Back to Journals » International Journal of Nanomedicine » Volume 12
PEG-b-(PELG-g-PLL) nanoparticles as TNF-α nanocarriers: potential cerebral ischemia/reperfusion injury therapeutic applications
Authors Xu G ![]() , Gu H, Hu B
, Gu H, Hu B ![]() , Tong F
, Tong F ![]() , Liu D, Yu X, Zheng Y
, Liu D, Yu X, Zheng Y ![]() , Gu J
, Gu J
Received 21 December 2016
Accepted for publication 3 March 2017
Published 23 March 2017 Volume 2017:12 Pages 2243—2254
DOI https://doi.org/10.2147/IJN.S130842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Guangtao Xu,1,2 Huan Gu,1,3 Bo Hu,4 Fei Tong,2 Daojun Liu,1 Xiaojun Yu,1 Yongxia Zheng,1,2 Jiang Gu1
1Department of Pathology and Chemistry, Provincial Key Laboratory of Infectious Diseases and Immunopathology, Collaborative and Creative Center, Molecular Diagnosis and Personalized Medicine, Shantou University Medical College, Shantou, Guangdong, 2Department of Pathology, Provincial Key Discipline of Pharmacology, Jiaxing University Medical College, Jiaxing, Zhejiang, People’s Republic of China; 3Department of Physics, University of Maryland, College Park, Annapolis, MD, USA; 4Department of Chemical Pathology, Jiaxing Hospital of Traditional Chinese Medicine, Zhejiang Chinese Medical University, Jiaxing, Zhejiang, People’s Republic of China
Abstract: Brain ischemia/reperfusion (I/R) injury (BI/RI) is a leading cause of death and disability worldwide. However, the outcome of pharmacotherapy for BI/RI remains unsatisfactory. Innovative approaches for enhancing drug sensitivity and recovering neuronal activity in BI/RI treatment are urgently needed. The purpose of this study was to evaluate the protective effects of tumor necrosis factor (TNF)-α-loaded poly(ethylene glycol)-b-(poly(ethylenediamine l-glutamate)-g-poly(l-lysine)) (TNF-α/PEG-b-(PELG-g-PLL)) nanoparticles on BI/RI. The particle size of PEG-b-(PELG-g-PLL) and the loading and release rates of TNF-α were determined. The nanoparticle cytotoxicity was evaluated in vitro using rat cortical neurons. Sprague Dawley rats were preconditioned with free TNF-α or TNF-α/PEG-b-(PELG-g-PLL) polyplexes and then subjected to 2 hours ischemia and 22 hours reperfusion. Brain edema was assessed using the brain edema ratio, and the antioxidative activity was assessed by measuring the superoxide dismutase (SOD) activity and the malondialdehyde (MDA) content in the brain tissue. We further estimated the inflammatory activity and apoptosis level by determining the levels of interleukin-4 (IL-4), IL-6, IL-8, IL-10, and nitric oxide (NO), as well as the expression of glial fibrillary acidic protein (GFAP), intercellular adhesion molecule-1 (ICAM-1), and cysteine aspartase-3 (caspase-3), in the brain tissue. We provide evidence that TNF-α preconditioning attenuated the oxidative stress injury, the inflammatory activity, and the apoptosis level in I/R-induced cerebral injury, while the application of block copolymer PEG-b-(PELG-g-PLL) as a potential TNF-α nanocarrier with sustained release significantly enhanced the bioavailability of TNF-α. We propose that the block copolymer PEG-b-(PELG-g-PLL) may function as a potent nanocarrier for augmenting BI/RI pharmacotherapy, with unprecedented clinical benefits. Further studies are needed to better clarify the underlying mechanisms.
Keywords: PEG-b-(PELG-g-PLL), TNF-α, ischemia/reperfusion, brain
Introduction
The number of people who experience cerebral ischemic stroke or stroke-related permanent brain damage is increasing worldwide.1 Brain ischemia/reperfusion (I/R) injury (BI/RI) is a common, major life-threatening complication resulting from several clinical diseases that seriously harm human health.2 Inflammatory response, oxidative stress, and apoptosis have been reported to contribute to the pathogenesis of BI/RI.2–4 Perturbation of blood flow by transient ischemia and reperfusion damages brain structures and component cells, including neurons and glial cells, through hypoxia, production of ROS and promotion of inflammation.1,5
Proinflammatory cytokines, such as ICAM-1 and GFAP, are marker proteins of astrocytes, which are often used for identifying astrocytes and are considered targets that reflect the activity of astrocytes. These cytokines have been previously found to be major contributors of BI/RI.6–10 Studies show that the expression of the apoptosis-related protein caspase-3 is elevated in BI/RI.6,11 Various pharmacotherapies for BI/RI have been developed, such as the collaborative application of ligustrazine and borneol, inhalation of water electrolysis-derived hydrogen, lithium injection, or administration of safranal nanoemulsion, mitochondrial division inhibitor-1, and nanoliposomal cyclosporin A.12–17 However, the outcome of BI/RI treatment remains unsatisfactory despite these pharmacotherapies. It is generally accepted that TNF-α is an inflammatory cytokine involved in systemic inflammation and is one of the cytokines that make up the acute phase reaction.18 It is produced chiefly by activated macrophages, neutrophils, mast cells, eosinophils, and neurons. TNF-α can mediate the ischemic tolerance in human stroke, whereby TNF-α aids in the preservation of mitochondrial membrane integrity, thereby blocking the onset of apoptosis.19–21 Research shows that TNF-α can prevent the death/apoptosis of neurons by a mechanism involving activation of transcription factor NF-κB, which induces the expression of Mn-SOD and Bcl-2.22–24 The pharmacokinetics of TNF-α is characterized by an extremely short half-life (15–30 min) and low bioavailability.25,26 Reports have concluded that it is difficult to control the dose of TNF-α preconditioning for treatment of BI/RI, and a large dose of TNF-α can result in significant side effects, including shock and death.27–30
Therefore, innovative approaches to the screening of more effective strategies for enhancing drug sensitivity and brain function in the treatment of BI/RI are highly desirable. To overcome the limitation of TNF-α in therapeutic application, in this study, we developed a PEG-b-(PELG-g-PLL) polyplex as a TNF-α nanocarrier, composed of a linear PEG block polymer and a brush-like PLL block polymer, wherein the positively charged PLL brush block polymer combines with the negatively charged TNF-α through electrostatic interactions, so that the circulation life of TNF-α in vivo can be enhanced. We attempted to investigate the sustained pharmacological effects of TNF-α/PEG-b-(PELG-g-PLL) particles in protecting the brain against I/R-induced cerebral injury.
Materials and methods
Materials
Regenerated cellulose membrane tubings (MWCO, 100 kDa, 3.5 kDa and 7 kDa) were purchased from Union Carbide (Danbury, CT, USA). Rat recombinant TNF-α and rabbit IgG antibodies to the proteins GFAP, ICAM-1 and caspase-3 were from Sigma-Aldrich (St Louis, MO, USA). Antioxidative activity assay kit and inflammatory factors kit were from Roche Diagnostics (Mannheim, Germany). Other organic chemicals were from Aladdin (Shanghai, China).
Preparation of PEG-b-(PELG-g-PLL)
Preparation of PEG-b-(PELG-g-PLL) was performed as previously described.31 Briefly, PEG-b-PBLG was synthesized by the ring-opening polymerization of BLG-NCA with PEG-NH2 monomethyl ether as the macroinitiator, followed by aminolysis with ethylenediamine to obtain PEG-b-PELG. PEG-b-PELG was used as the macroinitiator to initiate the ring-opening polymerization of ZLL-NCA to yield PEG-b-(PELG-g-PZLL). The benzyl groups of PZLL were then digested in the presence of HBr to obtain PEG-b-(PELG-g-PLL).
