")
Back to Journals » Cancer Management and Research » Volume 11
Peptidylarginine deiminase 4 overexpression resensitizes MCF-7/ADR breast cancer cells to adriamycin via GSK3β/p53 activation
Authors Zhou Q, Song C, Liu X, Qin H, Miao L, Zhang X
Received 18 October 2018
Accepted for publication 10 December 2018
Published 10 January 2019 Volume 2019:11 Pages 625—636
DOI https://doi.org/10.2147/CMAR.S191353
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Qianqian Zhou,1,* Chao Song,1,* Xiaoqiu Liu,2,3,* Hao Qin,1 Lixia Miao,1 Xuesen Zhang1
1State Key Laboratory of Reproductive Medicine, Nanjing Medical University, Nanjing, China; 2Key Laboratory of Pathogen Biology of Jiangsu Province, Nanjing Medical University, Nanjing, China; 3Department of Microbiology, Nanjing Medical University, Nanjing, China
*These authors contributed equally to this work
Background: Adriamycin (ADR) is widely used in the clinical chemotherapy against breast cancer. But its efficacy is strongly limited due to the acquisition of multidrug resistance (MDR). Therefore, acquisition of the resistance to ADR is still a major cause of chemotherapy failure in breast cancer patients. Peptidylarginine deiminase IV (PAD4) is reported to target non-histone proteins for citrullination, regulate their substrate activities, and thereby play critical roles in maintaining cell phenotype in breast cancer cells. However, whether PAD4 is involved in the development of MDR in breast cancer is poorly understood.
Materials and methods: We examined the expression of PAD family members, including PAD4 in ADR-resistant MCF-7 cells compared with the parental control cells by real-time PCR and Western blotting analyses. Rescue of PAD4 expression in MCF-7/ADR cells was performed to assess whether PAD4 could restore the sensitivity of MCF-7/ADR cells to ADR treatment with cell counting kit-8, flow cytometry, TUNEL, nuclear and cytoplasmic extract preparations, and immunofluorescence staining analyses.
Results: Both PAD2 and PAD4 were significantly decreased in ADR-resistant cells. However, only PAD4 overexpression can increase the sensitivity of MCF-7/ADR cells to ADR treatment and decrease MDR1 gene expression. Overexpression of PAD4 in MCF-7/ADR cells inhibited cell proliferation by inducing cell apoptosis. Under ADR treatment, overexpression of PAD4 promoted nuclear accumulation of glycogen synthase kinase-3β and p53, which further activated proapoptotic gene expression and downregulated MDR1 expression. Moreover, PAD4 activity was required for activating proapoptotic gene transcripts.
Conclusion: We demonstrate the previously unappreciated role of PAD4 in reversing ADR resistance in MCF-7/ADR cells and help establish PAD4 as a candidate biomarker of prognosis and chemotherapy target for MDR in breast cancers.
Keywords: PAD4, apoptosis, MDR, breast cancer, GSK3β
Introduction
Breast cancer is one of the most prevalent malignant tumors and the first leading cause of cancer-related death in women worldwide.1 About 266,120 new cases of breast cancer in US women are expected to be diagnosed, and 40,920 will die of this disease in 2018.2 Currently, chemotherapy is still the major treatment for breast cancer. However, the development of multidrug resistance (MDR) against the anticancer agents used is the key factor leading to the chemotherapy failure.3–5 Adriamycin (ADR), also known as doxorubicin, is one of the most widely used traditional chemotherapeutic drugs, and is generally used to treat solid tumors, including breast cancer, especially in advanced or metastatic breast cancer patients.6–9 Unfortunately, continuous ADR treatment usually induces development of MDR, which impedes its clinical application. Therefore, it remains imperative to discover novel molecular targeting agents or re-sensitize the existing drugs to ADR therapy, with the hope of effectively improving the prognosis and treatment of these patients.10
Mechanically, it has been widely accepted that drug resistance in cancer cells is associated with the overexpression of transmembrane transporter proteins P-glycoprotein (P-gp) and multidrug resistance-associated protein-1 (MRP1), encoded by MDR1 and MRP1 genes, respectively.11–14 These proteins lead to reduced intracellular drug concentration and decreased cytotoxicity through their ability to pump the drugs out of the cells.15,16 In recent years, it has become increasingly urgent to identify effective MDR reversal agents to increase the efficacy of chemotherapies. One of the effective ways to overcome P-gp-mediated drug resistance is either to block its drug-pump function or inhibit its expression by some specific agents.17 A couple of P-gp modulators, chemosensitizers, and P-gp inhibitors have been designed to overcome P-gp-mediated MDR, but their intrinsic toxicity at doses is necessary for drug activity, and drug interaction profiles of these chemosensitizers need to be optimized. Therefore, development of rational therapeutic strategies targeting MDR cells is still a critical clinical challenge for successful chemotherapy.
Peptidylarginine deiminases (PADs) are a family of calcium-dependent enzymes that convert positively charged arginine and methylarginine residues in substrate proteins to the neutral, non-standard residue citrulline and this enzymatic activity is alternatively referred to as citrullination or deamination.18,19 This alteration of protein charge results in changes in protein structure, function, and molecular interactions.19 For example, PAD2 and PAD4 can target histone H3 and H4 at gene promoters for citrullination, leading to local changes in chromatin structure and modulation of tumor-associated gene transcription in human breast cancer cells.20–22 Besides, PAD4 also targets non-histone proteins for citrullination in breast cancer cells, including glycogen synthase kinase-3β (GSK3β), and Ets-like protein-1 (Elk-1), to regulate their substrate activities and thereby play critical roles in maintaining cell phenotype in breast cancer cells.23,24 In addition, we recently found that PAD1 is the only PAD highly upregulated in triple-negative breast cancer cells. PAD1 interacts with and citrullinates MEK1 to regulate extracellular signal–regulated kinase 1/2– matrix metallopeptidase 2 signaling, thus promoting tumorigenesis.25 Furthermore, accumulating evidences showing that overexpression of PAD2 and PAD4 were detected in a wide range of human malignant cancers,26 and that PAD inhibitors suppress the proliferation of cancer cell lines both in vitro and in vivo,25,27 supports the notion that PADs play important roles in tumorigenesis. However, whether PADs are involved in the development of MDR in cancers is poorly understood. Thus, we aimed to investigate whether PADs might play roles in MDR of breast cancer cells and further explored possible mechanisms for this.
Materials and methods
Cell culture and treatment
The MCF-7 human breast cancer cell line was purchased from the Cell Bank at Shanghai Institute of Cell Biology (Shanghai, China). MCF-7 and MCF-7/ADR cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% FBS at 37°C in a humidified 5% CO2 atmosphere. The ADR-resistant MCF-7/ADR cells were successfully selected from drug-sensitive MCF-7 cells by exposure to increasing concentration of ADR (5 µg/mL) on the parental MCF-7 cells. PAD2 or PAD4 cDNA was sub-cloned to retroviral vector pQXCIP (Clontech Laboratories, Inc., Palo Alto, CA, USA), yielding PAD2-pQXCIP or PAD4-pQXCIP plasmids. Stable PAD2- or PAD4-overexpression MCF-7/ADR cells were generated by transduction with retroviral particles containing PAD2- or PAD4-pQXCIP construct. In the control group, MCF-7/ADR cells were transduced with an empty pQXCIP construct. Cells were selected by medium containing 1 µg/mL puromycin (Sigma-Aldrich Co., St Louis, MO, USA).
RNA extraction and quantitative real-time PCR (qRT-PCR) assay
Total RNA was isolated from cells using the Qiagen RNeasy Mini Kit in combination with on-column DNase treatment (Applied Biosystems, Foster City, CA, USA). A High Capacity RNA-to-cDNA Kit (Applied Biosystems) was used to synthesize the first strand of cDNA. qRT-PCR was performed using the Power SYBR Green PCR Master Mix (Applied Biosystems) with gene-specific primers. Primer sequences are listed in the Supplementary material (Table S1).
