Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Pediatric-onset multiple sclerosis in Egypt: a multi-center registry of 186 patients
Authors Hamdy SM, Abdel-Naseer M ![]() , Shalaby NM, Elmazny A
, Shalaby NM, Elmazny A ![]() , Girgis M, Nada MA
, Girgis M, Nada MA ![]() , Hassan A
, Hassan A ![]() , Mourad HS
, Mourad HS ![]() , Hegazy MI
, Hegazy MI ![]() , Abdelalim A
, Abdelalim A ![]() , Kishk NA
, Kishk NA ![]() , Abokrysha NT, Genedy SA, Essawy EA
, Abokrysha NT, Genedy SA, Essawy EA ![]() , Shehata HS
, Shehata HS ![]()
Received 16 December 2017
Accepted for publication 15 January 2018
Published 23 February 2018 Volume 2018:14 Pages 631—640
DOI https://doi.org/10.2147/NDT.S160060
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Sherif M Hamdy,1 Maged Abdel-Naseer,1 Nevin M Shalaby,1 Alaa Elmazny,1 Marian Girgis,2 Mona A Nada,1 Amr Hassan,1 Husam S Mourad,1 Mohamed I Hegazy,1 Ahmed Abdelalim,1 Nirmeen A Kishk,1 Noha T Abokrysha,1 Shaimaa A Genedy,1 Ehab A Essawy,3 Hatem S Shehata1
1Neurology Department, Cairo University, Cairo, Egypt; 2Pediatric Department, Cairo University, Cairo, Egypt; 3Division of Biochemistry, Chemistry Department, Faculty of Science, Helwan University, Helwan, Egypt
Introduction: Although the frequency of pediatric-onset multiple sclerosis (POMS) has increased in recent decades, it is still highly uncommon, which creates a need for the involvement of more registries from various clinical centers.
Objective: To characterize the demographic, clinical, and paraclinical features of Egyptian patients with POMS.
Patients and methods: A retrospective chart review study was undertaken on 237 Egyptian patients with demyelinating events which started before the age of 18 years who attended one of five tertiary referral centers in Cairo, Egypt.
Results: Multiple sclerosis was diagnosed in 186 patients, 47 (25.27%) patients had disease onset before the age of 12 years; “early-onset pediatric multiple sclerosis (EOPMS)”. The mean age of disease onset was (14.13±2.49 years), with a female:male ratio of 1.62:1, none of the enrolled patients had a primary progressive course (PPMS), whereas 10 patients (5.38%) had a secondary progressive form. Approximately two-thirds of the patients had monofocal disease onset, and less than 10% presented with encephalopathy; most of them had EOPMS. Motor weakness was the presenting symptom in half of the patients, whereas cerebellar presentation was detected in 34.95%, mainly in EOPMS. Seizures (not related to encephalopathy) were more frequent in those with EOPMS. Initial brain magnetic resonance images were positive in all patients, with detected atypical lesions in 29.03%, enhanced lesions in 35.48%, black holes in 13.98%, and infratentorial in 34.41%. Cervical cord involvement was found in 68.28%. More than two-thirds of the patients received either immunomodulatory or immunosuppressant (IS) treatment throughout their disease course, and about half of them received their treatment within the first year from symptoms onset, with a more favorable outcome, and patients with highly active disease received natalizumab, fingolimod, or other IS.
Conclusion: The results from this registry – the largest for MS in the Arab region to date – are comparable to other registries. Immunomodulatory therapies in POMS are well tolerated and efficacious and they can improve the long-term outcome in children.
