")
Back to Journals » The Application of Clinical Genetics » Volume 11
Pena-Shokeir syndrome: current management strategies and palliative care
Authors Adam S , Coetzee M , Honey EM
Received 31 May 2018
Accepted for publication 19 July 2018
Published 25 October 2018 Volume 2018:11 Pages 111—120
DOI https://doi.org/10.2147/TACG.S154643
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Sumaiya Adam,1 Melantha Coetzee,2 Engela Magdalena Honey3
1Department of Obstetrics and Gynaecology, Steve Biko Academic Hospital, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa; 2Division of Neonatology, Department of Pediatrics and Child Health, Steve Biko Academic Hospital, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa; 3Department of Biochemistry, Genetics and Microbiology, Faculty of Natural and Agricultural Sciences, University of Pretoria, Pretoria, South Africa
Abstract: Pena–Shokeir syndrome (PSS) type 1, also known as fetal akinesia deformation sequence, is a rare genetic syndrome that almost always results in intrauterine or early neonatal death. It is characterized by markedly decreased fetal movements, intrauterine growth restriction, joint contractures, short umbilical cord, and features of pulmonary hypoplasia. Antenatal diagnosis can be difficult. Ultrasound features are varied and may overlap with those of Trisomy 18. The poor prognosis of PSS is due to pulmonary hypoplasia, which is an important feature that distinguishes PSS from arthrogryposis multiplex congenital without pulmonary hypoplasia, which has a better prognosis. If diagnosed in the antenatal period, a late termination of pregnancy can be considered following ethical discussion (if the law allows). In most cases, a diagnosis is only made in the neonatal period. Parents of a baby affected with PSS require detailed counseling that includes information on the imprecise recurrence risks and a plan for subsequent pregnancies.
Keywords: fetal akinesia deformation sequence, ultrasound, comfort care
Introduction
Pena–Shokeir syndrome (PSS) is a lethal form of multiple congenital contractures with autosomal recessive inheritance implicated in 50% of cases.1 However, heterogeneity makes recurrence risk calculations difficult.
Two types of PSS have been described:
- Type 1: It is a fetal akinesia/hypokinesia sequence that is characterized by multiple joint contractures, facial anomalies, and pulmonary hypoplasia. It has a prevalence of <1/1,000,000 births, with an autosomal recessive mode of inheritance.2 About 100 cases have been described in the literature, with about 30% of fetuses dying in utero, and the vast majority of neonates succumbing in the early neonatal period due to pulmonary hypoplasia.
- Type 2: It is also known as cerebro-oculo-facio-skeletal (COFS) syndrome. It is a rapidly progressive neurological disorder resulting in brain atrophy, characterized by intracerebral calcifications, cataracts, microcornea, optic atrophy, progressive joint contractures, and growth failure. It is a very rare autosomal recessive disorder caused by mutations in ERCC6/CSB, although mutations in ERCC1, ERCC2, and ERCC5 have been linked to some cases. Death usually occurs by 5 years of age.3
Further discussion in this review will be restricted to PSS type 1.
Pathogenesis
PSS is a heterogeneous group of disorders characterized by a decrease or absence of intrauterine fetal movement. It is also called “fetal akinesia deformation sequence” (FADS). Arthrogryposis multiplex congenital (AMC), multiple pterygium syndrome (MPS), and lethal congenital contracture syndromes are conditions that have overlapping phenotypes and etiologies. The clinical phenotype initially described by Pena and Shokeir (1974, 1976) included camptodactyly; multiple contractures; facial anomalies consisting of a high nasal bridge, micrognathia, and a cleft palate; and pulmonary hypoplasia resulting in death in utero or shortly thereafter.4,5 The initial description included multiple consanguineous families, and an autosomal recessive inheritance pattern was suggested. Sporadic and familial occurrences have been described subsequently depending on the underlying etiology. This has led to the identification of up to 30 different subgroups.6,7 X-linked dominant inheritance has also been suggested.8,9
The underlying etiology is dysfunction of the neuromuscular system resulting in decreased intrauterine fetal movements. It includes the brain and spinal cord, motor neuron, neuromuscular junction, and neurotransmitter defects.6 Muscle myopathic changes leading to the failure of development of normal mature muscle, including dystrophies and dysplasias, have also been implicated.7 Other causes include connective tissue abnormalities such as chondrodysplasias, conditions associated with joint limitation and laxity, as well as restricted skin (restrictive dermopathy).7 Environmental causes include antibodies to neurotransmitters and fetal acetylcholine receptor (AChR), as well as reduced intrauterine space such as that seen in multiple births, oligohydramnios, and uterine abnormalities. The earlier the effect, the more severe the phenotype. Maternal illness and drug use, as well as fetal ischemia, have also been described in isolated cases.6
A possible relation to maternal myasthenia gravis was described in 1994. Maternal anti-AChR antibody titers were increased in the absence of any clinical symptoms of myasthenia gravis in the mothers. In these cases, the recurrence risk was very high and all subsequent pregnancies were affected.10,11 Other case reports have confirmed this and have suggested that the antibodies were different from those usually associated with myasthenia gravis. Adult anti-AChR antibodies replace fetal anti-AChR by 33 weeks of gestation. This explains why the fetus is markedly affected compared with their mothers.10,12
Genetics
Advances in molecular genetic research have improved our knowledge of the genetic causes of PSS and suggest that many cases are at the severe end of the spectrum of other recognized conditions involving the neuromuscular system. Alternatively, autosomal dominant neuromuscular conditions can present with PSS when the gene defect is in the homozygous state.7
Variations in the genes involved in the neuromuscular pathways for fetal movement cause PSS. Variations in RAPSN (Online Mendelian Inheritance in Man [OMIM] 601592), DOK7 (OMIM 610285), and MUSK (OMIM 601296) are also implicated in congenital myasthenic syndrome (CMS), but have been identified as some of the major genes involved in the etiology of PSS through deficient interaction in the neuromuscular junction.