Purification of the synthesized nanocopolymers was performed as previously described.31 Briefly, 1) the reaction mixture of PEG-b-PBLG was stirred at 40°C for 3 days and then dialyzed for 3 days (MWCO, 7 kDa); 2) the reaction mixture of PEG-b-PELG was stirred at 40°C for 2 days and then dialyzed for 3 days (MWCO, 3.5 kDa); 3) the reaction mixture of PEG-b-(PELG-g-PZLL) was stirred at 40°C for 3 days and then dialyzed for 3 days (MWCO, 7 kDa); and 4) the reaction mixture of PEG-b-(PELG-g-PLL) was dialyzed for 5 days (MWCO, 3.5 kDa). The aforementioned copolymers were obtained by freeze-drying using a vacuum freeze-dryer (SYHX, Beijing, China) (input–output mass ratio, 67.15%; purity, ≥95.0%) and then characterized by 1H-NMR spectroscopy (Varian, Walnut Creek, CA, USA), gel permeation chromatography (GPC) (Shimadzu, Tokyo, Japan), FTIR spectroscopy (Nicolet; Thermo Electron Scientific Instruments Corporation, Madison, WI, USA), TEM (Jeol, Tokyo, Japan), and particle size/zeta potential determination (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK).31
Cytotoxicity assay in vitro
To assess the cytotoxic effects of PEG-b-(PELG-g-PLL) and TNF-α/PEG-b-(PELG-g-PLL), an MTT colorimetric assay was performed.31,32 Rat cortical neural cells (obtained from embryonic day 18 embryos and incubated for 2 weeks) were seeded at a density of 5×103 cells per well in a 96-well poly-D-lysine-coated plate.33 The cells were treated with a concentration gradient of PEG-b-(PELG-g-PLL) or TNF-α/PEG-b-(PELG-g-PLL) diluted with saline (0.9%), and the cells were then cultured for an additional 24 h at 37°C. To determine cell viability, a 20 μL MTT solution was added to each well and incubated for 2 h, and subsequently, the wells were treated with ethanol–dimethyl sulfoxide solution (1:1, 100 μL) to solubilize the dark blue crystals of MTT formazan completely (~20 min). The absorbance or OD of each well at 570 nm was measured on a microplate multimode reader (Turner Biosystems, Sunnyvale, CA, USA). The blank control was taken from wells without cells, which were also cultured with the MTT solution. Cells incubated in the wells without polymers were used as a negative control. PEG-b-(PELG-g-PLL) and TNF-α/PEG-b-(PELG-g-PLL) were used as potential cytotoxicity samples. The percentage of relative cell viability was calculated by comparing the values of treated cells with those of untreated cells, calculated as (ODsample − ODblank)/(ODcells − ODblank) ×100%.
Encapsulation and release of TNF-α in PEG-b-(PELG-g-PLL)
To evaluate the encapsulation of TNF-α by PEG-b-(PELG-g-PLL), a given volume of TNF-α (0.2 mg/mL) in PB (0.01 mmol/L, pH 7.4) was mixed with PEG-b-(PELG-g-PLL) in PB. After 30 min, the mixed solution was transferred to a dialysis tubing (MWCO, 100 kDa) and dialyzed against 0.1 mol/L PB under sink conditions. The dialysis of free TNF-α in PB was performed under the same conditions to serve as controls. When the free TNF-α in the control experiment was completely dialyzed, the quantification of TNF-α in the outer dialysate of the polymer solution was measured using ELISA according to the manufacturer’s instructions. The TNF-α/PEG-b-(PELG-g-PLL) copolymers were characterized using a nanoparticle size analyzer and TEM.31
TNF-α release from TNF-α/PEG-b-(PELG-g-PLL) was measured using dialysis method (MWCO, 100 kDa) at room temperature with a TNF-α/polymer complex solution (5 mL) against PB (0.1 mol/L, 100 mL, pH 7.4). At set time intervals, a given volume of release medium was withdrawn and replenished with an equal volume of fresh release medium.31 Finally, the released TNF-α was quantified by ELISA according to the manufacturer’s instructions (Roche, Berlin, Germany).
Blood TNF-α concentration evaluation
TNF-α (1 μg/kg body weight [bw]) or TNF-α/PEG-b-(PELG-g-PLL) (10.92 μg/kg bw, containing 1 μg/kg TNF-α) was administered by cisterna magna injection to Sprague Dawley male rats (180–200 g). The blood (0.2 mL) was collected from the tail vein at specified time intervals and then centrifuged at 4°C at 3,600× g for 15 min to harvest the serum. The concentration of TNF-α in rat blood was measured using a double-antibody sandwich ELISA kit.
Animal models
Sprague Dawley male rats (180–200 g) were generally anesthetized by intraperitoneal injection with 2% (w/w) pentobarbital sodium (2.5 mL/kg bw). A total of 100 Sprague Dawley rats were randomized into 5 equal groups: 1) sham-operated group (Sham group), wherein rats were only treated with separation of the bilateral common carotid arteries; 2) I/R group, wherein I/R was achieved by clamping the bilateral common carotid arteries for 2 h, followed by 22 h reperfusion; 3) PEG-b-(PELG-g-PLL) + I/R group (P group), wherein PEG-b-(PELG-g-PLL) (9.92 μg/kg bw [based on loading capacity of copolymer], dissolved in 2 μL of saline) was administered via a single-dose injection into the cisterna magna at 48 h prior to the I/R procedure; 4) TNF-α + I/R group (TNF-α group), wherein TNF-α (1 μg/kg bw, dissolved in 2 μL of saline) was administered via a single-dose injection into the cisterna magna at 48 h prior to the I/R procedure; and 5) TNF-α/PEG-b-(PELG-g-PLL) + I/R group (TNF-α/P group), wherein the TNF-α/polymer complexes (10.92 μg/kg bw [containing 1 μg/kg TNF-α, based on loading capacity of copolymer], dissolved in 2 μL of saline) were administered via a single-dose injection into the cisterna magna at 48 h prior to the I/R procedure. The CBF was measured with a transcranial Doppler (Kejin, Nanjing, China). A greater than 90% reduction in the CBF of the bilateral common carotid arteries and internal carotid arteries was confirmed during the vessel occlusion before reperfusion. The rectal temperature was also monitored with a digital thermometer and maintained at 36.5°C–37.5°C during the surgical procedure and up to 22 h after reperfusion. Finally, all rats were sacrificed by decapitation. A total of 10 whole brains per group were removed to weigh the wet brain and then placed in a vacuum-drying oven at 80°C until constant weight (W, in grams; ~48 h). The remaining 10 whole brain tissues in each group were homogenized in ice-cold normal saline and then centrifuged at 4°C at 3,600× g for 15 min to harvest the supernatant. The supernatant was stored at −80°C for molecular biological detections.
Compliance with ethical standards
Sprague Dawley rats were obtained from the Laboratory Animal Unit of Shantou University Medical College (Shantou, China). All the animal experiments were carried out in accordance with the rules of the IACUC and the research protocol was approved by the Ethics Committee for the Use of Animal Subjects of Shantou University Medical College.
Assessment of brain edema after I/R
The intensity of brain edema (the so-called brain water content), calculated as an edema ratio (%) = (Ww − Wd)/Ww ×100%, was used to assess I/R-induced brain injury, in which Ww was the wet weight of the brain (in grams) and Wd was the dry weight of the brain (grams).
Measurement of the antioxidative activity in the rat brain tissue
The antioxidative activity was detected from the activity of SOD and the MDA content in the rat brain tissue. SOD activity and MDA content were measured at 550 nm and 532 nm following the xanthine oxidase and thiobarbituric acid method (Roche).34,35
ELISA study
The levels of IL-4, IL-6, IL-8, IL-10, and NO in the brain tissue of different groups after reperfusion were measured with the double-antibody sandwich ELISA kits according to the manufacturer’s instructions.