Keywords: multiple sclerosis, pediatric onset, early onset, registry, Egypt
Introduction
Multiple sclerosis (MS) ranks among the most common causes of disability in young and middle-aged adults, that typically manifests between the ages of 20 and 40 years; however, up to 10% of the MS population develop their first clinical symptoms before the age of 18.1,2
In recent years, pediatric onset multiple sclerosis (POMS) has been increasingly recognized; however, because it is a comparatively rare disorder (frequency of 0.2–0.64/100,000), most investigations have been limited by relatively small sample sizes, retrospective data, or both. Indeed, the clinical and magnetic resonance imaging (MRI) presentations of POMS may differ from the adult onset, especially at disease onset and in earlier-onset pediatric MS (EOPMS) patients.3
Several studies were conducted to characterize MS patients in Egypt; however, the characteristics were defined globally to patients with a wide range of age at onset, including both with pediatric and adult onset MS.4,5 Consequently, it was important to establish a study comprising only POMS patients, in order to obtain more accurate and robust data about this understudied age group.
Thus, this study aimed to depict the demographic, clinical neurophysiological, and biochemical characteristics of Egyptian POMS patients.
Patients and methods
This study was registered with ClinicalTrials.gov (NCT03360188). It was conducted in accordance with the principles established by the 18th World Medical Assembly (Helsinki, 1964) and all applicable amendments laid down by it, International Council for Harmonization guidelines for good clinical practice, and in compliance with all national and international laws and regulations. Written informed consent was obtained from all participants or their parents in this study.
Study design
This was an observational, retrospective analysis of historic and current information on 237 patients attending five tertiary referral centers [Kasr Al-Ainy Multiple Sclerosis Research Unit (KAMSU) – Cairo University Hospitals, Abo El Reesh Pediatric Hospital, and three private centers] in Cairo, Egypt between 2011 and December 2015.
Study population
Initially, medical records of 251 patients with their first acquired demyelinating events starting before the age of 18 years were reviewed. Fourteen patients (5.58%) were excluded due to missing data that could not be obtained.
Inclusionary criteria
The included patients had the onset of the first symptoms before the age of 18 years; and they were further classified into 1) below 12 years old or early-onset pediatric multiple sclerosis (EOPMS), and 2) ≥12 years old or late-onset pediatric MS (LOPMS).
Diagnostic criteria
The International Pediatric Multiple Sclerosis Study Group (IPMSSG) criteria6 were applied on all included patients who were stratified into the following groups: 1) those with ≥2 non-encephalopathic clinical central nervous system (CNS) events (NEE) separated by more than 30 days; 2) one non-encephalopathic episode typical of MS associated with MRI findings consistent with 2010 Revised McDonald criteria for dissemination in space (DIS) and in which a follow up MRI shows at least one new lesion, consistent with dissemination in time (DIT);7 3) one acute disseminated encephalomyelitis (ADEM) attack followed ≥3 months by a non-encephalopathic clinical event associated with new MRI lesions fulfilling 2010 Revised McDonald DIS criteria; or 4) a first, single non-encephalopathic acute event associated with MRI findings consistent with the 2010 Revised McDonald criteria for DIS and DIT (applies only to children ≥12 years old). These criteria were applied retrospectively in patients diagnosed before 2013.
Pediatric clinically isolated syndrome (CIS) was defined as a first monofocal or multifocal CNS demyelinating event without encephalopathy, unless due to fever.6
ADEM was diagnosed as the first polyfocal clinical CNS event with encephalopathy that cannot be explained by fever and with diffuse, poorly demarcated, large white matter lesions (by MRI).6
Secondary progressive multiple sclerosis (SPMS) was diagnosed according to a consensus definition.8,9 Neuromyelitis optica (NMO) was diagnosed according to the 2015 diagnostic criteria;10 and patients with a diagnosis made before 2015 (n=4) were reassessed by on-site coordinators.