RAPSN
RASPN is located on chromosome 11p11.2. It codes for a postsynaptic protein (rapsyn) that links the AChR to the agrin-binding dystrophin-associated glycoprotein complex and stabilizes the AChR at the neuromuscular junction.13 Homozygosity or compound heterozygosity for mutations results in AChR deficiency and FADS or CMS. Michalk et al reported the case of a brother and a sister in a non-consanguineous Pakistani family.14 Both siblings were born with severe respiratory problems, contractures, and subtle dysmorphisms. The male infant died at 10 months due to respiratory failure. He also had cryptorchidism. The female infant had a cleft palate but was still alive at 10 months. They both had compound heterozygous mutations of c.416T→C/c.566C→T. Vogt et al reported a homozygous mutation of c.1177_1178delAA in a consanguineous family (monozygotic twin males and a female).15 All the affected fetuses were terminated and showed FADS upon examination after delivery. A homozygous c.484G>A (p.Glu162Lys) variant was identified by Winters et al in consanguineous parents in their fifth pregnancy. Their fourth pregnancy was also affected, but they refused testing during the previous pregnancy.16
DOK7
DOK7 is involved in AChR clustering in synaptogenesis and binds with and phosphorylates MUSK. It is located at 4p16.3. In a consanguineous family with three affected children, Vogt et al identified a homozygous splice-site mutation (IVS3+1G>T or c.331+1G>T) in DOK7 causing lethal FADS.17 DOK7 variations are usually associated with CMS and with limb girdle weakness.18 This model illustrates that a partial loss of DOK7 function causes CMS but a complete loss of DOK7 function causes a lethal FADS phenotype.
MUSK
MUSK is located at 9q31.3. It forms and maintains the neuromuscular junction. Muscle cells produce and express AChR. Agrin, a proteoglycan released by motor nerves, binds to LRP4 and activates MUSK, which then signals via DOK7 and RAPSN to stabilize the AChR.19 Wilbe et al reported that a complete loss of MUSK causes autosomal recessive FADS.20 A homozygous frameshift mutation (c.40dupA) in exon 1 leading to a premature stop codon was found in a non-consanguineous Swedish family with five affected fetuses. The mutation was identified through whole-exome sequencing followed by Sanger sequencing. In the same year, Tan-Sindhunata et al identified a homozygous Dutch family with FADS.21
Other variations in the genes involved in the neuromuscular pathways for fetal movement can cause PSS. There are, however, a large percentage of unknown cases. Next-generation sequencing (NGS) technologies will improve the diagnosis and enable accurate prognosis prediction, recurrence risk estimation, as well as genetic counseling. NGS panels are being developed to investigate heterogeneous conditions such as PSS where variants in different genes are causative. Whole-exome sequencing earlier in the diagnostic process will be a useful tool to identify new genes not previously described.
Details on the anatomical areas involved, genes (Mendelian Inheritance in Man [MIM] number), inheritance pattern, phenotype (MIM number), and references of all known gene variations associated with FADS are given in Table 1.