Western blot analysis
The cells were washed twice with cold PBS and then harvested for Western blotting. Cells were lysed in cold RIPA buffer containing protease inhibitors. Approximately 40 µg of total protein was separated by 10% SDS-PAGE, and then transferred to polyvinylidene difluoridemembranes. The membranes were then blocked and incubated with the following primary antibodies overnight at 4°C: PAD2 (Proteintech, Rosemont, IL, USA); PAD4 (Sigma-Aldrich); GSK3β (Cell Signaling Technology, Danvers, MA, USA), p53 (Bioworld Technology, Nanjing, China), caspase 3, cleaved caspase 3, caspase 8, cleaved caspase 8, caspase 9, and cleaved caspase 9 (Cell Signaling Technology), and glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as a loading control. The membranes were washed and then incubated with horseradish peroxidase-conjugated secondary antibodies. The signals were visualized using an Enhanced Chemiluminescence Detection Kit (Pierce Biotechnology, Rockford, IL, USA).
Apoptosis evaluation by flow cytometry and TUNEL assays
Apoptotic cells were detected using the Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (Yeasen, Shanghai, China). Briefly, cells were cultured in RPMI-1640 with 10% FBS overnight in 60-mm dishes. After treatment with 10 µg/mL ADR for 48 hours, cells were washed and then cell pellets were re-suspended in ice-cold binding buffer. Subsequently, 5 µL Annexin V-FITC solution and 5 µL dissolved propidium iodide (PI) were added to the cell suspension. After gentle mixing, samples were incubated for 10 minutes in the dark at room temperature. A FACScan flow cytometer was applied to quantify cellular apoptosis. Cells were considered apoptotic if they were Annexin V-positive/PI-negative, while double-positive cells either reflected necrosis or late apoptosis. For TUNEL, cells were grown on glass slides in 12-well plates, followed by fixation with 4% paraformaldehyde. TUNEL staining was performed with a TUNEL Apoptosis Detection Kit (FITC) (Yeasen) according to the manufacturer’s instructions. Following TUNEL staining, cells were washed and then blocked with 4% BSA for 5 minutes at room temperature. Nuclei were visualized by DNA staining with Hoechst stain (1 µg/mL). The images were captured using a Carl Zeiss (Oberkochen, Germany) lens. TUNEL positive signal was counted from randomly selected fields.
Immunofluorescence staining
Cells were grown on glass slides in 12-well plates, then fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. After blocking, cells were incubated with primary antibodies against Flag and GSK3β. Then Fluor 555-conjugated goat anti-mouse secondary antibody (Invitrogen, Carlsbad, CA, USA) or Fluor 488-conjugated goat anti-rabbit secondary antibody (Invitrogen) were employed to detect fluorescence. The nuclei were stained with DAPI (Vector Laboratories, Cambridgeshire, UK). Representative images were collected with LSM 510 laser scanning confocal microscope (Carl Zeiss).
Cell proliferation assay
Cells were seeded into 96-well plates (5,000 cells/well), incubated overnight, and then treated with 30 µg/mL of ADR at indicated times for 24, 48, 72, 96, and 120 hours. A total of 10 µL of cell counting kit-8 (CCK-8) reagent (Yeasen) was added to each well, and plates were incubated for 4 hours at 37°C in accordance with the CCK-8 kit protocol. OD values were measured at 450 nm using a plate reader (Thermo Scientific Multiskan GO, Thermo Fisher Scientific, Vantaa, Finland).
Nuclear and cytoplasmic extract preparations
Cells were washed twice with cold PBS and then lysed in cold cell lysis buffer (1 M Tris–HCl, pH =7.9; 1 M KCl, 10%NP40, 1× proteinase inhibitors) for 60 minutes on ice. The lysates were then centrifuged and supernatants were collected as cytoplasmic fraction. The pellets were washed and then lysed in cold lysis buffer (1 M Tris–HCl, pH =7.9; 0.5 M EDTA, 10%SDS, 1× proteinase inhibitors). The supernatants were collected as nuclear fraction.
Statistical analyses
All experiments were independently repeated at least three times. The results are presented as the mean ± SD. Data were analyzed using Student’s t-test to determine the level of significance between control and treatment groups. P<0.05 was considered to be statistically significant.
Results
PAD2 and PAD4 are downregulated in MCF-7/ADR cells
MDR1 gene was reported to be upregulated and mediate MDR in multidrug resistant breast cancer cells.11–14 We, therefore, first tested MDR1 gene expression by qRT-PCR analysis in our MCF-7/ADR cells and the parental MCF-7 cells. Results showed that MDR1 gene was dramatically induced in our ADR-resistant human breast cancer cells (Figure 1A). We next showed that the control MCF-7 cells completely died under 1 µg/mL ADR treatment for 24 hours, while MCF-7/ADR cells still survived even exposed to ADR as high as 8 µg/mL in the culture media (Figure 1B), confirming that MCF-7/ADR cells were resistant to ADR.