Western blot analysis
Western blot analysis was conducted following previous reports.36–38 The rat brain tissue was homogenized in protein lysate buffer. The homogenate was subjected to 10% SDS-PAGE and then electrophoretically transferred to a nitrocellulose membrane. After blocking with 5% skim milk in TBST for 1 h, the membrane was incubated with primary antibody (against active GFAP, ICAM-1, and caspase-3) at 4°C overnight and subsequently treated with alkaline phosphatase-conjugated secondary antibodies. GFAP, ICAM-1, and caspase-3 were developed using BCIP/NBT. Blots were stained with anti-β-actin antibody, and the quantification of proteins was normalized based on β-actin band density. The antigen–antibody products were detected with Thermo Scientific Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA). The results were analyzed with a FluorChem™ system (Alpha Innotech, San Leandro, CA, USA).
Statistical analysis
All measurements were carried out in triplicate. Data were expressed as the mean ± standard deviation and analyzed by analysis of variance with post hoc testing. Microsoft Excel 2013 database and the SPSS 19.0 software were used to record and analyze all the data. A P-value <0.05 was considered statistically significant.
Results
Synthesis of PEG-b-(PELG-g-PLL)
The block copolymer PEG-b-(PELG-g-PLL) was composed of a linear PEG block and a brush-like PLL block, wherein the molecular weight of the PEG block was 5 kDa, the degree of polymerization of PELG brush backbone was 50, and the degree of polymerization of PLL brush side chain was 3. The synthesis route and structure of PEG-b-(PELG-g-PLL) are demonstrated in Figure 1.
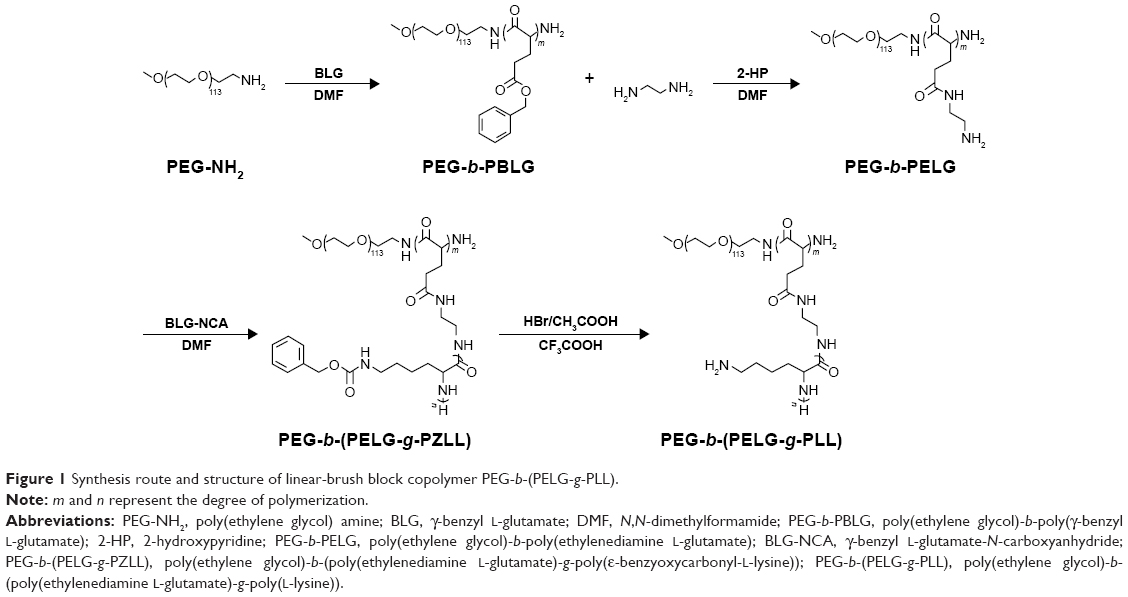 | Figure 1 Synthesis route and structure of linear-brush block copolymer PEG-b-(PELG-g-PLL). |
Characterization of TNF-α/PEG-b-(PELG-g-PLL)
The molecular weight of PEG-b-(PELG-g-PLL) obtained from 1H-NMR (31.3 kDa) was greater than the value from the GPC measurement (20.8 kDa), possibly because of the smaller hydrodynamic volumes of brush polymers compared with linear polymers. The TNF-α/polymer complex was visualized using TEM. The TNF-α/polymer complex was further characterized with granulometry. The TNF-α/polymer complex presented a spherical structure with a diameter of ~83 nm. The mean diameters of the block copolymer PEG-b-(PELG-g-PLL) and TNF-α/polymer complex were ~7 nm and ~26 nm, respectively. The larger diameter observed by TEM might be due to the aggregation of the complex during the drying process. The zeta potential (at pH 7.4 at 25°C) was 27.4 mV (PEG-b-(PELG-g-PLL)) and 21.1 mV (TNF-α/PEG-b-(PELG-g-PLL)). The higher positive charge of the polymer and the lower positive charge of the TNF-α/polymer were due to the presence of the brush-like PLL amino groups and the electrostatic neutralizations between PLL and TNF-α, respectively (Table 1, Figure 2).
 | Table 1 Particle size, TEM size, molecular weight, PDI, loading capacity, and zeta potential of PEG-b-(PELG-g-PLL) |
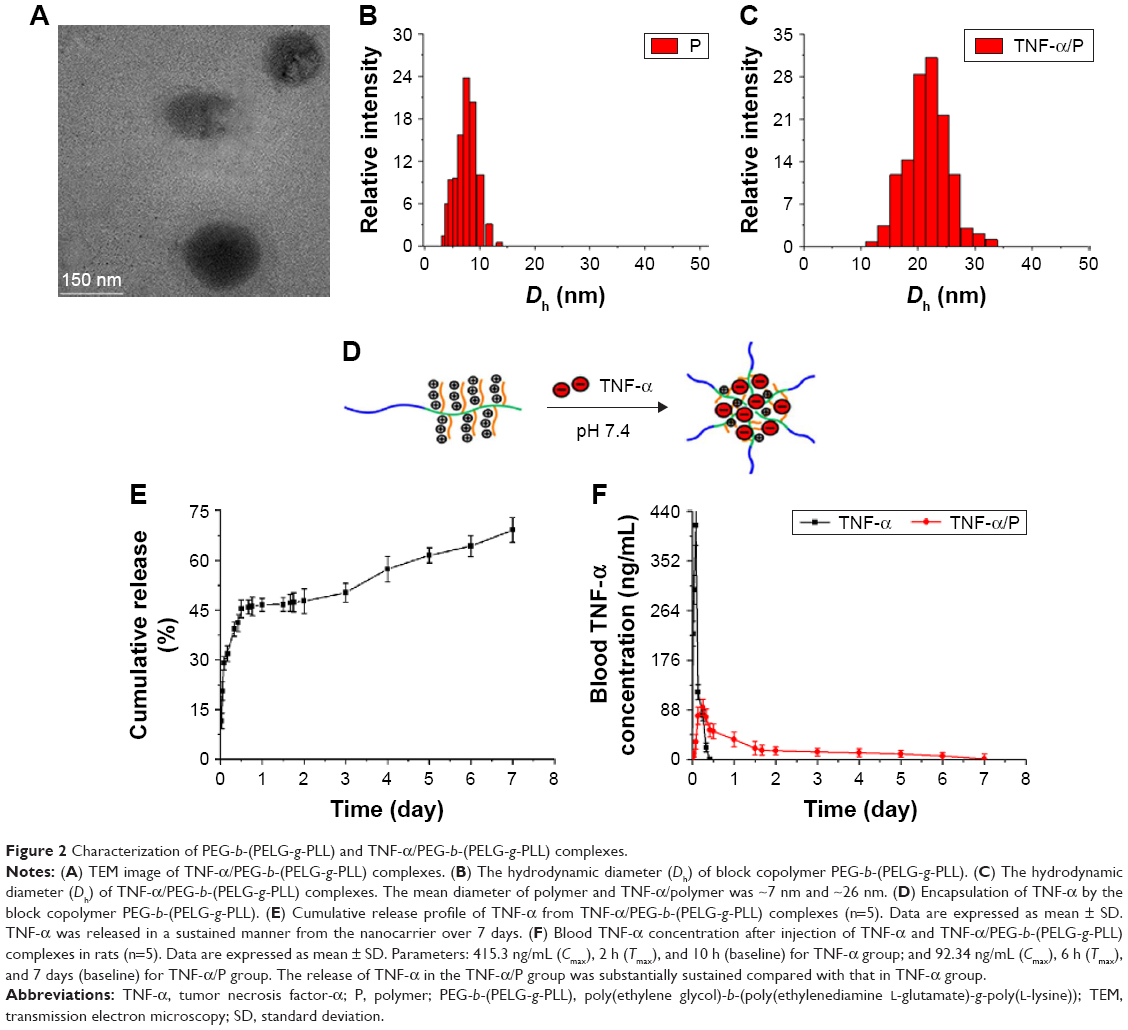 | Figure 2 Characterization of PEG-b-(PELG-g-PLL) and TNF-α/PEG-b-(PELG-g-PLL) complexes. |
Encapsulation of TNF-α in and release of TNF-α from PEG-b-(PELG-g-PLL)
TNF-α was efficiently entrapped by the block copolymer PEG-b-(PELG-g-PLL) at physiological pH (pH 7.4) via electrostatic interactions between the positively charged PLL and the negatively charged TNF-α (the isoelectric point of TNF-α is ~3.9) (Figure 2). TNF-α was mixed with PEG-b-(PELG-g-PLL) at a mass ratio of 1:5 and dialyzed (MWCO, 100 kDa) against PB solution (pH 7.4, 0.01 mmol/L). The loading capacity of TNF-α in PEG-b-(PELG-g-PLL) was expressed as the mass ratio (mTNF-α/mpolymer) of encapsulated TNF-α to polymer host and was determined to be ~10.08% (Table 1).