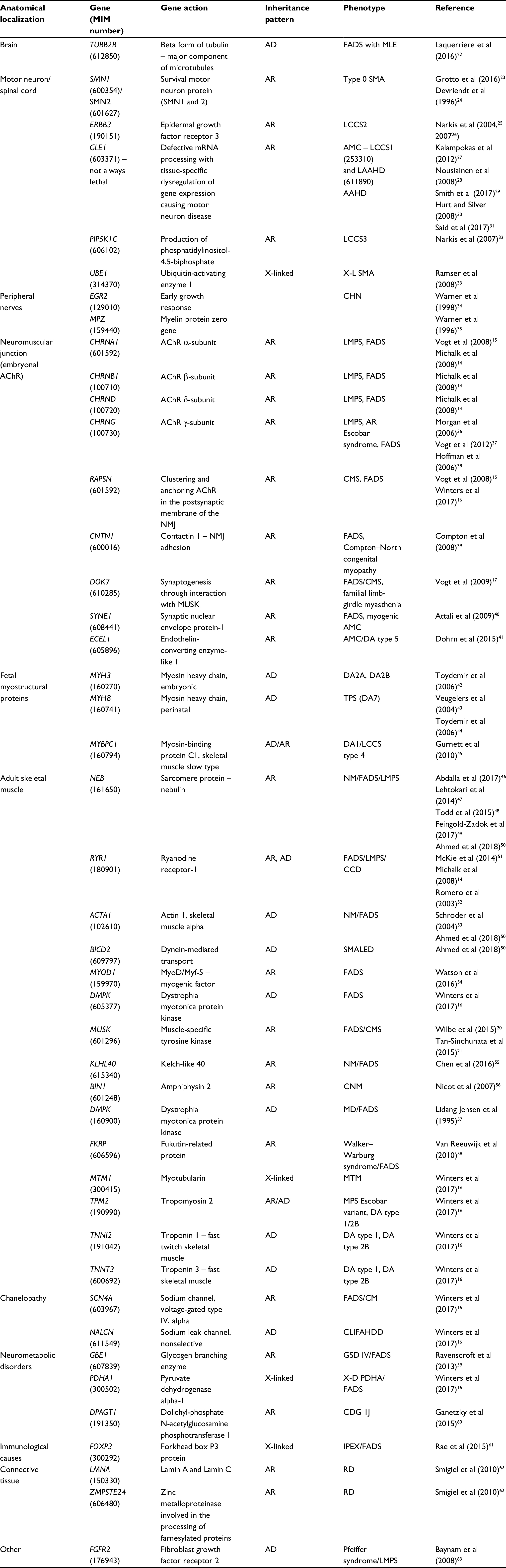 | Table 1 Summary of the genetic causes of Pena–Shokeir phenotype Abbreviations: MIM, Mendelian Inheritance in Man; AD, autosomal dominant; FADS, fetal akinesia deformation sequence; MLE, microlissencephaly; AR, autosomal recessive; SMA, spinal muscular atrophy; LCCS, lethal congenital contracture syndrome; AMC, arthrogryposis multiplex congenital; LAAHD, lethal arthrogryposis with anterior horn cell disease; AAHD, arthrogryposis with anterior horn cell disease; X-L SMA, X-linked spinal muscular atrophy (301830); CHN, congenital hypomyelination neuropathy; AChR, acetylcholine receptor; LMPS, lethal multiple pterygium syndrome; NMJ, neuromuscular junction; CMS, congenital myasthenic syndrome; DA, distal arthrogryposis; TPS, trismus-pseudocamptodactyly syndrome; NM, nemalin myopathy; CCD, central core disease; SMALED, autosomal dominant spinal muscular atrophy with lower extremity predominance; CNM, centronuclear myopathy; MD, myotonic dystrophy; MTM, myotubular myopathy; MPS, multiple pterygium syndrome; CM, congenital myopathy; CLIFAHDD, congenital contractures of the limbs and face with hypotonia and developmental delay; GSD, glycogen storage disease; X-D PDHA, X-linked dominant pyruvate dehydrogenase E1-alpha deficiency; CDG IJ, Congenital disorder of glycosylation type Ij; IPEX, immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome; RD, rare disease; FADS, fetal akinesia deformation sequence; AMC, arthrogryposis multiplex congenital; DA, distal arthrogryposis. |
Antenatal diagnosis
It is not easy to diagnose PSS as it has a variable phenotype and shares many ultrasonographic features with Trisomy 18 and other syndromes (Table 2).6
 | Table 2 Differential diagnosis of conditions with markedly decreased intrauterine fetal movements6 |
The presence of pulmonary hypoplasia is helpful in distinguishing PSS from AMC which has a better prognosis. Invasive testing for chromosomal analysis is advised when a diagnosis is suspected. If the karyotype is normal, and other conditions have been excluded, a presumptive diagnosis of PSS can be made.
Antenatal diagnosis of PSS can be made on ultrasound as early as 12 weeks of gestation if decreased intrauterine fetal movements, fetal edema, and fixed limb posturing are observed. Polyhydramnios is invariably present in the latter half of pregnancy, with a proportion of fetuses developing hydrops fetalis, and becoming more prone to death in utero.