An Expanded Disability Status Scale (EDSS) of 4 is used as an outcome variable that indicates a limited walking ability, as it is often reached simultaneously with the transition to the secondary progression which is considered a reliable measure of an irreversible course, particularly in studies over a long time span.9
Procedures
All medical records were analyzed from June 2016 to April 2017 to collect the following data: 1) demographic characteristics; 2) clinical data; including age at onset, age at diagnosis, disease course, time to conversion to secondary progression (SP), duration of follow-up, initial and last (EDSS) score (after 3 months of last relapse), time to EDSS score of 4, disease-modifying treatment (DMT), the annualized relapse rate (ARR); 3) laboratorial data: oligoclonal bands (OCBs) in the cerebrospinal fluid (CSF) and IgG index; 4) characteristics of the initial MRI of the brain and spinal cord including: typicality of lesions on T2 weighted sequence, presence of brain black holes in T1 weighted sequence, presence of spinal cord T2 lesions, presence of gadolinium (Gd) enhancing lesions, and distribution of brain lesions (supratentorial and/or infratentorial).
Data quality
To ensure high-quality data, the accuracy and completeness were systemically assessed. The medical records were screened independently by two authors (NS and AE) and relevant data was extracted to a standard electronic form according to the KAMSU registry. An assigned coordinator from each center was requested to facilitate data collection from site-specific records; an audit site-specific data abstractor was continuously monitoring the records. Data accuracy was checked by separate authors (MA, HS, AH, MN) who reviewed the electronic forms from the two authors for their resemblance. Scheduled monthly meetings between abstractors were held to resolve any data conflicts. In the case of incomplete documentation or unclear information, verification was done by the on-site data coordinator either by phone or during a face-to-face interview with the patients or their caregivers during routine follow-up or unscheduled visits. In the case of failure to complete missing documentation, the entire case was deleted and excluded from analyses (n=14); participants disposition is shown in Figure 1.
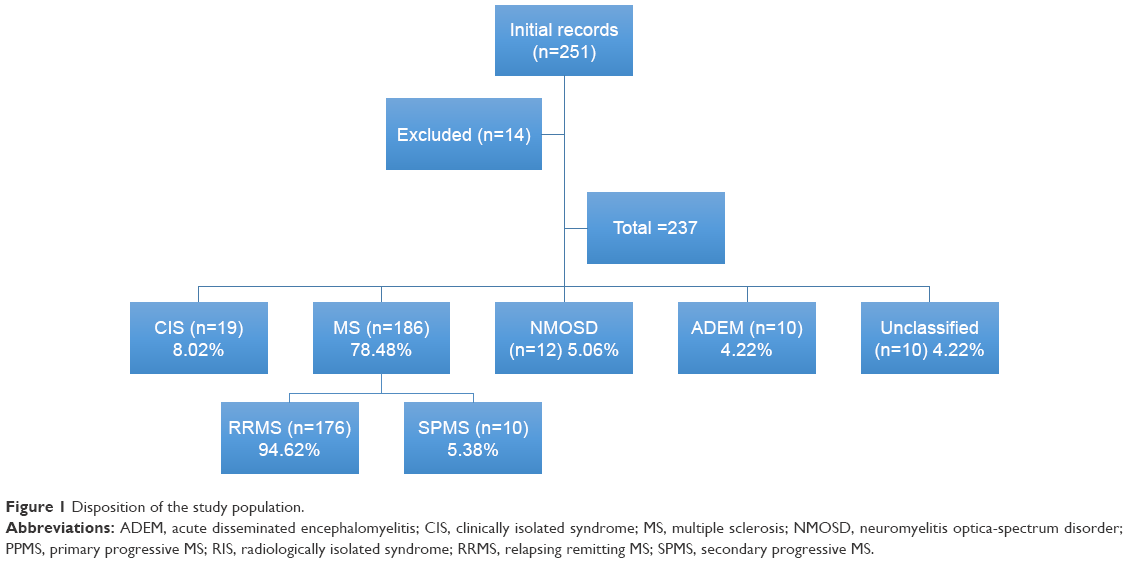 | Figure 1 Disposition of the study population. |
Several measures were taken to ensure the confidentiality of the collected information. Patient names were not included in the database presented by the site coordinator and there was no direct contact between patients and site-specific auditors. The study staff signed a confidentiality agreement for non-disclosure. The study protocol was approved by the Neurology Department Review Board at Cairo University.