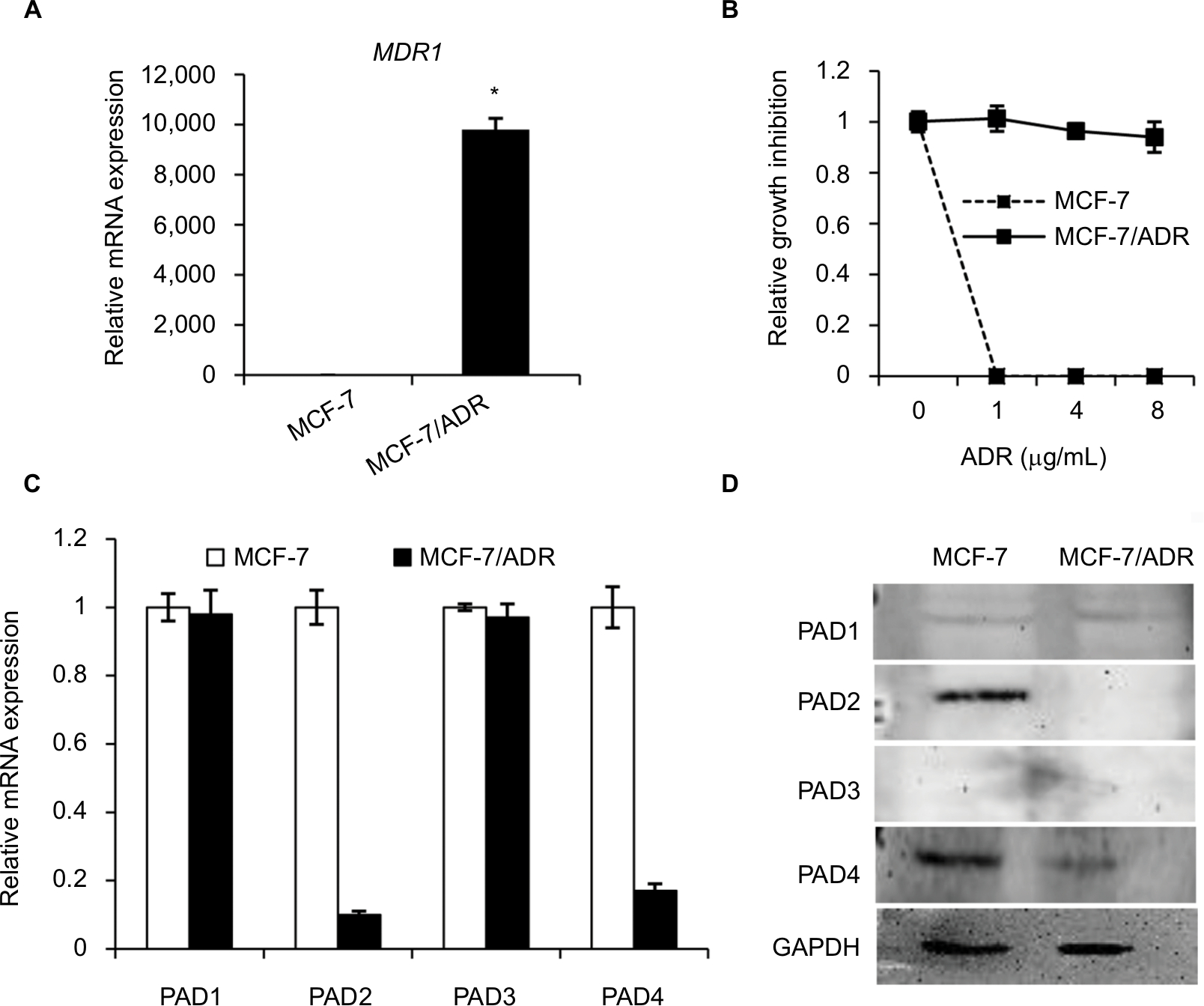 | Figure 1 The expression levels of PAD2 and PAD4 are downregulated in ADR-resistant breast cancer MCF-7 cells. Notes: (A) Expression levels of MDR1 mRNA in MCF-7 and MCF-7/ADR cells were determined by qRT-PCR. GAPDH was used as an internal control. (B) MCF-7 and MCF-7/ADR cells were cultured in regular medium with different ADR concentrations (1, 4, and 8 µg/mL), cell number was counted 24 hours post ADR treatment. Relative growth inhibition was normalized to the cell number of MCF-7 cells without ADR treatment. (C, D) Endogenous PAD1 and PAD4 mRNA levels in MCF-7 and MCF-7/ADR cells determined by qRT-PCR (C) and Western blot (D). The mRNA levels of PAD1 and PAD4 in MCF-7/ADR cells were normalized to that in MCF-7 cells. GAPDH served as a control. The representative immunoblots show that PAD2 and PAD4 are significantly downregulated in MCF-7/ADR cells compared with MCF-7 cells. GAPDH served as loading control. *P<0.05. Abbreviations: ADR, adriamycin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MDR1, multidrug resistance 1; PAD, peptidylarginine deiminase; qRT, quantitative real-time. |
To determine the expression pattern of PAD family members in ADR-resistant breast cancer cells, we first examined their mRNA levels in MCF-7/ADR cells and the parental MCF-7 cells. Results showed that both PAD2 and PAD4 transcript levels were significantly downregulated in MCF-7/ADR cells compared with the control cells, while PAD1 and PAD3 mRNA levels were not affected (Figure 1C). To further confirm these findings, we compared PADs’ protein levels in these two cell lines using Western blotting (Figure 1D). We found that, consistent with their mRNA level changes, both PAD2 and PAD4 protein levels were sharply decreased in MCF-7/ADR cells. We also noted that PAD1 and PAD3 proteins were hardly detected in either MCF-7 or the ADR-resistant cells. These results suggested us that the decreased expression of PAD2 and PAD4 may be associated with the ADR resistance in breast cancer cells, but not the other PADs.
Overexpression of PAD4 increases the sensitivity of MCF-7/ADR cells to ADR and inhibits proliferation of MCF-7/ADR cells
To further explore the role of PAD2 and PAD4 in ADR resistance in breast cancer cells, we first stably overexpressed PAD2 and PAD4 in MCF-7/ADR cells separately. As shown in Figure 2A, PAD2 protein level was highly elevated in PAD2 overexpression cells compared with either the empty vector (EV) control or PAD4 overexpression cells, and PAD4 protein was also overexpressed specifically in PAD4 overexpression line. Then we evaluated the effect of PAD2 and PAD4 overexpression on the proliferation of ADR-resistant MCF-7 cells. Results showed that, without ADR treatment, overexpression of either PAD2 or PAD4 in MCF-7/ADR cells did not affect cell growth, compared with the control cells (Figure 2B and C). However, as evidenced by the results of the CCK-8 assay (Figure 2D), ADR treatment on the three cell lines resulted in IC50 values of 101, 116, and 29.78 µg/mL in EV control, PAD2 overexpression, and PAD4 overexpression MCF-7/ADR cancer cells, respectively, indicating that MCF-7/ADR cells with PAD4 overexpression were less resistant to ADR than the control or PAD2 overexpression lines. We then gave all the three cell lines 30 µg/mL of ADR treatment and examined the cell viability at the indicated time. Results showed that PAD4 overexpression significantly resensitized MCF-7/ADR cells to ADR as early as 48 hours post ADR treatment (Figure 2E). Meanwhile, we found that the expression level of MDR1 gene was also significantly decreased in PAD4 overexpression MCF-7/ADR cells in the presence of 10 µg/mL of ADR, but this decrease was not observed in the other two lines or the cells without ADR exposure (Figure 2F). These results indicated that PAD4 may regulate P-gp expression and potentiate ADR-induced cytotoxicity in MCF-7/ADR cells.