Antenatal ultrasonographic features include decreased fetal movements, intrauterine growth restriction, joint contractures, short umbilical cord, and features of pulmonary hypoplasia. Limbs may be in contraction or extension – knees are usually extended and elbows are flexed. Feet may have severe equinovarus or rocker bottom deformity. Facial features include hypertelorism, low-set ears, depressed tip of the nose, and micrognathia. A disproportionately large head in relation to the body is usually described.6 Three-dimensional ultrasound and fetal magnetic resonance imaging (MRI) may be considered as an adjunct to fetal imaging.64
Obstetric management
The ideal way to convey bad news to a patient remains controversial. Adequate counseling often requires multiple consultations with various health care professionals, including maternal and fetal specialists, geneticists, and neonatologists. The consultation process should include the patient’s partner or family who can assist with the decision-making process, as well as offering emotional support. Counseling should include information on the diagnosis and prognosis (antenatal, intrapartum, postnatal, and long term), and information regarding future pregnancies.65,66
The fetus diagnosed with PSS has a poor prognosis. Close to term, the obstetric management would usually continue to be supportive, with delivery via cesarean section reserved for obstetric indications. The patient should be counseled that no intrapartum fetal heart rate monitoring to be carried out due to the poor prognosis, and that the neonate will be evaluated after birth by a neonatologist and geneticist. Palliative care following secondary postnatal assessment should be discussed with the patient. Due to the dismal prognosis of PSS, late termination of pregnancy can be offered as a management option in countries/states where it is legal and acceptable.
Postnatal care
PSS must be differentiated from other causes of FADS as the level of care offered after delivery may be dependent on the expected prognosis. If the diagnosis of PSS is made during the antenatal period, the parents may choose to terminate the pregnancy. Alternatively, the patient may opt for comfort care postdelivery or may choose full resuscitative intervention postdelivery. However, not all patients will have an antenatal diagnosis, and decision making regarding lifesaving interventions in the immediate postdelivery period may be difficult.
The postnatal diagnosis of PSS can be made when there is a combination of prenatal-onset growth restriction, multiple ankyloses (elbows, knees, hips, and ankles), typical facial features, and pulmonary hypoplasia.67 Additional findings may include camptodactyly, absent palmar creases, rocker bottom feet, talipes equinovarus, cryptorchidism, short neck, small mouth, and a cleft palate.67 The craniofacial abnormalities, skeletal abnormalities, and pulmonary hypoplasia are all secondary to decreased or absent fetal movement in utero.68–70 The differential diagnosis includes Trisomy 18, MPS, and COFS. A normal karyotype, and the absence of pterygia causing flexion contractures, microphthalmia, and microcephaly allow differentiation of PSS from Trisomy 18, MPS, and COFS.71,72
Despite the poor survival rate, most patients with PSS receive complete resuscitation after delivery and full postnatal care, including antimicrobials, inotropes/vasopressors, and repeated cardiopulmonary resuscitation until death.69 There are, however, cases describing redirection of care from life-sustaining treatment (invasive ventilation) to comfort care after discussion with the parents.72,73
If full resuscitative efforts are agreed upon, the resuscitation team must anticipate that the newborn will have respiratory distress secondary to pulmonary hypoplasia and be prepared for a difficult intubation74 due to edema of the head,75,76 a short neck,76 and micrognathia.75,76 Various airway devices must be available, including a face mask, oral and nasal airways, laryngeal mask airways, and endotracheal tubes with the addition of a fiber optic or video laryngoscope, if possible.2
No specific treatment is available for those with PSS, and management is largely supportive.77 A karyotype should be done on all neonates with this phenotype to exclude Trisomy 18 as the two conditions share many common phenotypic features.73,76,78 For those undergoing continued care, prolonged mechanical ventilation can be expected and a tracheostomy may have to be considered if extubation is not possible.74 In addition, pulmonary hypertension may be present and may require treatment with sildenafil or iloprost to decrease the pulmonary pressures.74 Echocardiography should be performed to exclude congenital cardiac abnormalities.79,80
As PSS may be associated with central nervous system abnormalities,72,77 it is recommended to perform an MRI of the brain and to screen for endocrine abnormalities such as central hypothyroidism.74 The presence of cerebral malformations may also increase the risk of seizures, which require anticonvulsive therapy.69,73,75 These patients may also sustain fractures postnatally as the bones are hypoplastic with decreased calcification.78
Enteral feeding is often delayed due to short-gut syndrome67,70 and intestinal malrotation,79 necessitating prolonged parenteral nutrition. In addition, oral feeding may be impossible due to impaired swallowing,80 and patients may require the surgical placement of a feeding gastrostomy tube.