In vitro release of TNF-α from PEG-b-(PELG-g-PLL) was performed using dialysis (MWCO, 100 kDa). PEG-b-(PELG-g-PLL) entrapped TNF-α with negligible release in a releasing medium of 0.01 mol/L PB solution. When the concentration of PB was increased to 0.1 mol/L (close to physiological ionic strength), sustained release of TNF-α was observed, with 28.94% of TNF-α released over a period of 2 h, 50.24% of TNF-α released over a period of 3 days, and 69.16% of TNF-α released over a period of 7 days (Figure 2).
Blood TNF-α concentration
After a single cisterna magna injection at a dose of 1 μg/kg bw, the plasma concentration of TNF-α in Sprague Dawley rats of the TNF-α group rapidly reached a maximum concentration (Cmax) (415.3 ng/mL) at 2 h (time of maximum concentration, Tmax) and declined to baseline values within 10 h. It reached a maximum value of 92.34 ng/mL in the TNF-α/polymer group at 6 h and declined to baseline values within 7 days of administration. These results showed that the release of TNF-α in TNF-α/polymer group was substantially sustained compared with that in the TNF-α group (Figure 2).
Cell viability
The cytotoxicity of the block copolymer PEG-b-(PELG-g-PLL) and TNF-α/PEG-b-(PELG-g-PLL) on neuronal cells was monitored within 24 h of culturing. The results indicated that the block copolymer PEG-b-(PELG-g-PLL) was noncytotoxic at the concentrations of ≤0.5 μg/mL (P>0.05). At the concentration of >0.5 μg/mL, the block copolymer exhibited a low cytotoxicity against neuronal cells and high relative cell viability (minimum mean was ~85.5%), even when administered at a maximum dose of 1.0 μg/mL (P<0.05) (Figure 3).
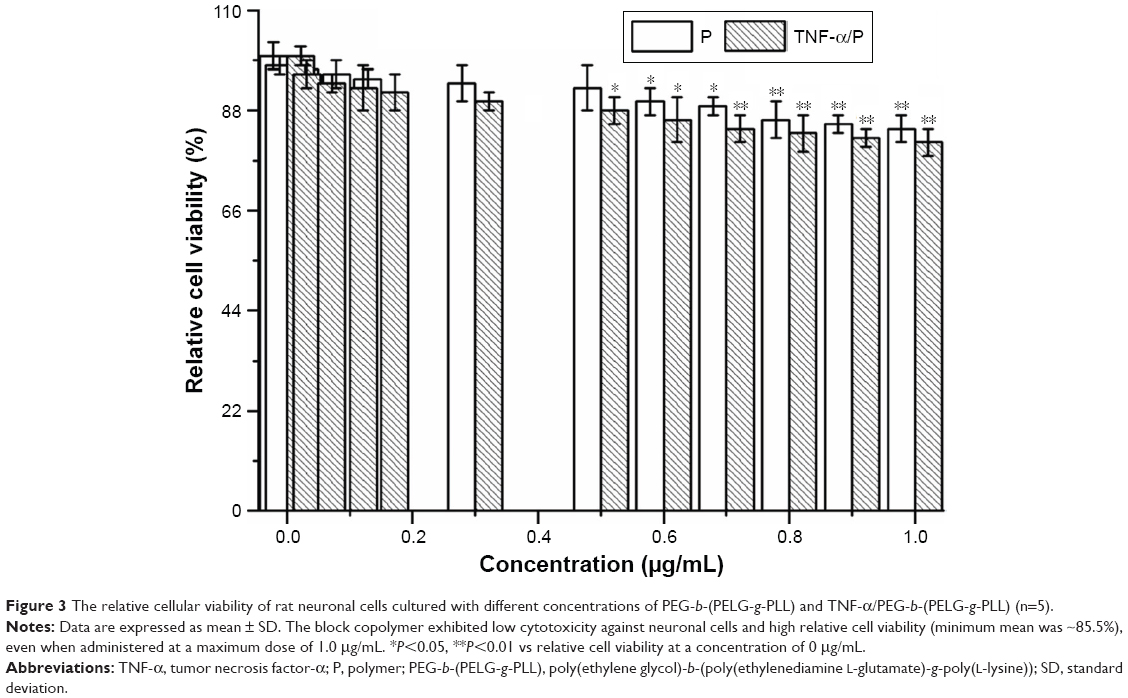 | Figure 3 The relative cellular viability of rat neuronal cells cultured with different concentrations of PEG-b-(PELG-g-PLL) and TNF-α/PEG-b-(PELG-g-PLL) (n=5). |
Brain edema ratio
The brain edema ratio in the I/R group was significantly higher than that in the sham-operated group (P<0.01). Pretreatment with TNF-α prior to I/R significantly decreased the brain edema ratio (P<0.05). TNF-α/PEG-b-(PELG-g-PLL) polyplexes significantly reduced the brain edema ratio compared to free TNF-α (P<0.05) (Figure 4).