Statistical analysis
All statistical analyses were performed using IBM SPSS statistics software (Version 21.0 for Windows, SPSS Inc., NY, USA). The Shapiro-Wilk test was used to determine the normality of distributions. Baseline characteristics of the cohorts were summarized as frequencies (%) or median with interquartile range (IQR) in non-uniform values, or mean ± SD when presenting the center and dispersion of normally distributed variables. Comparisons between EOPMS (<12 years) and LOPMS (≥12 years) were done for non-normally distributed variables using the Log-rank test or Mann–Whitney U-test as appropriate; while the chi-squared test was used for group comparisons of proportions between groups in clinical and para-clinical characteristics. The annual relapse rate was calculated as follows:11
|
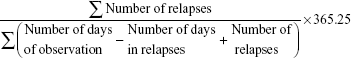
A Kaplan–Meier survival curve was used to compare the EDSS 4.0-free experience during the follow-up period of those who started their neuromodulation within 1 year from onset (early treatment) and those with delayed treatment; comparisons between early treatment and delayed treatment groups were made by the log-rank test. In all analyses, statistical significance was defined by an alpha level of 0.05.
Results
Baseline characteristics
A total of 237 patients were finally analyzed in the current study, 64.56% (153) were females, and 35.44% (84) were males. At time of data collection, MS was diagnosed in 186 patients (78.48%), and the diagnoses of 10 patients (4.22%) were not yet settled. We identified 47/186 patients (25.27%) with EOPMS. A positive family history of MS (at least one first and/or second degree relative) was reported in 15/186 patients (8.06%). The median (IQR) time between the first manifestation and confirmation of the diagnosis was 6 months (3–9) months. The demographics of current series by diagnostic category are summarized in Table 1.
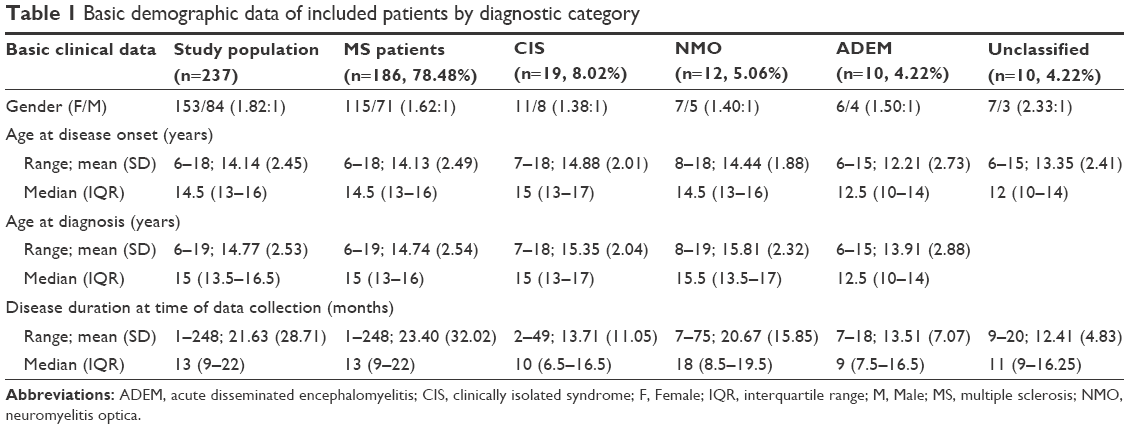 | Table 1 Basic demographic data of included patients by diagnostic category |
Clinical characteristics of MS (n=165)
Diagnostic categories and disease course
None of our 186 MS patients had PPMS; whereas 10 patients (5.38%) had SPMS; other patients (n=176) had relapsing remitting multiple sclerosis (RRMS) form and were classified according to clinical features at the time of first assessment into four groups, as shown in Table 2. The mean duration from symptoms onset to developing a secondary progressive course was 142.56±39.06 months (median =140, IQR =120.5–155), whereas mean time to develop EDSS 4 from symptoms onset was 92.21±22.79 months (median =91, IQR =73.5–101.5); which was reported in 55/186 patients (29.57%).