Because of the wide array of physical abnormalities, a multidisciplinary approach is appropriate for those patients surviving the neonatal period who may require repeated surgical procedures for various craniofacial and skeletal deformities. Members of the team should include a pediatrician/neonatologist, occupational therapist, physiotherapist, speech therapist, and surgeons from various disciplines, including pediatric surgery, orthopedic surgery, maxillofacial surgery, and ENT surgery.
Patients with PSS are at an increased risk of malignant hyperthermia and bronchospasm during anesthesia. However, the use of sevoflurane for induction and maintenance, and rocuronium or vecuronium for muscle relaxation has been successful without adverse events reported in many patients.74,75
Prognosis
Although the ultimate prognosis of PSS is dependent on the underlying cause, this condition has been described as almost uniformly lethal, with 30% of fetuses being stillborn and live-born infants usually dying within the first month of life.68,72,77 However, survival beyond 12 months of age has been described, indicating that early mortality is not always inevitable. Of interest is a 9-year-old girl who underwent repeated surgical procedures for skeletal deformities77 and a 21-year-old pregnant female who underwent cesarean section at 38 weeks of gestation with a normal fetus.81 Early death is usually a result of primary cerebral malformations68 or acute respiratory failure secondary to pulmonary hypoplasia.68,72 The degree of lung hypoplasia and the ability of the lungs to sustain life are dependent on the timing of onset, with early-onset akinesia associated with severe hypoplasia and demise.78 After death, it is essential to request a postmortem as this will help describe the features of this rare condition even further.69
Subsequent pregnancies
Patients who have delivered a fetus with PSS should have close fetal surveillance in subsequent pregnancies. This will allow the early recognition of anomalies. However, since the phenotypic condition is derived from heterogeneous causes, the counseling and recurrence risk calculation are imprecise. It has been estimated that the recurrence risk varies between 0% and 25%.82
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. SA was responsible for the introduction, differential diagnosis, and obstetric management sections, and compiled the final manuscript. EMH was responsible for sections on pathophysiology and genetics. MC was responsible for sections on postnatal care and prognosis.
Disclosure
The authors report no conflicts of interest in this work.
References
Kowalczyk B, Felus´ J. Arthrogryposis: an update on clinical aspects, etiology, and treatment strategies. Arch Med Sci. 2016;12(1):10–24. | ||
OrphanAnesthesia. Anesthesia recommendations for patients suffering from Pena-Shokeir syndrome. Available from: www.orphananesthesia.eu. Accessed on May 9, 2018. | ||
Orphanet. Pena Shokeir syndrome type 2. Available from: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=1649&Disease_Disease_Search_diseaseType=ORPHA&Disease_Disease_Search_diseaseGroup=1466&Disease(s)/group%20of%20diseases=Pena-Shokeir-syndrome-type-2&title=Pena-Shokeir- syndrome-type-2. Accessed September 10, 2018. | ||
Pena SD, Shokeir MH. Syndrome of camptodactyly, multiple ankyloses, facial anomalies, and pulmonary hypoplasia: a lethal condition. J Pediatr. 1974;85(3):373–375. | ||
Pena SD, Shokeir MH. Syndrome of camptodactyly, multiple ankyloses, facial anomalies and pulmonary hypoplasia – further delineation and evidence for autosomal recessive inheritance. Birth Defects Orig Artic Ser. 1976;12(5):201–208. | ||
Hall JG. Pena-Shokeir phenotype (fetal akinesia deformation sequence) revisited. Birth Defects Res A Clin Mol Teratol. 2009;85(8):677–694. | ||
Ravenscroft G, Sollis E, Charles AK, North KN, Baynam G, Laing NG. Fetal akinesia: review of the genetics of the neuromuscular causes. J Med Genet. 2011;48(12):793–801. | ||
Tolmie JL, Patrick A, Yates JR. A lethal multiple pterygium syndrome with apparent X-linked recessive inheritance. Am J Med Genet. 1987;27(4):913–919. | ||