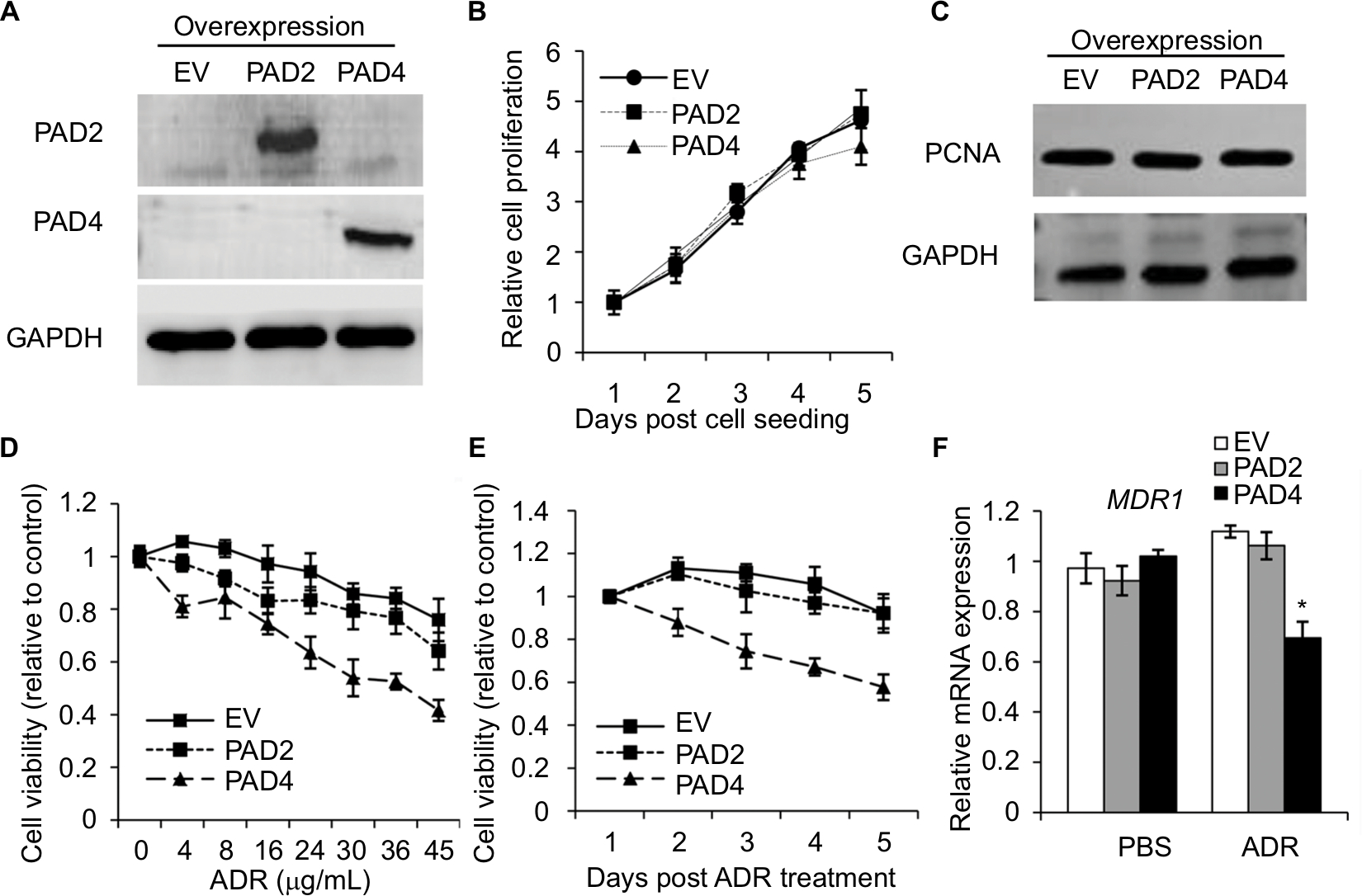 | Figure 2 PAD4 overexpression in MCF-7/ADR cells increases the chemosensitivity to ADR in vitro. Notes: (A) The representative immunoblots showing PAD2 and PAD4 were stably overexpressed in MCF-7/ADR cells, separately. GAPDH served as loading control. (B) Cell proliferation was not affected with both PAD2 and PAD4 overexpression in MCF-7/ADR cells by CCK-8 assay, compared with the EV control overexpression cells. (C) Western blot analysis showed that PAD2 and PAD4 overexpression did not affect PCNA expression in MCF-7/ADR cells. (D) CCK-8 assay was performed to determine the IC50 for EV control, PAD2 overexpression, and PAD4 overexpression MCF-7/ADR cancer cells. Relative growth inhibition was normalized to the cell number of MCF-7 cells without ADR treatment. (E) Cells were cultured in regular medium under treatment with 30 µg/mL ADR at indicated times, cell proliferation was tested by CCK-8 analysis and normalized to EV control cells. (F) qRT-PCR analysis of MDR1 mRNA expression in cells with or without 10 µg/mL ADR treatment for 48 hours. *P<0.05. Abbreviations: ADR, adriamycin; CCK-8, cell counting kit 8; EV, empty vector; IC50, half maximal inhibitory concentration; PAD, peptidylarginine deiminase; PCNA, proliferating cell nuclear antigen; qRT, quantitative real-time. |
ADR activates cell apoptosis in PAD4 overexpressed MCF-7/ADR
Resistance to anticancer drugs is typically accompanied by low susceptibility to apoptosis.28 Since enforced PAD4 expression can induce ADR sensitivity in MCF-7/ADR cells, we were wondering whether PAD4 overexpression may also affect cell apoptosis. To test this hypothesis, we first carried out fluorometric TUNEL analyses in the three cell lines we generated as mentioned previously, and rarely found apoptotic cell signals in each cell line in the absence of ADR (PBS treatment as a control). However, in the presence of 10 µg/mL of ADR, PAD4 overexpression significantly increased cell apoptosis (Figure 3A). Next, flow cytometric analysis showed that the apoptotic rate was significantly increased in MCF-7/ADR cells with PAD4 overexpression in the presence of ADR, compared with the PAD2 overexpression or EV control cells (Figure 3B). Furthermore, qRT-PCR analysis for the proapoptotic molecules, such as p21, Bax, and Bad confirmed that ADR activated cell apoptosis in PAD4 overexpressed MCF-7/ADR cells through induction of proapoptotic genes (Figure 3C).29–32 As controls, these genes could not be activated either in MCF-7/ADR cells with overexpressed PAD2 or the control MCF-7/ADR cells. Finally, examination of the activation of caspases induced by ADR showed significant activation of caspase-3 and 8, but not caspase-9 in PAD4 overexpressed MCF-7/ADR cells, indicating that ADR treatment would have an antiproliferative effect on PAD4 overexpressed MCF-7/ADR cells via induction of the extrinsic caspase-mediated apoptotic pathway (Figure 3D and E).
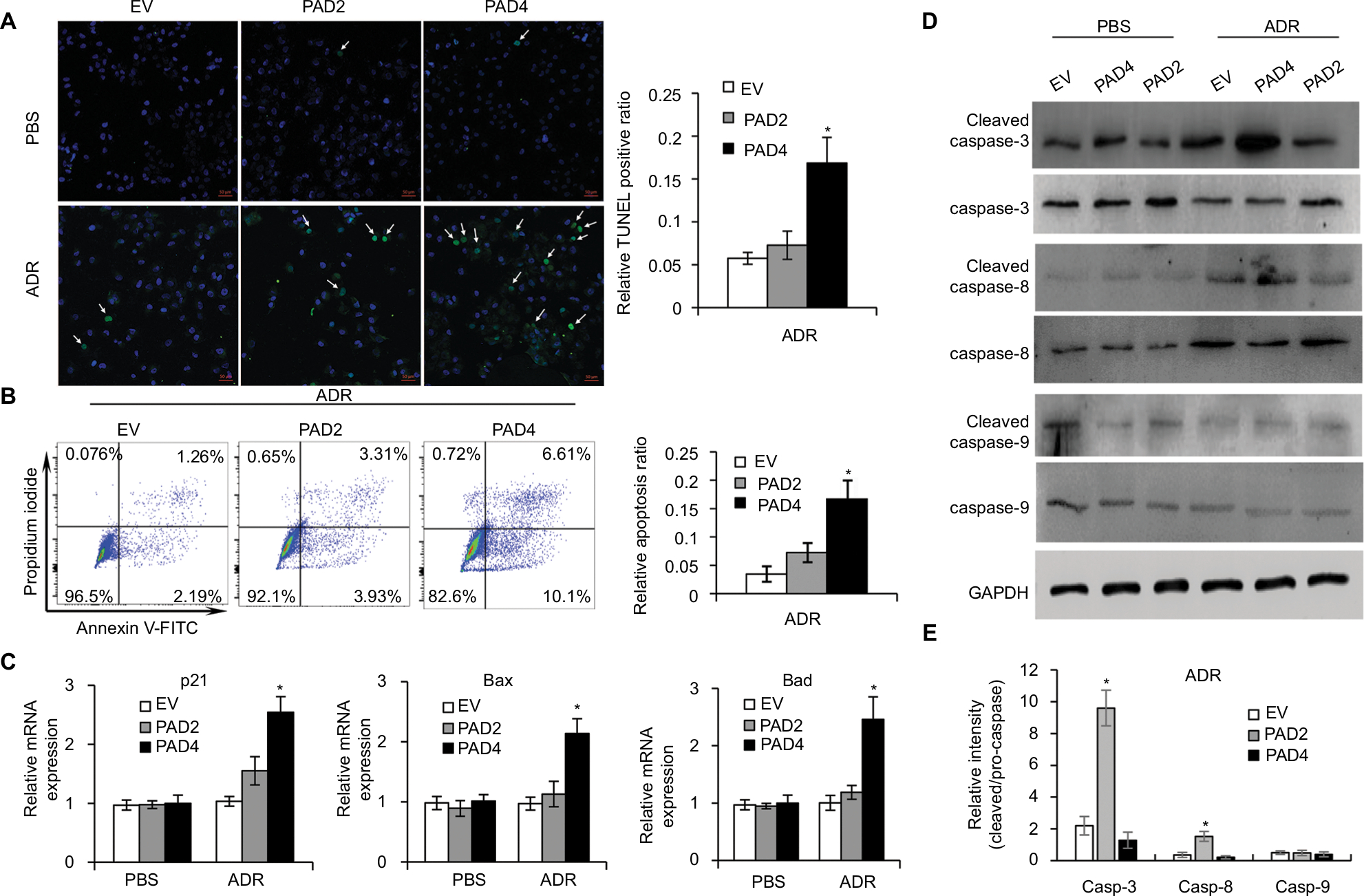 | Figure 3 Adriamycin increases cell apoptosis in MCF-7/ADR cells with PAD4 overexpression. Notes: (A) TUNEL staining in cells treated with PBS or 10 µg/mL ADR. Arrow indicating the TUNEL positive signal. Quantitative analysis of TUNEL-positive signals is shown on the right. (B) Flow cytometric analysis of ADR-induced apoptosis of the PAD4 overexpressed MCF-7/ADR cells compared with EV or PAD2 overexpression cells. Relative apoptosis ratio is quantified on the right. (C) qRT-PCR analysis of gene expression for p21, Bax, and Bad in cells treated with PBS (control) or 10 µg/mL ADR for 48 hours. Relative mRNA expression was normalized to GAPDH. (D) Western blot analysis of the activation of caspase 3, 8, and 9 in cells treated with PBS (control) or 10 µg/mL ADR for 48 hours. GAPDH was used as an internal control. (E) The relative expression levels of proteins were analyzed by ImageJ software. *P<0.05. Abbreviations: ADR, adriamycin; EV, empty vector; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PAD, peptidylarginine deiminase; qRT, quantitative real-time. |
Overexpressed PAD4 leads to increased nuclear levels of p53 in MCF-7/ADR cells in response to ADR
The transcription activator p53, plays a pivotal role in controlling cell cycle progression and apoptosis in response to genotoxic and cellular stress.33 It was also known that PAD4 increases tumor suppressor p53 and its downstream target genes to control cell cycle and induces apoptosis in multiple cancer cells.29,34 To test whether p53 was also activated in PAD4 overexpressed MCF-7/ADR cells in the presence of ADR treatment, we first compared total p53 protein level in the three MCF-7/ADR cell lines in the absence or presence of ADR. Results showed that total p53 protein levels were not significantly changed under these conditions (Figure 4A). As p53 is known to undergo nuclear accumulation in response to DNA damage,35,36 we next separated nuclear and cytoplasmic fractions of the cells and subjected them to Western blotting. We found that p53 protein level was significantly increased in the nuclear fraction of PAD4 overexpressed MCF-7/ADR cells with ADR treatment, however, this accumulation was not observed in PAD2 overexpressed or control cells (Figure 4B and C). As a control, PBS treatment did not induce p53 to undergo accumulation in the nuclei of the cells (Figure 4D), which is consistent with the previous reports.