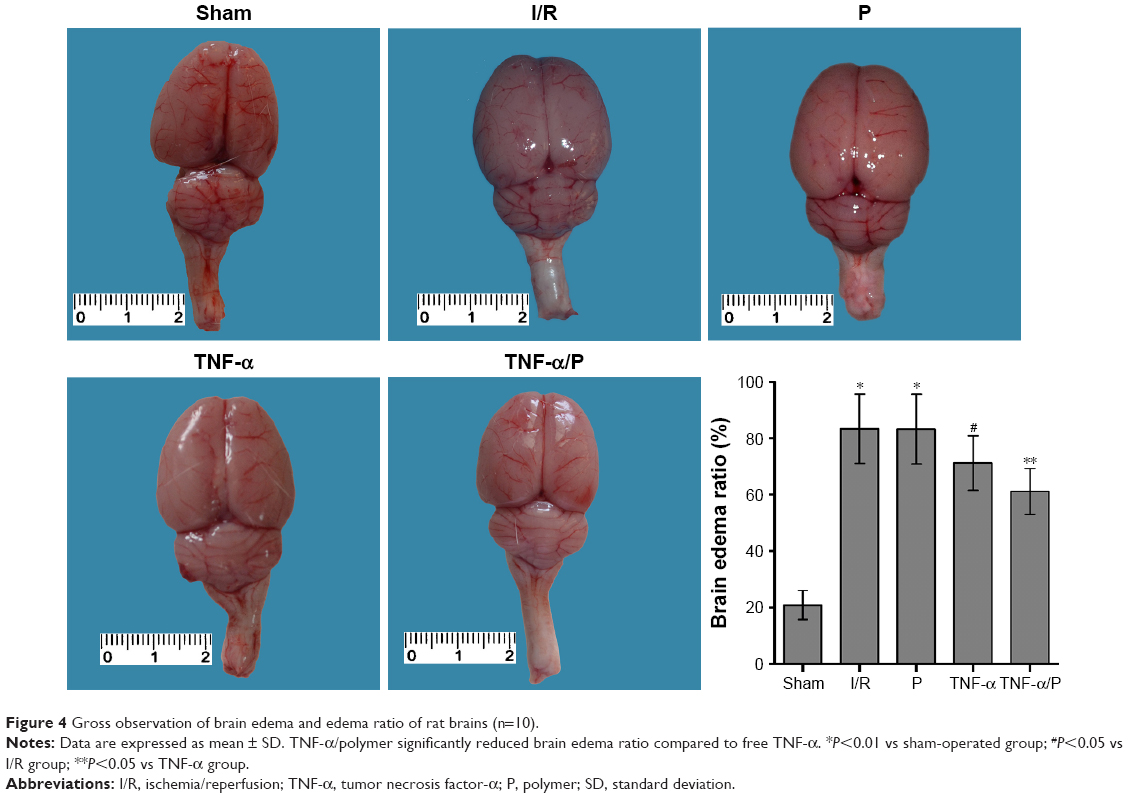 | Figure 4 Gross observation of brain edema and edema ratio of rat brains (n=10). |
SOD activity and MDA content in the brain tissue
Cerebral I/R resulted in reduced SOD activity and increased MDA content in the brain tissue. Administration of TNF-α increased the activity of SOD and reduced the level of MDA, compared with the results in the I/R group (P<0.05). Furthermore, preconditioning with TNF-α/PEG-b-(PELG-g-PLL) polyplexes significantly improved SOD activity and reduced MDA content, compared to preconditioning with free TNF-α (P<0.05) (Figure 5).
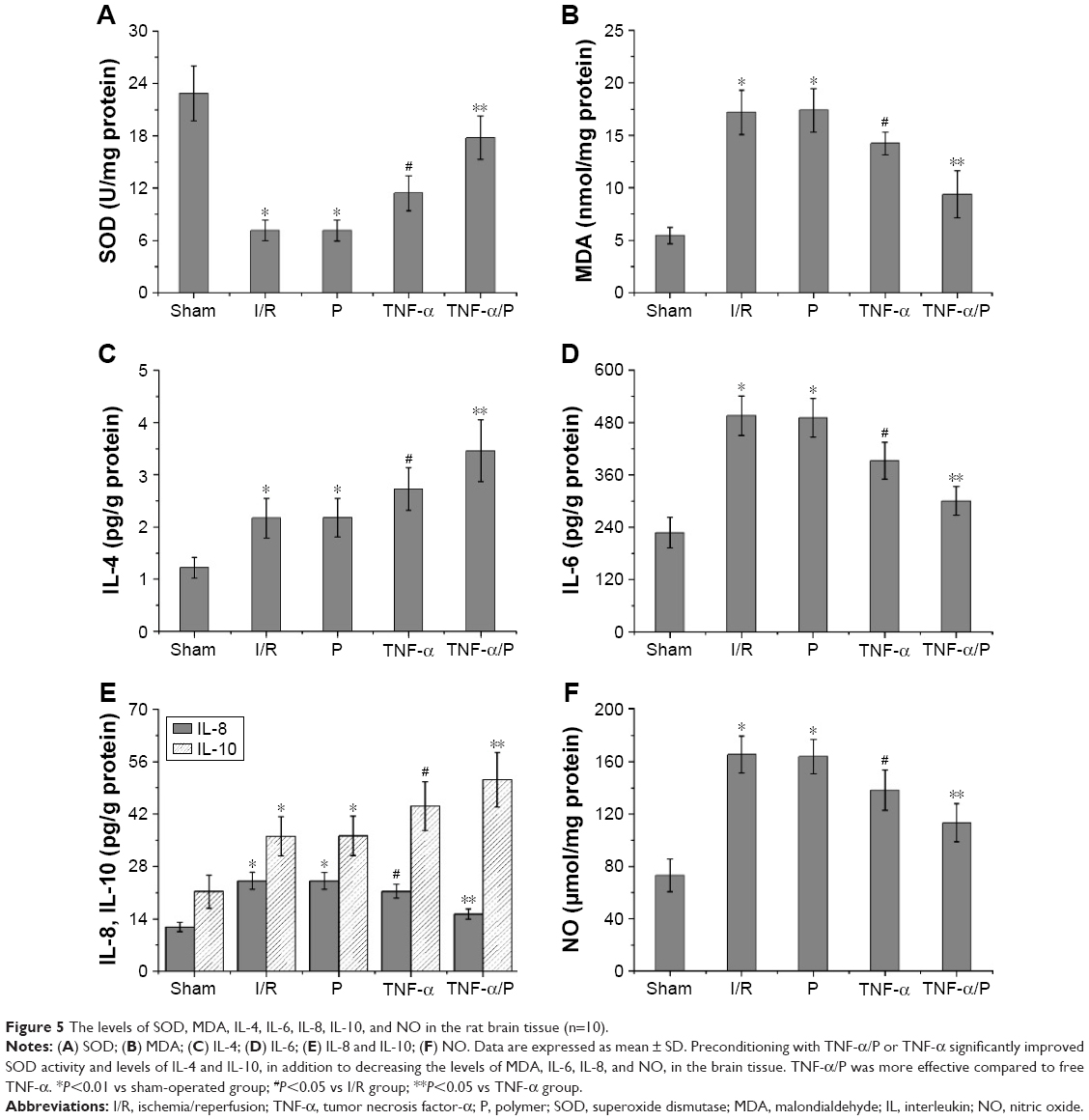 | Figure 5 The levels of SOD, MDA, IL-4, IL-6, IL-8, IL-10, and NO in the rat brain tissue (n=10). |
Levels of IL-4, IL-6, IL-8, IL-10, and NO in the brain tissue
BI/RI led to increased levels of IL-4, IL-6, IL-8, IL-10, and NO in the brain tissue. After administration of TNF-α, increased levels of IL-4 and IL-10 (anti-inflammatory factors) and decreased levels of IL-6, IL-8 (proinflammatory factors), and NO were observed compared with those in the I/R group (P<0.05). Similarly, TNF-α/PEG-b-(PELG-g-PLL) polyplexes were more effective compared to free TNF-α (P<0.05) (Figure 5).
Expression of GFAP, ICAM-1, and caspase-3 in the brain tissue
The expression levels of GFAP, ICAM-1, and caspase-3 in the brain tissue of the I/R group were significantly higher than those in the sham-operated group (P<0.01). Administration of TNF-α significantly downregulated the expression of GFAP, ICAM-1, and caspase-3 in the brain tissue compared with that in the I/R group (P<0.05). Similarly, TNF-α/PEG-b-(PELG-g-PLL) polyplexes enhanced the effects compared to free TNF-α (P<0.05) (Figure 6).