McKeown CM, Harris R. An autosomal dominant multiple pterygium syndrome. J Med Genet. 1988;25(2):96–103. | ||
Brueton LA, Huson S, Thompson E, et al. Myasthenia gravis: an important cause of the Pena-Shokeir phenotype. J. Med Genet. 1994;31:167. | ||
Brueton LA, Huson SM, Cox PM, et al. Asymptomatic maternal myasthenia as a cause of the Pena-Shokeir phenotype. Am J Med Genet. 2000;92:1–6. | ||
Hesselmans LF, Jennekens FG, Van den Oord CJ, Veldman H, Vincent A. Development of innervation of skeletal muscle fibers in man: relation to acetylcholine receptors. Anat Rec. 1993;236(3):553–562. | ||
Apel ED, Roberds SL, Campbell KP, Merlie JP. Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex. Neuron. 1995;15(1):115–126. | ||
Michalk A, Stricker S, Becker J, et al. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am J Hum Genet. 2008;82(2):464–476. | ||
Vogt J, Harrison BJ, Spearman H, et al. Mutation analysis of CHRNA1, CHRNB1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am J Hum Genet. 2008;82(1):222–227. | ||
Winters L, Van Hoof E, De Catte L, et al. Massive parallel sequencing identifies RAPSN and PDHA1 mutations causing fetal akinesia deformation sequence. Eur J Paediatr Neurol. 2017;21(5):745–753. | ||
Vogt J, Morgan NV, Marton T, et al. Germline mutation in DOK7 associated with fetal akinesia deformation sequence. J Med Genet. 2009;46(5):338–340. | ||
Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313(5795):1975–1978. | ||
Witzemann V. Development of the neuromuscular junction. Cell Tissue Res. 2006;326(2):263–271. | ||
Wilbe M, Ekvall S, Eurenius K, et al. MuSK: a new target for lethal fetal akinesia deformation sequence (FADS). J Med Genet. 2015;52(3):195–202. | ||
Tan-Sindhunata MB, Mathijssen IB, Smit M, et al. Identification of a Dutch founder mutation in MUSK causing fetal akinesia deformation sequence. Eur J Hum Genet. 2015;23(9):1151–1157. | ||
Laquerriere A, Gonzales M, Saillour Y, et al. De novo TUBB2B mutation causes fetal akinesia deformation sequence with microlissencephaly: An unusual presentation of tubulinopathy. Eur J Med Genet. 2016;59(4):249–256. | ||
Grotto S, Cuisset JM, Marret S, et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J Neuromuscul Dis. 2016;3(4):487–495. | ||
Devriendt K, Lammens M, Schollen E, et al. Clinical and molecular genetic features of congenital spinal muscular atrophy. Ann Neurol. 1996;40(5):731–738. | ||
Narkis G, Landau D, Manor E, et al. Homozygosity mapping of lethal congenital contractural syndrome type 2 (LCCS2) to a 6 cM interval on chromosome 12q13. Am J Med Genet A. 2004;130A(3):272–276. | ||
Narkis G, Ofir R, Manor E, Landau D, Elbedour K, Birk OS. Lethal congenital contractural syndrome type 2 (LCCS2) is caused by a mutation in ERBB3 (Her3), a modulator of the phosphatidylinositol-3-kinase/Akt pathway. Am J Hum Genet. 2007;81(3):589–595. | ||
Kalampokas E, Kalampokas T, Sofoudis C, Deligeoroglou E, Botsis D. Diagnosing arthrogryposis multiplex congenita: a review. ISRN Obstet Gynecol. 2012;2012:264918. | ||
Nousiainen HO, Kestilä M, Pakkasjärvi N, et al. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40(2):155–157. | ||
Smith C, Parboosingh JS, Boycott KM, et al. Expansion of the GLE1-associated arthrogryposis multiplex congenita clinical spectrum. Clin Genet. 2017;91(3):426–430. | ||
Hurt JA, Silver PA. mRNA nuclear export and human disease. Dis Model Mech. 2008;1(2–3):103–108. | ||
Said E, Chong JX, Hempel M, et al. Survival beyond the perinatal period expands the phenotypes caused by mutations in GLE1. Am J Med Genet A. 2017;173(11):3098–3103. | ||
Narkis G, Ofir R, Landau D, et al. Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKI gamma of the phophatidylinositol pathway. Am J Hum Genet. 2007;81(3):530–539. | ||
Ramser J, Ahearn ME, Lenski C, et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am J Hum Genet. 2008;82(1):188–193. | ||
Warner LE, Mancias P, Butler IJ, et al. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet. 1998;18(4):382–384. | ||
Warner LE, Hilz MJ, Appel SH, et al. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron. 1996;17(3):451–460. | ||