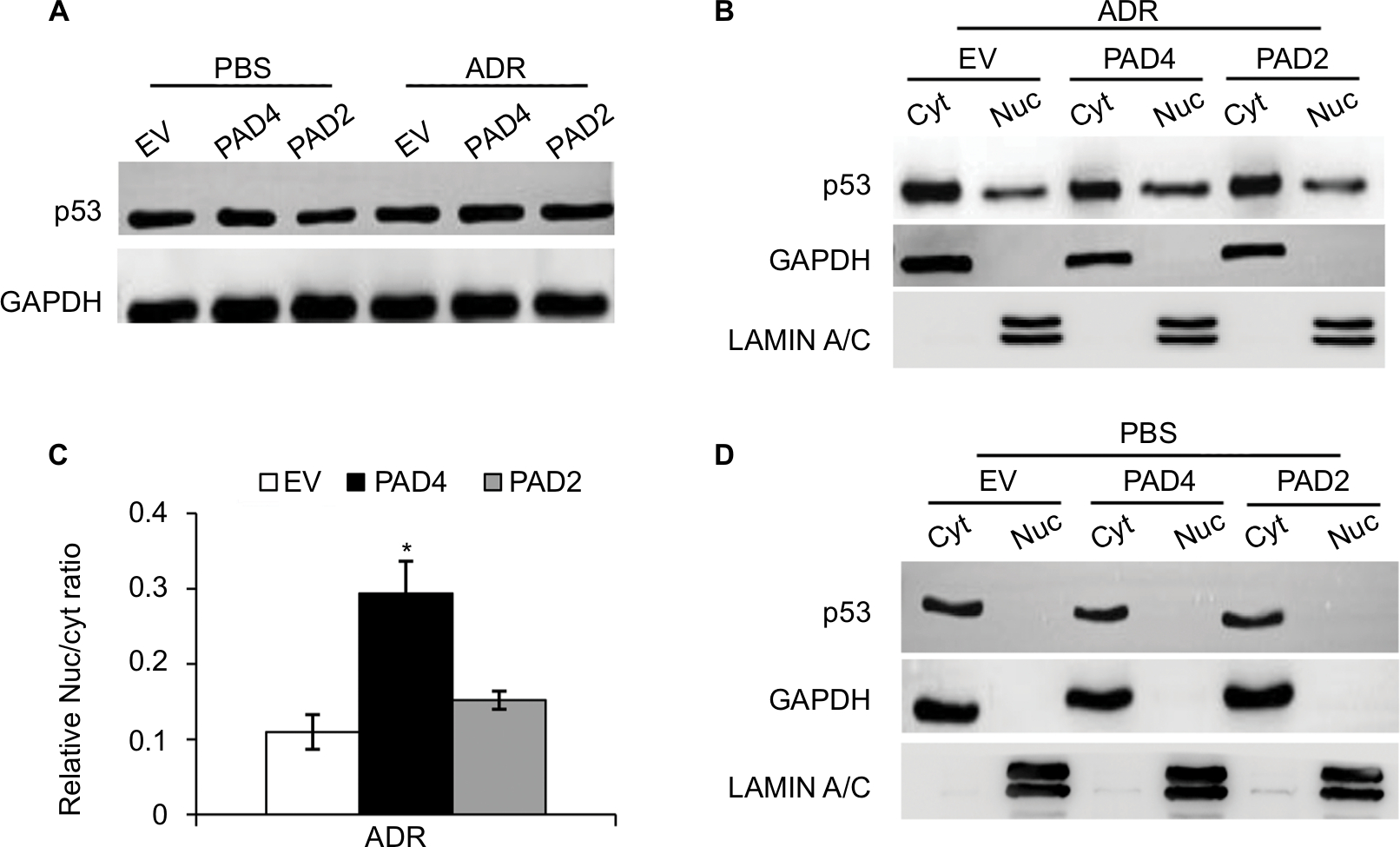 | Figure 4 ADR treatment potentiates nuclear levels of p53 in MCF-7/ADR cells with PAD4 overexpression. Notes: (A) Immunoblot analysis showing that p53 expression levels were not affected in MCF-7/ADR cells with EV, PAD2, and PAD4 overexpression. GAPDH served as loading control. (B) Cellular proteins after ADR treatment were separated into cytoplasmic and nuclear pools by fractionation methods and examined by Western blot with anti-p53 antibody. Cleanliness of fractionation was determined by probing with antibodies for LAMIN A/C (nuclear) and GAPDH (cytoplasmic) proteins. (C) Relative Nuc/cyt ratio of p53 was quantified. *P<0.05 (D) Indicated cell lysates from cells treated with PBS were analyzed by SDS/PAGE and probed for antibodies against p53, GAPDH, and LAMIN A/C. Abbreviations: ADR, adriamycin; Cyt, cytoplasmic; EV, empty vector; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Nuc, nuclear; PAD, peptidylarginine deiminase. |
Nuclear accumulation of GSK3β is regulated by PAD4 with ADR treatment
GSK3β has been reported to phosphorylate p53 and regulate the intracellular localization of p53.37–39 Several apoptotic stimuli also induce nuclear accumulation of GSK3β, where it co-localizes with p53.40,41 In addition, it is confirmed that there exists a direct, activating interaction between GSK3β and p53 after DNA damage,40 and that GSK3β participates in regulating p53 activity in the nucleus.42 These findings prompted us to test whether GSK3β may also be activated in the nucleus in PAD4 overexpression cells after ADR treatment. As expected, we found a similar activation pattern of GSK3β in the nucleus to that of p53 protein (Figure 5A and B). These results suggested that the activation of GSK3β/p53 signaling may mediate the accelerated apoptosis in PAD4 overexpressed MCF-7/ADR cells after ADR treatment.