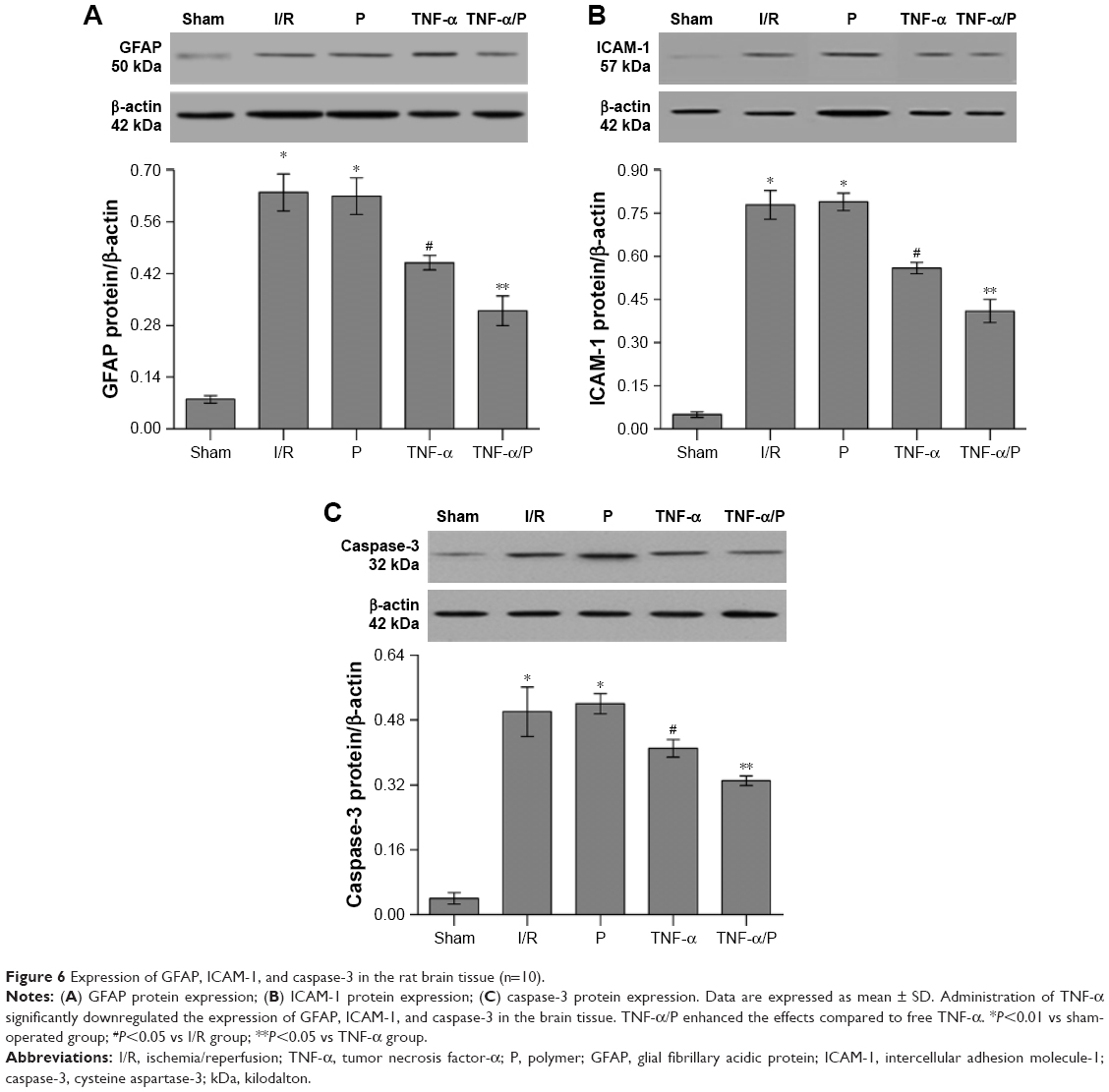 | Figure 6 Expression of GFAP, ICAM-1, and caspase-3 in the rat brain tissue (n=10). |
Discussion
In our previous work,31 we reported the design of PEG-b-(PELG-g-PLL) as a potential insulin nanocarrier to prolong the in vivo half-life of insulin. This is the first study to demonstrate that preconditioning with TNF-α and TNF-α/PEG-b-(PELG-g-PLL) is capable of attenuating brain I/R-induced cerebral injury. The block copolymer is composed of a linear PEG block polymer and a brush-like PLL block polymer. At pH 7.4, the positively charged PLL brush block polymer combines with the negatively charged TNF-α through electrostatic interactions. The loaded TNF-α can be released in a sustained manner; thus, the bioavailability of free TNF-α is significantly improved. TNF-α, a well-known proinflammatory cytokine present in neurons and the glia, is involved in physiological and pathophysiological processes of the brain.23,33,37,38 TNF-α acts as an effector molecule in brain development and is often involved in different signaling pathways to elicit the activation of macrophages and glial cells for neurotoxin production and to initiate the apoptosis/death process in neurons.39–41 Until now, the overall role of TNF-α has been constantly debated, and there are few studies focusing on the application of TNF-α-loaded nanocarriers for the treatment of BI/RI. In the current study, we delineated an important neuroprotective role of TNF-α, through TNF-α-loaded nanocarrier preconditioning, in BI/RI.
Reperfusion of ischemic tissues is often associated with inflammatory microvascular injury, particularly due to increased permeability of capillaries and arterioles that leads to an increase of diffusion and fluid filtration across the tissues.42 Previous studies have determined that cerebral ischemic stroke can result in BBB breakdown and increase in injury volume and edema by activation of the inflammation and immune system. Substances produced by immune responses (such as inflammatory mediators) are able to cross the BBB and affect central nervous system function.43–45 These data indicate that cytokines often cross the BBB with relatively high efficacy. IL-4 can promote activation of type 2 macrophages, which is usually coupled with secretion of IL-10, which in turn results in diminution of pathological inflammation.46 Conversely, IL-6 and IL-8 act as proinflammatory cytokines during tissue injury, leading to inflammation.47 Oxidative stress, reflecting a disturbance between the systemic manifestation of ROS and the ability of a biological system to detoxify the reactive intermediates, can trigger cell apoptosis, cell death, and even cell necrosis. SOD is an important antioxidant defense protein in nearly all living cells, whereby it can catalyze the conversion of the superoxide radical into molecular oxygen or hydrogen peroxide. The results described in this study demonstrate that the increased levels of cerebral I/R-induced oxidative stress and inflammation were significantly inhibited by the upregulation of SOD, IL-4, and IL-10 and by the downregulation of MDA, IL-6, IL-8, and NO after pretreatment with TNF-α or TNF-α/PEG-b-(PELG-g-PLL) polyplexes. Therefore, TNF-α/PEG-b-(PELG-g-PLL) may potentially be useful in the treatment of I/R-induced cerebral injury through its ability to modulate the oxidative stress status and inflammation cascades.
ICAM-1, an endothelial- and leukocyte-associated transmembrane protein, can stabilize cell–cell interactions and facilitate leukocyte–endothelial transmigration.48 ICAM-1 can produce proinflammatory effects, such as recruitment of inflammatory leukocytes by signaling cascades involving a number of kinases. A high level of ICAM-1 in acute cerebral ischemia patients has been linked to an increased risk of mortality.49,50 Therefore, ICAM-1 can be considered an indicator of inflammation in brain ischemic shock patients. ICAM-1 and GFAP contribute to the inflammatory process by facilitating the adhesion of monocytes to endothelial cells. Exposure to local cerebral ischemia induces protein expression of ICAM-1 and GFAP.37,51 Our data reveal that the expression levels of ICAM-1 and GFAP after I/R-induced cerebral injury were remarkably increased, while they were depressed after pretreatment with TNF-α or TNF-α/PEG-b-(PELG-g-PLL). TNF-α or TNF-α/PEG-b-(PELG-g-PLL) pretreatment had a neuroprotective effect against BI/RI, and this effect was related to the inhibition of oxidative stress, inflammation, and apoptosis via reduction in both the expression of ICAM-1, GFAP, and caspase-3 and the levels of IL-6, IL-8, NO, and MDA, as well as increases in SOD activity and the levels of IL-4 and IL-10. The TNF-α nanocarrier PEG-b-(PELG-g-PLL) exhibited a powerful capacity to enhance the bioactivity of TNF-α.