 | Table 2 Diagnostic categories and disease course of patients with multiple sclerosis |
Initial presentation
Out of 186 MS patients, 62.37% (n=116) had monofocal neurological presentation, and less than 10% presented with encephalopathy that could not be explained by fever or systemic illness; most of them had EOPMS. The difference in initial clinical presentation between those aged <12 years (younger age group; n=47) compared with those aged ≥12 years (LOPMS; n=139) is shown in Table 3.
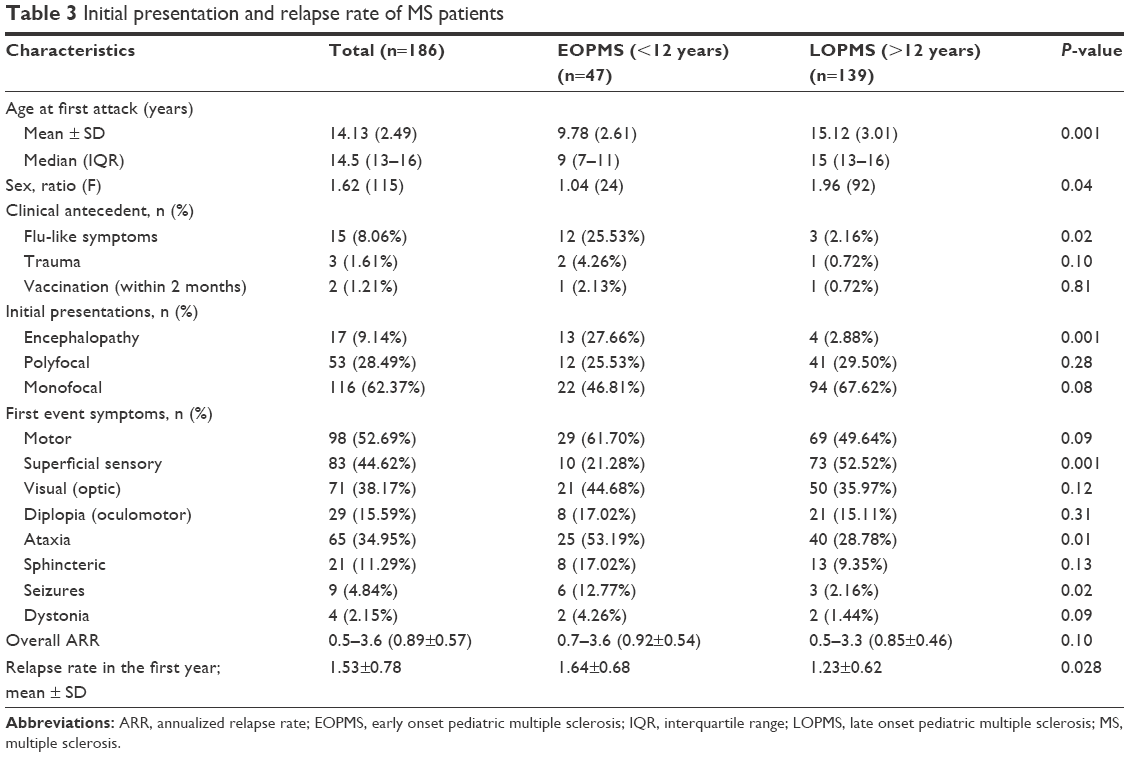 | Table 3 Initial presentation and relapse rate of MS patients |
Symptoms developing during the course of the disease
Motor weakness was the most commonly experienced symptom, followed by sensory symptoms; which were more frequently encountered in patients with onset above 12 years (LOPMS) (P<0.02), whereas seizures (not related to encephalopathy) were more frequent in those with EOPMS (<12 years) (P<0.001) (Figure 2).
 | Figure 2 Symptoms developing during the course of the disease. |
Cognitive functions were explored in 53 patients (32.12%) through an interview-based assessment for patients and their parents (which is not routinely assessed in our registry). It was affected in 28 patients (based on parental subjective reporting deterioration in scholastic achievements, with developing difficulties in engagement and planning of activities of daily living).