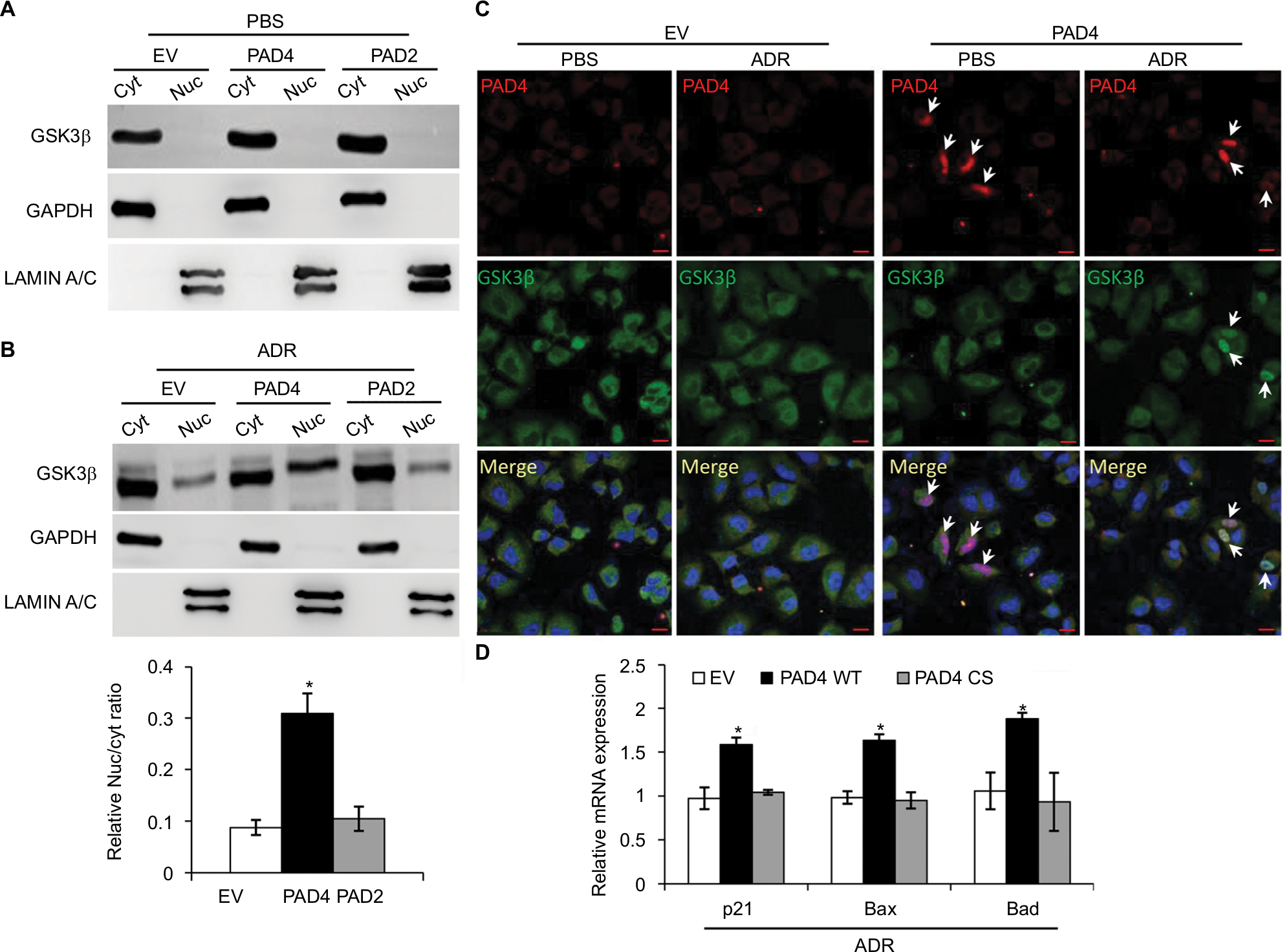 | Figure 5 ADR treatment increases nuclear accumulation of GSK3β in MCF-7/ADR cells with PAD4 overexpression. Notes: (A, B) Cellular fractionation analysis from MCF-7/ADR cells with EV, PAD2, and PAD4 overexpression under either PBS (A) or 10 µg/mL ADR (B) treatment, followed by Western blot with anti- GSK3β, GAPDH, and LAMIN A/C antibodies. Relative Nuc/cyt ratio of GSK3β was shown in the lower panel of (B). *P<0.05 (C) Immunofluoresce analysis showing that overexpressed PAD4 (Flag-tagged) leads to accumulation of GSK3β in the nucleus after ADR treatment. All cells were stained with antibodies against the Flag-tag (red) and GSK3β (green). Nuclei were stained with DAPI. Scale bar, 20 µm. Arrows indicate the PAD4 overexpressed signals in the nuclei, colocalizing with GSK3β. (D) qRT-PCR analysis of p21, Bax, and Bad in cells with control (EV), catalytic PAD4 WT, or PAD4 CS overexpression under 10 µg/mL ADR for 48 hours. Relative mRNA expression was normalized to GAPDH. *P<0.05. Abbreviations: ADR, adriamycin; CS, catalytic mutant; Cyt, cytoplasmic; EV, empty vector; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GSK3β, glycogen synthase kinase-3β; Nuc, nuclear; PAD, peptidylarginine deiminase; WT, wild type |
A previous study has shown that PAD4 recognizes, binds, and citrullinates GSK3β in breast cancer cells, and nuclear accumulation of GSK3β was observed only in cells transfected with wild type PAD4, suggesting that this unique posttranslational modification in GSK3β is crucial for the nuclear localization of the kinase.23 Therefore, we investigated whether PAD4 may exert the similar function to GSK3β and regulate its nuclear accumulation. As shown in Figure 5C, in the absence of PAD4 overexpression, GSK3β was mainly localized in the cytoplasm of cells with either PBS or ADR treatment. However, PAD4 overexpression induced a substantial amount of co-localization between PAD4 and GSK3β in the nuclei under ADR treatment. As a control, nuclear GSK3β accumulation was not observed in the cells with PBS treatment, even though PAD4 was overexpressed in the same cells (Figure 5C). These results indicated that ADR may promote PAD4 enzyme activity to activate GSK3β/p53 pathway in regulation of cell apoptosis. To further test this hypothesis, we transfected MCF-7/ADR cells with wild type PAD4, catalytic mutant (C645S) PAD4, or EV as a control, and then performed qRT-PCR analysis to examine the expression of the proapoptotic molecules (Figure 5D). A significant increase in the proapoptotic gene transcripts was observed in the cells with exogenous expression of wild type PAD4, whereas C645S mutant did not cause such gene activation above the basal level, thus supporting our hypothesis.
Discussion
A range of potential factors have been proposed to contribute to ADR chemoresistance, among which is the overexpression of a superfamily of energy-dependent ATP binding cassette transporters, including P-gp, that extrude anticancer drugs out of the cell.11,12,14 To achieve better clinical outcome and restore the cytotoxicity of antitumor drugs in ADR-resistant breast cancer cells, strategies for overcoming P-gp-dependent drug resistance have been proposed.43,44 Although significant progress has been made in reversing MDR, the clinical challenge of MDR for breast cancer patients still exists. Therefore, there is still an urgent need to explore new therapeutic approaches to potentially reverse drug resistance in breast cancers.
In this study, we first evaluated PADs expression in MCF-7/ADR cells compared with the parental MCF-7 cells and found that both PAD2 and PAD4 are dramatically downregulated in MCF-7/ADR cells, while PAD1 and PAD3 are undetectable in both cell lines. These results raise a possibility that decreased PAD2 and PAD4 may be involved in drug resistance in MCF-7/ADR cells. We, therefore, overexpressed PAD2 or PAD4 in MCF-7/ADR cells and demonstrated that only PAD4 overexpression in MCF-7/ADR cells has the ability to reverse MDR through inducing cell apoptosis, confirming our hypothesis that PAD4 is involved in MDR in MCF-7/ADR cells. Regarding the molecular mechanisms underlying restoration of the sensitivity of MCF-7/ADR cells to ADR, we then demonstrated that ADR treatment potentiates PAD4 to facilitate increased nuclear levels of GSK3β and p53, which further activates proapoptotic gene expression. These results suggest that ADR may promote PAD4 enzyme activity to activate GSK3β/p53 pathway in regulation of cell apoptosis (Figure 6).