The results of this study indicate that oxidative stress, depletion of antioxidant defense, and increased lipid peroxidation are of critical importance in the pathogenesis of BI/RI. The encapsulation of TNF-α in the block copolymer PEG-b-(PELG-g-PLL) can prolong the in vivo circulation time of TNF-α and enhance its bioavailability. This suggests that utilization of the block copolymer PEG-b-(PELG-g-PLL) as a TNF-α nanocarrier can improve the protective effect of TNF-α in BI/RI compared to preconditioning with free TNF-α.
Conclusion
Preconditioning with TNF-α/PEG-b-(PELG-g-PLL) demonstrated a protective effect against cerebral I/R injury through reducing brain damage, decreasing the levels of IL-6, IL-8, NO, and MDA, decreasing the expression of ICAM-1, GFAP, and caspase-3, and increasing the levels of IL-4, IL-10, and SOD. The use of the block copolymer PEG-b-(PELG-g-PLL) as a TNF-α nanocarrier significantly improved this protective effect as a result of sustained release and prolonged the in vivo circulation time, thus enhancing the bioactivity of TNF-α. Detailed studies are currently under way to explore the underlying mechanisms of the protective effect of TNF-α/PEG-b-(PELG-g-PLL) polyplexes in BI/RI.
Abbreviations
BBB, blood–brain barrier; Bcl-2, B-cell lymphoma 2; BCIP/NBT, 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium; BI/RI, brain ischemia/reperfusion injury; BLG, γ-benzyl L-glutamate; BLG-NCA, γ-benzyl L-glutamate-N-carboxyanhydride; bw, body weight; caspase-3, cysteine aspartase-3; CBF, cerebral blood flow; Cmax, maximum concentration; DMF, N, N-dimethylformamide; ELISA, enzyme-linked immunosorbent assay; FTIR, Fourier transform infrared; GFAP, glial fibrillary acidic protein; GPC, gel permeation chromatography; HBr, hydrogen bromide; 1H-NMR, 1H-nuclear magnetic resonance spectroscopy; IACUC, Institutional Animal Care and Use Committee; ICAM-1, intercellular adhesion molecule-1; Ig, immunoglobulin; IL, interleukin; I/R, ischemia/reperfusion; MDA, malondialdehyde; Mn, number-average molecular weight; Mn-SOD, manganese-dependent superoxide dismutase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MWCO, molecular weight cutoff; NF-κB, nuclear factor kappa B; NO, nitric oxide; OD, optical density; PB, phosphate buffer; PEG, polyethylene glycol; PEG-NH2, PEG amine; PEG-b-PBLG, PEG-b-poly(γ-benzyl L-glutamate); PEG-b-PELG, PEG-b-poly(ethylenediamine L-glutamate); PEG-b-(PELG-g-PZLL), PEG-b-(poly(ethylenediamine L-glutamate)-g-poly(ε-benzyoxycarbonyl-L-lysine)); PEG-b-(PELG-g-PLL), PEG-b-(poly(ethylenediamine L-glutamate)-g-poly(L-lysine)); PLL, poly(L-lysine); ROS, reactive oxygen species; SOD, superoxide dismutase; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; TBST, Tris-buffered saline containing 0.2%–0.4% Tween-20; TEM, transmission electron microscopy; Tmax, time of maximum concentration; TNF-α, tumor necrosis factor; Ww, wet weight of the brain; Wd, dry weight of the brain; ZLL-NCA, ε-benzyoxycarbonyl-L-lysine-N-carboxyanhydride.
Acknowledgments
We greatly appreciate the editors and three anonymous peer reviewers for their critical reading and insightful comments, which have improved our manuscript substantially. This study was funded by grants from the Science and Technology Planning Project (Laboratory Animal Project) of Zhejiang Province (2015C37130), the Zhejiang Provincial Natural Science Foundation (LQ14H230001), and the National Natural Science Foundation of China (81400654). The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, or manuscript writing.
Author contributions
Xu G and Gu J conceived and designed the experiments. Tong F and Liu D synthesized the nanocarrier. Xu G, Gu H, Hu B, and Tong F performed the experiments. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work. All authors have read, and confirm that they meet, ICMJE criteria for authorship.
Disclosure
The authors report no conflicts of interest in this work.
References
Amadatsu T, Morinaga J, Kawano T, et al. Macrophage-derived angiopoietin-like protein 2 exacerbates brain damage by accelerating acute inflammation after ischemia-reperfusion. PLoS One. 2016;11(11):e0166285. | ||
Huang T, Gao DK, Hei Y, Zhang X, Chen XY, Fei Z. D-allose protects the blood brain barrier through PPAR gamma-mediated anti-inflammatory pathway in the mice model of ischemia reperfusion injury. Brain Res. 2016;1642:478–486. | ||
Wen XR, Tang M, Qi DS, et al. Butylphthalide suppresses neuronal cells apoptosis and inhibits JNK-caspase3 signaling pathway after brain ischemia/reperfusion in rats. Cell Mol Neurobiol. 2016;36(7):1087–1095. | ||
Hu B, Wu Y, Liu J, et al. GSK-3beta inhibitor induces expression of Nrf2/TrxR2 signaling pathway to protect against renal ischemia/reperfusion injury in diabetic rats. Kidney Blood Press Res. 2016;41(6):937–946. | ||
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. | ||
Bai S, Hu Z, Yang Y, et al. Anti-inflammatory and neuroprotective effects of triptolide via the NF-kappaB signaling pathway in a rat MCAO model. Anat Rec (Hoboken). 2016;299(2):256–266. | ||
Bernardi A, Frozza RL, Hoppe JB, et al. The antiproliferative effect of indomethacin-loaded lipid-core nanocapsules in glioma cells is mediated by cell cycle regulation, differentiation, and the inhibition of survival pathways. Int J Nanomedicine. 2013;8:711–728. | ||
Huang XP, Ding H, Lu JD, Tang YH, Deng BX, Deng CQ. Effects of the combination of the main active components of astragalus and panax notoginseng on inflammation and apoptosis of nerve cell after cerebral ischemia-reperfusion. Am J Chin Med. 2015;43(7):1419–1438. | ||
Mane V, Muro S. Biodistribution and endocytosis of ICAM-1-targeting antibodies versus nanocarriers in the gastrointestinal tract in mice. Int J Nanomedicine. 2012;7:4223–4237. | ||
Kretzer IF, Maria DA, Guido MC, Contente TC, Maranhao RC. Simvastatin increases the antineoplastic actions of paclitaxel carried in lipid nanoemulsions in melanoma-bearing mice. Int J Nanomedicine. 2016;11:885–904. | ||
Niizuma K, Yoshioka H, Chen H, et al. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim Biophys Acta. 2010;1802(1):92–99. | ||
Yu B, Ruan M, Zhang ZN, Cheng HB, Shen XC. Synergic effect of borneol and ligustrazine on the neuroprotection in global cerebral ischemia/reperfusion injury: a region-specificity study. Evid Based Complement Alternat Med. 2016;2016:4072809. | ||
Cui J, Chen X, Zhai X, et al. Inhalation of water electrolysis-derived hydrogen ameliorates cerebral ischemia-reperfusion injury in rats – a possible new hydrogen resource for clinical use. Neuroscience. 2016;335:232–241. | ||
Ahmad N, Ahmad R, Abbas Naqvi A, et al. The effect of safranal loaded mucoadhesive nanoemulsion on oxidative stress markers in cerebral ischemia. Artif Cells Nanomed Biotechnol. 2016. Epub 2016 Sep 9. | ||
Partoazar A, Nasoohi S, Rezayat SM, et al. Nanoliposome containing cyclosporin A reduced neuroinflammation responses and improved neurological activities in cerebral ischemia/reperfusion in rat. Fundam Clin Pharmacol. 2016. Epub 2016 Sep 12. | ||
Takahashi T, Steinberg GK, Zhao H. Lithium treatment reduces brain injury induced by focal ischemia with partial reperfusion and the protective mechanisms dispute the importance of akt activity. Aging Dis. 2012;3(3):226–233. | ||
