanocapsules as drug carrier. Diabetes. 1988;37(2):246–251. | ||
Vandamme TF, Lenourry A, Charrueau C, Chaumeil JC. The use of polysaccharides to target drugs to the colon. Carbohydr Polym. 2002;48:219–231. | ||
Maroni A, Del Curto MD, Serratoni M, et al. Feasibility, stability and release performance of a time-dependent insulin delivery system intended for oral colon release. Eur J Pharm Biopharm. 2009;72(1):246–251. | ||
Lopes MA, Abrahim BA, Cabral LM, et al. Intestinal absorption of insulin nanoparticles: contribution of M cells. Nanomedicine. 2014;10(6):1139–1151. | ||
Takenaga M, Yamaguchi Y, Kitagawa A, et al. Optimum formulation for sustained-release insulin. Int J Pharm. 2004;271(1–2):85–94. | ||
Hofmann C, Goldfine ID, Whittaker J. The metabolic and mitogenic effects of both insulin and insulin-like growth factor are enhanced by transfection of insulin receptors into NIH3T3 fibroblasts. J Biol Chem. 1989;264(15):8606–8611. | ||
Ziv E, Bendayan M. Intestinal absorption of peptides through the enterocytes. Microsc Res Tech. 2000;49(4):346–352. | ||
Wissing SA, Kayser O, Müller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56(9):1257–1272. | ||
Sarmento B, Martins S, Ferreira D, Souto EB. Oral insulin delivery by means of solid lipid nanoparticles. Int J Nanomedicine. 2007;2(4):743–749. | ||
Hu FQ, Yuan H, Zhang HH, Fang M. Preparation of solid lipid nanoparticles with clobetasol propionate by a novel solvent diffusion method in aqueous system and physicochemical characterization. Int J Pharm. 2002;239(1–2):121–128. | ||
Cui FD, Tao AJ, Cun DM, Zhang LQ, Shi K. Preparation of insulin loaded PLGA-Hp55 nanoparticles for oral delivery. J Pharm Sci. 2007;96(2):421–427. | ||
Yoo MK, Kang SK, Choi JH, et al. Targeted delivery of chitosan nanoparticles to Peyer’s patch using M cell-homing peptide selected by phage display technique. Biomaterials. 2010;31(30):7738–7747. | ||
Wang XQ, Zhang Q. pH-sensitive polymeric nanoparticles to improve oral bioavailability of peptide/protein drugs and poorly water-soluble drugs. Eur J Pharm Biopharm. 2012;82(2):219–229. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.