Morgan NV, Brueton LA, Cox P, et al. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am J Hum Genet. 2006;79(2):390–395. | ||
Vogt J, Morgan NV, Rehal P, et al. CHRNG genotype-phenotype correlations in the multiple pterygium syndromes. J Med Genet. 2012;49(1):21–26. | ||
Hoffmann K, Muller JS, Stricker S, et al. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am J Hum Genet. 2006;79(2):303–312. | ||
Compton AG, Albrecht DE, Seto JT, et al. Mutations in contactin-1, a neural adhesion and neuromuscular junction protein, cause a familial form of lethal congenital myopathy. Am J Hum Genet. 2008;83(6):714–724. | ||
Attali R, Warwar N, Israel A, et al. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum Mol Genet. 2009;18(18):3462–3469. | ||
Dohrn N, Le VQ, Petersen A, et al. ECEL1 mutation causes fetal arthrogryposis multiplex congenita. Am J Med Genet A. 2015;167A(4):731–743. | ||
Toydemir RM, Rutherford A, Whitby FG, et al. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat Genet. 2006;38(5):561–565. | ||
Veugelers M, Bressan M, McDermott DA, et al. Mutation of perinatal myosin heavy chain associated with a Carney complex variant. N Engl J Med. 2004;351(5):460–469. | ||
Toydemir RM, Chen H, Proud VK, et al. Trismus-pseudocamptodactyly syndrome is caused by recurrent mutation of MYH8. Am J Med Genet A. 2006;140(22):2387–2393. | ||
Gurnett CA, Desruisseau DM, McCall K, et al. Myosin binding protein C1: a novel gene for autosomal dominant distal arthrogryposis type 1. Hum Mol Genet. 2010;19(7):1165–1173. | ||
Abdalla E, Ravenscroft G, Zayed L, Beecroft SJ, Laing NG. Lethal multiple pterygium syndrome: A severe phenotype associated with a novel mutation in the nebulin gene. Neuromuscul Disord. 2017;27(6):537–541. | ||
Lehtokari VL, Kiiski K, Sandaradura SA, et al. Mutation update: the spectra of nebulin variants and associated myopathies. Hum Mutat. 2014;35(12):1418–1426. | ||
Todd EJ, Yau KS, Ong R, et al. Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet J Rare Dis. 2015;10:148. | ||
Feingold-Zadok M, Chitayat D, Chong K, et al. Mutations in the NEB gene cause fetal akinesia/arthrogryposis multiplex congenita. Prenat Diagn. 2017;37(2):144–150. | ||
Ahmed AA, Skaria P, Safina NP, et al. Arthrogryposis and pterygia as lethal end manifestations of genetically defined congenital myopathies. Am J Med Genet A. 2018;176(2):359–367. | ||
McKie AB, Alsaedi A, Vogt J, et al. Germline mutations in RYR1 are associated with foetal akinesia deformation sequence/lethal multiple pterygium syndrome. Acta Neuropathol Commun. 2014;2(1):148. | ||
Romero NB, Monnier N, Viollet L, et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain. 2003;126(Pt 11):2341–2349. | ||
Schroder JM, Durling H, Laing N. Actin myopathy with nemaline bodies, intranuclear rods, and a heterozygous mutation in ACTA1 (Asp154Asn). Acta Neuropathol. 2004;108(3):250–256. | ||
Watson CM, Crinnion LA, Murphy H, et al. Deficiency of the myogenic factor MyoD causes a perinatally lethal fetal akinesia. J Med Genet. 2016;53(4):264–269. | ||
Chen TH, Tian X, Kuo PL, Pan HP, Wong LC, Jong YJ. Identification of KLHL40 mutations by targeted next-generation sequencing facilitated a prenatal diagnosis in a family with three consecutive affected fetuses with fetal akinesia deformation sequence. Prenat Diagn. 2016;36(12):1135–1138. | ||
Nicot AS, Toussaint A, Tosch V, Pan HP, Wong LC, Jong YJ. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39(9):1134–1139. | ||
Lidang Jensen M, Rix M, Schroder HD, Teglbjaerg PS, Ebbesen F. Fetal akinesia-hypokinesia deformation sequence (FADS) in 2 siblings with congenital myotonic dystrophy. Clin Neuropathol. 1995;14(2):105–108. | ||
Van Reeuwijk J, Olderode-Berends MJ, Van den Elzen C, et al. A homozygous FKRP start codon mutation is associated with Walker-Warburg syndrome, the severe end of the clinical spectrum. Clin Genet. 2010;78(3):275–281. | ||
Ravenscroft G, Thompson EM, Todd EJ, et al. Whole exome sequencing in foetal akinesia expands the genotype-phenotype spectrum of GBE1 glycogen storage disease mutations. Neuromuscul Disord. 2013;23(2):165–169. | ||