MRI findings and CSF analysis
Initial brain MRI scans were conducted for all patients; while initial cervical spine MRI scans were done for 144 patients with MS (87.27%). Atypical confluent lesions and the infratentorial location are significantly recognized in EOPMS. Initial cervical MRI was done for 144/165; and it was positive in 119 patients, all lesions were typically small, peripherally located, and involved less than three cord segments. The number of cervical cord lesions ranged from 1–6 with, median (IQR) of 3 (2–4).
CSF analysis was done for only 34/186 (18.29%) of our cohort (Table 4).
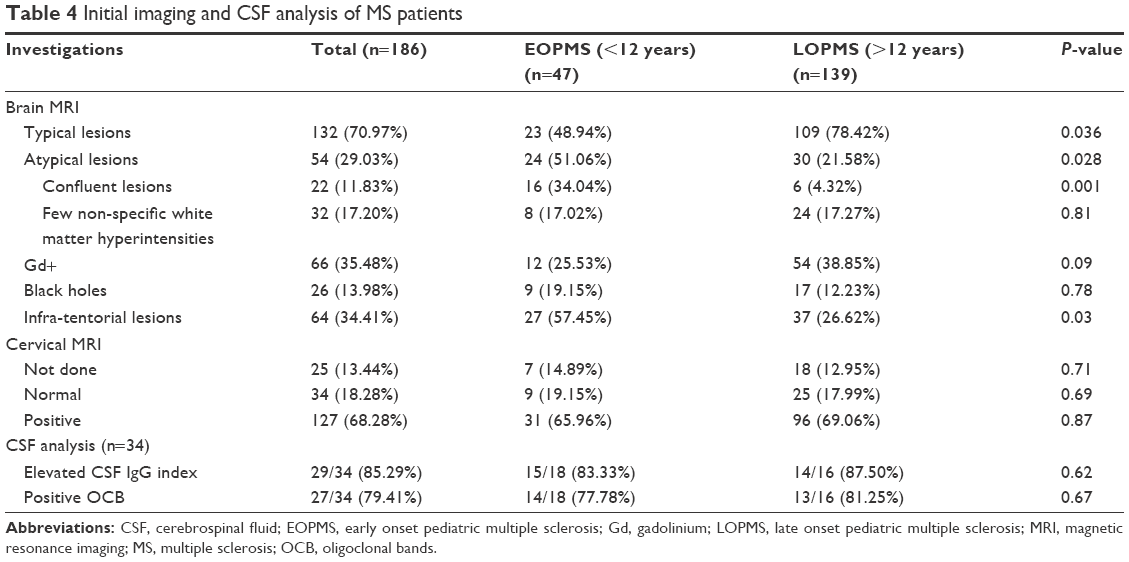 | Table 4 Initial imaging and CSF analysis of MS patients |
Therapeutic regimen
Relapse therapy
Throughout the follow-up period, a total of 504 relapses were recorded, all were treated with pulse glucocorticosteroids in the form of 25–30 mg/kg/day of intravenous Methylprednisolone “IVMP” for 5–10 days. An oral tapering over a 21- to 28-day period was used in 77 relapses (15.28%), which were associated with severe disability, or with recurrent symptoms after discontinuation of intravenous corticosteroids. In poorly responsive relapses to high doses of IVMP (n=14, 2.78%), intravenous Immunoglobulin “IVIG” has been given. Adverse events of IVMP included sleep disturbances (n=11 patients), behavioral changes (n=9 patients), appetite disorders (n=16 patients), gastritis (n=8 patients), and corticodependence (adrenal insufficiency) in two patients.