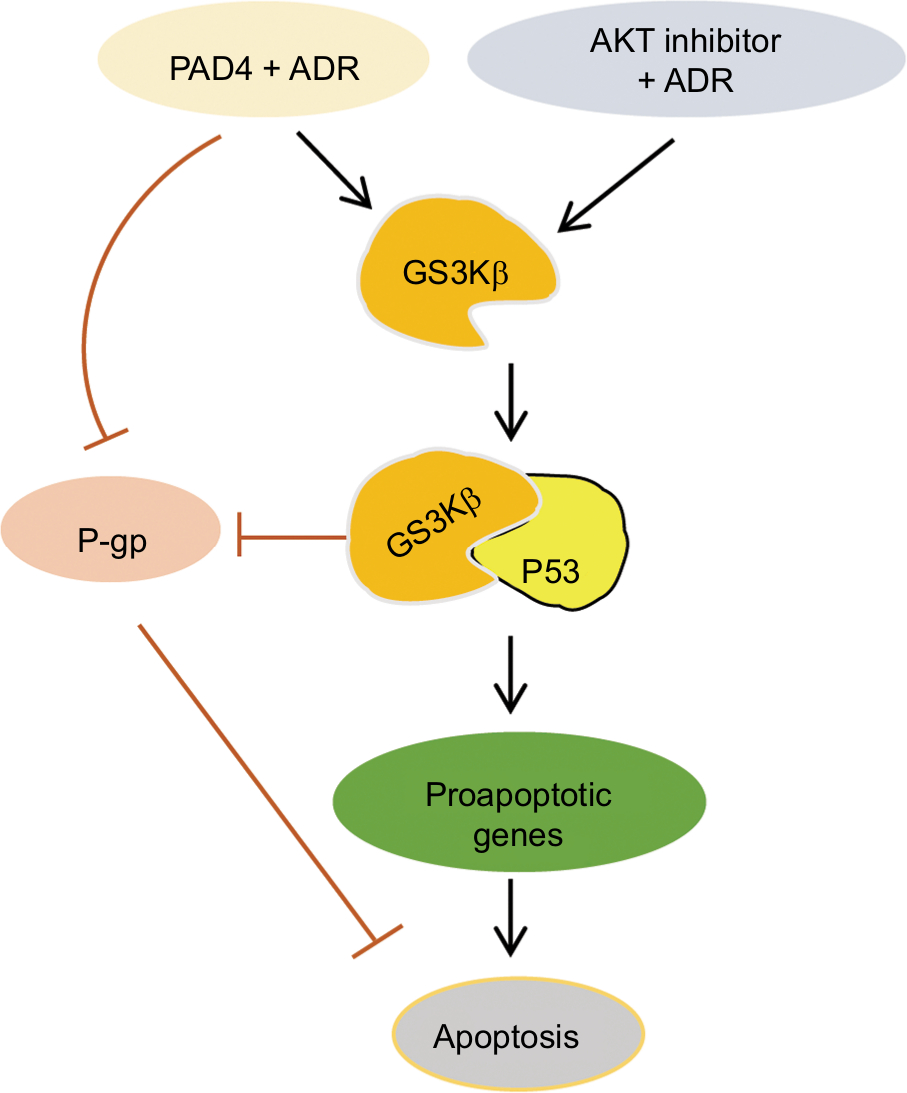 | Figure 6 Schematic of the effects of PAD4 on GSK3β/p53 signaling in regulation of proapoptotic gene expression under ADR treatment. Note: Arrows indicate activating events, whereas perpendicular lines indicate inhibitory events. Abbreviations: ADR, adriamycin; GSK3β, glycogen synthase kinase-3β; PAD, peptidylarginine deiminase; P-gp, P-glycoprotein. |
GSK3β is a serine/threonine kinase involved in several signal transduction cascades, and has been implicated in breast cancer development and drug resistance.45 Several apoptotic stimuli were found to cause the accumulation of GSK3β in the nucleus, forming a stable complex with p53 and contributing to p53-induced expression of proapoptotic molecules, such as p21, Bax, and Bad.29–32,41,45 Our data showing that ADR treatment potentiates PAD4 to facilitate nuclear accumulation of both GSK3β and p53 fit well with these reports that GSK3β and p53 interact synergistically and co-operate as partners in controlling cellular responses to DNA damage.46 Regarding the mechanisms of PAD4 facilitating GSK3β nuclear accumulation, a previous report has proved that PAD4 targets GSK3β for citrullination on its N-terminal domain and this posttranslational modification on GSK3β directly accelerates the nuclear protein levels of GSK3β in breast cancer cells, and thereby plays a critical role in regulating breast cancer initiation or progression. Additionally, PAD4 was also shown to citrullinate different non-histone proteins, including p300, nucleophosmin (NPM1), inhibitor of growth 4 (ING4), Ets-like protein-1 (Elk1) and antithrombin, thereby partially influencing the activity or localization of these proteins.24,47–51 Therefore, it is possible that in MCF-7/ADR cells, PAD4 overexpression may also citrullinate GSK3β to increase its nuclear accumulation and the activity of this kinase, which helps to resensitize the cells to ADR-induced apoptosis. To further prove this hypothesis, we showed that wild type PAD4 dramatically increased p53 target gene transcription in MCF-7/ADR cells upon ADR treatment, while catalytically inactive mutant PAD4 had no such effect, suggesting that mutant PAD4 may fail to citrullinate GSK3β.
The role of PAD4 in regulating cancer progression remains controversial. The high expression of PAD4 was observed in some malignant tumor tissues,51 suggesting that it may promote the process of tumorigenesis. However, a previous study has proved that overexpression of PAD4 enhances the apoptotic cell death in human leukemia HL-60 cells and human acute T leukemia Jurkat cells.29 Additionally, silencing PAD4 in breast cancer cells leads to induction of epithelial-to-mesenchymal transition and production of more invasive tumors in xenograft mouse model and clinical breast patients, supporting PAD4’s role as a tumor suppressor in breast cancer cells.23 Therefore, the role of PAD4 in regulation of tumorigenesis may be cell type- or cell context-dependent. Nevertheless, whether PAD4 is involved in regulation of MDR of breast cancer has not been reported before. To the best of our knowledge, this is the first study showing that PAD4 is significantly downregulated in MCF-7/ADR cells, and overexpression of PAD4 can reverse the MDR of MCF-7/ADR cells through inducing cell apoptosis. Therefore, rescue of PAD4 expression and in combination with ADR treatment might provide a clinical possibility to overcome the ADR resistance for breast cancer patients, especially for those who have already acquired drug resistance. Of note, whether presence of PAD4 expression in breast cancer tissues predicts a better responsibility to ADR awaits further clinical sample elucidation.
Conclusion
Our data highlight the importance of PAD4 in maintaining ADR sensitivity of MCF-7 breast cancer cells and indicate that rescue of PAD4 expression in ADR-resistant breast cancer may increase the success of chemotherapy by reversal of the ADR resistance.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81501797; 81372850), the Key Project of Science and Technology Innovation of Nanjing Medical University (2017NJMUCX007), and Jiangsu Six Talent Peaks Project.
Disclosure
The authors report no conflicts of interest in this work.