Ma X, Xie Y, Chen Y, Han B, Li J, Qi S. Post-ischemia mdivi-1 treatment protects against ischemia/reperfusion-induced brain injury in a rat model. Neurosci Lett. 2016;632:23–32. | ||
Eder P, Linke K, Witowski J. Update on the mechanisms of action of anti-TNF-alpha antibodies and their clinical implications in inflammatory bowel disease. Pol Arch Med Wewn. 2016;126(10):772–780. | ||
Liu J, Ginis I, Spatz M, Hallenbeck JM. Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. Am J Physiol Cell Physiol. 2000;278(1):C144–C153. | ||
Tanaka H, Yokota H, Jover T, et al. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci. 2004;24(11):2750–2759. | ||
Castillo J, Moro MA, Blanco M, et al. The release of tumor necrosis factor-alpha is associated with ischemic tolerance in human stroke. Ann Neurol. 2003;54(6):811–819. | ||
Shahid A, Ali R, Ali N, et al. Attenuation of genotoxicity, oxidative stress, apoptosis and inflammation by rutin in benzo(a)pyrene exposed lungs of mice: plausible role of NF-kappaB, TNF-alpha and Bcl-2. J Complement Integr Med. 2016;13(1):17–29. | ||
Marigo I, Zilio S, Desantis G, et al. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-synthase-producing dendritic cells. Cancer Cell. 2016;30(3):377–390. | ||
Wang ZL, Hu ZQ, Zhang DW, et al. Silencing tumor necrosis factor-alpha in vitro from small interfering RNA-decorated titanium nanotube array can facilitate osteogenic differentiation of mesenchymal stem cells. Int J Nanomedicine. 2016;11:3205–3214. | ||
Ma Y, Zhao S, Shen S, et al. A novel recombinant slow-release TNF alpha-derived peptide effectively inhibits tumor growth and angiogenesis. Sci Rep. 2015;5:13595. | ||
Gordon R, Anantharam V, Kanthasamy AG, Kanthasamy A. Proteolytic activation of proapoptotic kinase protein kinase Cdelta by tumor necrosis factor alpha death receptor signaling in dopaminergic neurons during neuroinflammation. J Neuroinflammation. 2012;9:82. | ||
Hrushesky WJ, Langevin T, Kim YJ, Wood PA. Circadian dynamics of tumor necrosis factor alpha (cachectin) lethality. J Exp Med. 1994;180(3):1059–1065. | ||
Welsh K, Milutinovic S, Ardecky RJ, et al. Characterization of potent SMAC mimetics that sensitize cancer cells to TNF family-induced apoptosis. PLoS One. 2016;11(9):e0161952. | ||
Song Y, Zhang C, Zhang J, et al. An injectable silk sericin hydrogel promotes cardiac functional recovery after ischemic myocardial infarction. Acta Biomater. 2016;41:210–223. | ||
Mocan L, Matea C, Tabaran FA, et al. Photothermal treatment of liver cancer with albumin-conjugated gold nanoparticles initiates Golgi Apparatus-ER dysfunction and caspase-3 apoptotic pathway activation by selective targeting of Gp60 receptor. Int J Nanomedicine. 2015;10:5435–5445. | ||
Tong F, Tang X, Li X, Xia W, Liu D. The effect of insulin-loaded linear poly(ethylene glycol)-brush-like poly(L-lysine) block copolymer on renal ischemia/reperfusion-induced lung injury through downregulating hypoxia-inducible factor. Int J Nanomedicine. 2016;11:1717–1730. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Saha RN, Ghosh A, Palencia CA, Fung YK, Dudek SM, Pahan K. TNF-alpha preconditioning protects neurons via neuron-specific up-regulation of CREB-binding protein. J Immunol. 2009;183(3):2068–2078. | ||
Abdel-Aleem GA, Khaleel EF, Mostafa DG, Elberier LK. Neuroprotective effect of resveratrol against brain ischemia reperfusion injury in rats entails reduction of DJ-1 protein expression and activation of PI3K/Akt/GSK3b survival pathway. Arch Physiol Biochem. 2016;122(4):200–213. | ||
Niska K, Pyszka K, Tukaj C, Wozniak M, Radomski MW, Inkielewicz-Stepniak I. Titanium dioxide nanoparticles enhance production of superoxide anion and alter the antioxidant system in human osteoblast cells. Int J Nanomedicine. 2015;10:1095–1107. | ||
Huang Z, Chen C, Li S, Kong F, Shan P, Huang W. Combined treatment with amlodipine and atorvastatin calcium reduces circulating levels of intercellular adhesion molecule-1 and tumor necrosis factor-alpha in hypertensive patients with prediabetes. Front Aging Neurosci. 2016;8:206. | ||
Guo C, Xia Y, Niu P, et al. Silica nanoparticles induce oxidative stress, inflammation, and endothelial dysfunction in vitro via activation of the MAPK/Nrf2 pathway and nuclear factor-kappaB signaling. Int J Nanomedicine. 2015;10:1463–1477. | ||
Cao JP, Xu JG, Li WY, Liu JA. Influence of selective brain cooling on the expression of ICAM-1 mRNA and infiltration of PMNLs and monocytes/macrophages in rats suffering from global brain ischemia/reperfusion injury. Biosci Trends. 2008;2(6):241–244. | ||
Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol. 2006;1(3):212–222. | ||
Garcia E, Aguilar-Cevallos J, Silva-Garcia R, Ibarra A. Cytokine and growth factor activation in vivo and in vitro after spinal cord injury. Mediators Inflamm. 2016;2016:9476020. | ||
Elnaggar YS, Etman SM, Abdelmonsif DA, Abdallah OY. Novel piperine-loaded Tween-integrated monoolein cubosomes as brain-targeted oral nanomedicine in Alzheimer’s disease: pharmaceutical, biological, and toxicological studies. Int J Nanomedicine. 2015;10:5459–5473. | ||
Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190(3):255–266. | ||
Lopalco A, Ali H, Denora N, Rytting E. Oxcarbazepine-loaded polymeric nanoparticles: development and permeability studies across in vitro models of the blood-brain barrier and human placental trophoblast. Int J Nanomedicine. 2015;10:1985–1996. | ||
Brown JA, Codreanu SG, Shi M, et al. Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit. J Neuroinflammation. 2016;13(1):306. | ||
Shityakov S, Salvador E, Pastorin G, Forster C. Blood-brain barrier transport studies, aggregation, and molecular dynamics simulation of multiwalled carbon nanotube functionalized with fluorescein isothiocyanate. Int J Nanomedicine. 2015;10:1703–1713. | ||
Mittal M, Tiruppathi C, Nepal S, et al. TNFalpha-stimulated gene-6 (TSG6) activates macrophage phenotype transition to prevent inflammatory lung injury. Proc Natl Acad Sci U S A. 2016;113(50):E8151–E8158. | ||
Lee H, Su YL, Huang BS, et al. Importance of the C-terminal histidine residues of Helicobacter pylori GroES for toll-like receptor 4 binding and interleukin-8 cytokine production. Sci Rep. 2016;6:37367. | ||
McDonald KK, Cooper S, Danielzak L, Leask RL. Glycocalyx degradation induces a proinflammatory phenotype and increased leukocyte adhesion in cultured endothelial cells under flow. PLoS One. 2016;11(12):e0167576. | ||
Rajput SK, Sharma AK, Meena CL, Pant AB, Jain R, Sharma SS. Effect of L-pGlu-(1-benzyl)-l-His-l-Pro-NH2 against in-vitro and in-vivo models of cerebral ischemia and associated neurological disorders. Biomed Pharmacother. 2016;84:1256–1265. | ||
Fumagalli S, De Simoni MG. Lectin complement pathway and its bloody interactions in brain ischemia. Stroke. 2016;47(12):3067–3073. | ||
Zhang L, Xu X, Yang R, et al. Paclitaxel attenuates renal interstitial fibroblast activation and interstitial fibrosis by inhibiting STAT3 signaling. Drug Des Devel Ther. 2015;9:2139–2148. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.