Ganetzky R, Izumi K, Edmondson A, et al. Fetal akinesia deformation sequence due to a congenital disorder of glycosylation. Am J Med Genet A. 2015;167A(10):2411–2417. | ||
Rae W, Gao Y, Bunyan D, et al. A novel FOXP3 mutation causing fetal akinesia and recurrent male miscarriages. Clin Immunol. 2015;161(2):284–285. | ||
Smigiel R, Jakubiak A, Esteves-Vieira V, et al. Novel frameshifting mutations of the ZMPSTE24 gene in two siblings affected with restrictive dermopathy and review of the mutations described in the literature. Am J Med Genet A. 2010;152A(2):447–452. | ||
Baynam G, Smith N, Goldblatt J. A c.1019A > G mutation in FGFR2, which predicts p.Tyr340Cys, in a lethally malformed fetus with Pfeiffer syndrome and multiple pterygia. Am J Med Genet A. 2008;146A(17):2301–2303. | ||
Tomai XH, Jasmine TX, Phan TH. Antenatal ultrasonography findings and magnetic resonance imaging in a case of Pena-Shokeir phenotype. Ultrasound. 2017;25(2):115–119. | ||
Louhiala P, Launis V. Directive or non-directive genetic counselling – Cutting through the surface. Int J Commun Health. 2013;2:28–33. | ||
Wilkinson D, de Crespigny L, Xafis V. Ethical language and decision-making for prenatally diagnosed lethal malformations. Semin Fetal Neonatal Med. 2014;19(5):306–311. | ||
Jones KL, Jones MC, Del Camp Casanelles M. Smiths Recognizable Patterns of Human Malformation: Expert Consult. Philadelphia: Saunders; 1997. | ||
Parlakgumus HA, Tarim E, Kucukgoz U. Fetal akinesia / hypokinesia deformation sequence (FADS): two and three dimensional ultrasound presentation. Turkiye Klinikleri J Gynecol Obst. 2008;18(5):336–339. | ||
Warda A, Abduljabbar H, Ghafouri H, Basalamah AH. Pena-Shokeir phenotype: a lethal pattern of multiple congenital anomalies. Ann Saudi Med. 1991;11(3):264–266. | ||
Adam S, Lombaard H, Spencer C. Discordant monoamniotic twins with Pena-Shokeir phenotype. Clin Case Rep. 2016;4(10):919–921. | ||
Paladini D, Tartaglione A, Agangi A, Foglia S, Martinelli P, Nappi C. Pena-Shokeir phenotype with variable onset in three consecutive pregnancies. Ultrasound Obstet Gynecol. 2001;17(2):163–165. | ||
Kho N, Czarnecki L, Kerrigan JF, Coons S. Pena-Shokier phenotype: case presentation and review. J Child Neurol. 2002;17(5):397–399. | ||
Knudtson EJ, Lorenz LB, Wilson PL, McDaniel B, Mulvihill JJ, Dannaway DC. Tonic-clonic seizures in a fetus with Pena-Shokeir syndrome. J Ultrasound Med. 2009;28(8):1121–1122. | ||
Aydin G, Gencay I, Colak S. Anesthesia management of a newborn with Pena-Shokeir Syndrome. J Clin Anesth. 2018;45:71–72. | ||
Tsujikawa S, Okutani R, Tsujii K, Oda Y. Anesthetic management of three pediatric cases with Pena-Shokeir syndrome. J Anesth. 2012;26(3):445–448. | ||
Anand DB, Suresh KP, Janak D, et al. Rare case of fetal akinesia deformation sequence (FADS): antenatal diagnosis at 13th weeks of gestation. Austin Gynecol Case Rep. 2017;2(2):1016. | ||
Boesen PV, French CE. Acute respiratory distress in Pena-Shokeir syndrome. Ear Nose Throat J. 2004;83(11):772–773. | ||
Hammond E, Donnenfeld AE. Fetal akinesia. Obstet Gynecol Surv. 1995;50(3):240–249. | ||
Chen CP. Prenatal diagnosis and genetic analysis of fetal akinesia deformation sequence and multiple pterygium syndrome associated with neuromuscular junction disorders: a review. Taiwan J Obstet Gynecol. 2012;51(1):12–17. | ||
Eguiliz I, Barber MA, Martin A, Plasencia W, Arencibia O. Fetal akinesia deformation sequence. Pena-Shokeir type 1 syndrome. New features of an un-common condition. J Obstet Gynaecol. 2009;26(8):818–820. | ||
Manimekalai N, Wasiluk I, Panni MK. Cesarean Section in an Obstetric Patient with Pena-Shokeir Syndrome Type-1, with Partially Corrected Scoliosis with Spinal Rod Placement and History of Malignant Hyperthermia. J Anesthe Clinic Res. 2013;4:315. | ||
Santana EF, Oliveira Serni PN, Rolo LC, Araujo Júnior E. Prenatal Diagnosis of Arthrogryposis as a Phenotype of Pena-Shokeir Syndrome using Two- and Three-dimensional Ultrasonography. J Clin Imaging Sci. 2014;4:20. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.