References
Dai X, Li T, Bai Z, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res. 2015;5(10):2929–2943. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Vtorushin SV, Khristenko KY, Zavyalova MV, et al. The phenomenon of multi-drug resistance in the treatment of malignant tumors. Exp Oncol. 2014;36(3):144–156. | ||
Li J, Liu J, Guo N, Zhang X. Reversal of multidrug resistance in breast cancer MCF-7/ADR cells by h-R3-siMDR1-PAMAM complexes. Int J Pharm. 2016;511(1):436–445. | ||
Wang X, Teng Z, Wang H, et al. Increasing the cytotoxicity of doxorubicin in breast cancer MCF-7 cells with multidrug resistance using a mesoporous silica nanoparticle drug delivery system. Int J Clin Exp Pathol. 2014;7(4):1337–1347. | ||
Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta. 1845;2014(1):84–89. | ||
Verma S, Dent S, Chow BJ, Rayson D, Safra T. Metastatic breast cancer: the role of pegylated liposomal doxorubicin after conventional anthracyclines. Cancer Treat Rev. 2008;34(5):391–406. | ||
Vatsyayan R, Chaudhary P, Lelsani PC, et al. Role of RLIP76 in doxorubicin resistance in lung cancer. Int J Oncol. 2009;34(6):1505–1511. | ||
Green AE, Rose PG. Pegylated liposomal doxorubicin in ovarian cancer. Int J Nanomedicine. 2006;1(3):229–239. | ||
Shen H, Li L, Yang S, et al. MicroRNA-29a contributes to drug-resistance of breast cancer cells to adriamycin through PTEN/AKT/GSK3β signaling pathway. Gene. 2016;593(1):84–90. | ||
Leslie EM, Deeley RG, Cole SP. Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters. Toxicology. 2001;167(1):3–23. | ||
Renes J, de Vries EG, Jansen PL, Müller M. The (patho)physiological functions of the MRP family. Drug Resist Updat. 2000;3(5):289–302. | ||
Lee CA, O’Connor MA, Ritchie TK, et al. Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: practical recommendations for clinical victim and perpetrator drug-drug interaction study design. Drug Metab Dispos. 2015;43(4):490–509. | ||
Leonessa F, Clarke R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr Relat Cancer. 2003;10(1):43–73. | ||
Dönmez Y, Gündüz U. Reversal of multidrug resistance by small interfering RNA (siRNA) in doxorubicin-resistant MCF-7 breast cancer cells. Biomed Pharmacother. 2011;65(2):85–89. | ||
Pan X, Yang X, Zang J, et al. Downregulation of eIF4G by microRNA-503 enhances drug sensitivity of MCF-7/ADR cells through suppressing the expression of ABC transport proteins. Oncol Lett. 2017;13(6):4785–4793. | ||
Chen JR, Jia XH, Wang H, Yi YJ, Wang JY, Li YJ. Timosaponin A-III reverses multi-drug resistance in human chronic myelogenous leukemia K562/ADM cells via downregulation of MDR1 and MRP1 expression by inhibiting PI3K/Akt signaling pathway. Int J Oncol. 2016;48(5):2063–2070. | ||
Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8(4):307–318. | ||
Zhang Y. Molecular biology: no exception to reversibility. Nature. 2004;431(7009):637–639. | ||
Zhang X, Bolt M, Guertin MJ, et al. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor target gene activation. Proc Natl Acad Sci U S A. 2012;109(33):13331–13336. | ||
Guertin MJ, Zhang X, Anguish L, et al. Targeted H3R26 deimination specifically facilitates estrogen receptor binding by modifying nucleosome structure. PLoS Genet. 2014;10(9):e1004613. | ||
Wang Y, Wysocka J, Sayegh J, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306(5694):279–283. | ||
Stadler SC, Vincent CT, Fedorov VD, et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3 activates TGF- signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc Natl Acad Sci U S A. 2013;110(29):11851–11856. | ||
Zhang X, Gamble MJ, Stadler S, et al. Genome-wide analysis reveals PADI4 cooperates with Elk-1 to activate c-Fos expression in breast cancer cells. PLoS Genet. 2011;7(6):e1002112. | ||
Qin H, Liu X, Li F, et al. PAD1 promotes epithelial-mesenchymal transition and metastasis in triple-negative breast cancer cells by regulating MEK1-ERK1/2-MMP2 signaling. Cancer Lett. 2017;409:30–41. | ||
Wang L, Song G, Zhang X, et al. PADI2-mediated citrullination promotes prostate cancer progression. Cancer Res. 2017;77(21):5755–5768. | ||
Qu Y, Olsen JR, Yuan X, et al. Small molecule promotes β-catenin citrullination and inhibits Wnt signaling in cancer. Nat Chem Biol. 2018;14(1):94–101. | ||
Mitani T, Ito Y, Harada N, et al. Resveratrol reduces the hypoxia-induced resistance to doxorubicin in breast cancer cells. J Nutr Sci Vitaminol (Tokyo). 2014;60(2):122–128. | ||
Liu GY, Liao YF, Chang WH, et al. Overexpression of peptidylarginine deiminase IV features in apoptosis of haematopoietic cells. Apoptosis. 2006;11(2):183–196. | ||
Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. | ||
Wei CL, Wu Q, Vega VB, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124(1):207–219. | ||
Franke TF, Yang SI, Chan TO, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81(5):727–736. | ||
Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–16. | ||
Li P, Yao H, Zhang Z, et al. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol Cell Biol. 2008;28(15):4745–4758. | ||
Esrig D, Spruck CH 3rd, Nichols PW, et al. p53 nuclear protein accumulation correlates with mutations in the p53 gene, tumor grade, and stage in bladder cancer. Am J Pathol. 1993;143(5):1389–1397. | ||
Wang X, Zalcenstein A, Oren M. Nitric oxide promotes p53 nuclear retention and sensitizes neuroblastoma cells to apoptosis by ionizing radiation. Cell Death Differ. 2003;10(4):468–476. | ||
Beurel E, Kornprobst M, Blivet-van Eggelpoël MJ, et al. GSK-3beta inhibition by lithium confers resistance to chemotherapy-induced apoptosis through the repression of CD95 (Fas/APO-1) expression. Exp Cell Res. 2004;300(2):354–364. | ||
Qu L, Huang S, Baltzis D, et al. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004;18(3):261–277. | ||
Pluquet O, Qu LK, Baltzis D, Koromilas AE. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta. Mol Cell Biol. 2005;25(21):9392–9405. | ||
Watcharasit P, Bijur GN, Zmijewski JW, et al. Direct, activating interaction between glycogen synthase kinase-3 and p53 after DNA damage. Proc Natl Acad Sci U S A. 2002;99(12):7951–7955. | ||
Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J Biol Chem. 2001;276(40):37436–37442. | ||
Tan J, Zhuang L, Leong HS, Iyer NG, Liu ET, Yu Q. Pharmacologic modulation of glycogen synthase kinase-3beta promotes p53-dependent apoptosis through a direct Bax-mediated mitochondrial pathway in colorectal cancer cells. Cancer Res. 2005;65(19):9012–9020. | ||
Dönmez Y, Akhmetova L, Işeri Özlem Darcansoy, Kars MD, Gündüz U. Effect of MDR modulators verapamil and promethazine on gene expression levels of MDR1 and MRP1 in doxorubicin-resistant MCF-7 cells. Cancer Chemother Pharmacol. 2011;67(4):823–828. | ||
Tsubaki M, Komai M, Itoh T, et al. By inhibiting Src, verapamil and dasatinib overcome multidrug resistance via increased expression of Bim and decreased expressions of MDR1 and survivin in human multidrug-resistant myeloma cells. Leuk Res. 2014;38(1):121–130. | ||
Farago M, Dominguez I, Landesman-Bollag E, et al. Kinase-inactive glycogen synthase kinase 3β promotes Wnt signaling and mammary tumorigenesis. Cancer Res. 2005;65(13):5792–5801. | ||
Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS. Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J Biol Chem. 2003;278(49):48872–48879. | ||
Lee YH, Coonrod SA, Kraus WL, Jelinek MA, Stallcup MR. Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc Natl Acad Sci U S A. 2005;102(10):3611–3616. | ||
Tanikawa C, Ueda K, Nakagawa H, Yoshida N, Nakamura Y, Matsuda K. Regulation of protein citrullination through p53/PADI4 network in DNA damage response. Cancer Res. 2009;69(22):8761–8769. | ||
Guo Q, Fast W. Citrullination of inhibitor of growth 4 (ING4) by peptidylarginine deminase 4 (PAD4) disrupts the interaction between ING4 and p53. J Biol Chem. 2011;286:17069–17078. | ||
Chang X, Yamada R, Sawada T, Suzuki A, Kochi Y, Yamamoto K. The inhibition of antithrombin by peptidylarginine deiminase 4 may contribute to pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2005;44(3):293–298. | ||
Ordóñez A, Yélamos J, Pedersen S, et al. Increased levels of citrullinated antithrombin in plasma of patients with rheumatoid arthritis and colorectal adenocarcinoma determined by a newly developed ELISA using a specific monoclonal antibody. Thromb Haemost. 2010;104(6):1143–1149. |
Supplementary materials
 | Table S1 The primer sequences were the following |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.