Immunomodulatory treatment
A total of 134/186 (72.04%) patients received either disease modifying treatment or immunosuppressants. About half of them (n=66) received their immunomodulatory treatment within the first year of symptoms onset (early DMT). The current therapeutic regimen is shown in Table 5. Gradual titration of the interferon to full doses unadjusted for age or body weight was done over 4–6 weeks.
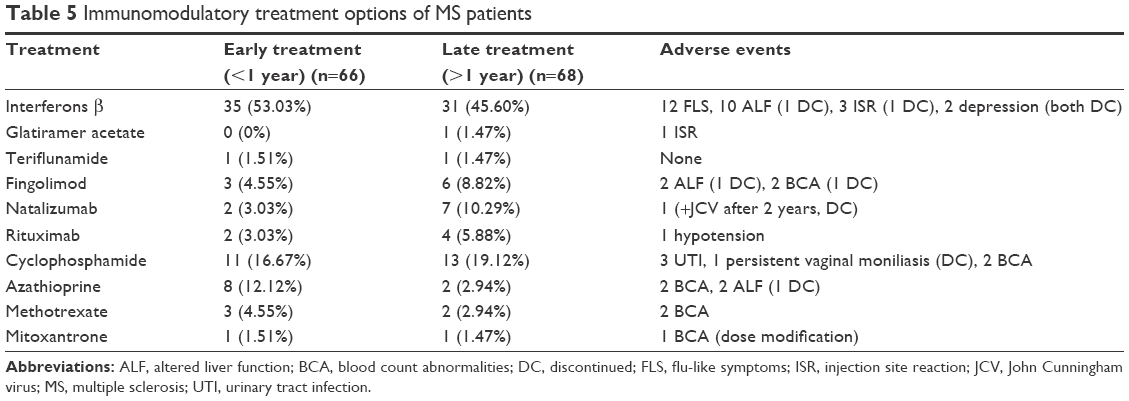 | Table 5 Immunomodulatory treatment options of MS patients |
The time to EDSS 4 from symptoms onset was significantly (P=0.011) shorter [mean (SD) =91.97 (30.16); Median (IQR) =85 (65.00–98.00) months] in those with delayed initiation of DMT (after 1 year of symptoms onset) than those with early DMT treatment [mean (SD) =118.85 (47.19); Median (IQR) =114 (89.00–145.00) months] (Figure 3).
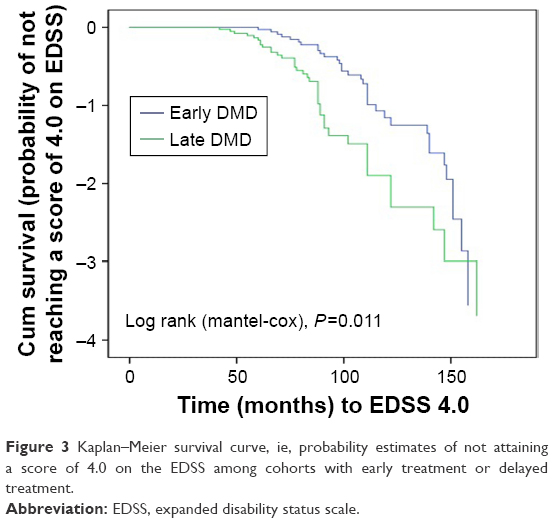 | Figure 3 Kaplan–Meier survival curve, ie, probability estimates of not attaining a score of 4.0 on the EDSS among cohorts with early treatment or delayed treatment. |
Discussion
This is one of the largest reported cohorts of POMS in the Middle East and North Africa; which included 186 POMS with disease onset before 18 years old patients. Mean age of onset in the current study was 14.14 years, which is in agreement with most pediatric MS series, where mean age of onset is 8–14 years.12–15 We noted that 25.27% (n=47) of patients seen at our centers had a young age of onset (<12 years), which is considerably higher than what was reported in other pediatric series; where EOPMS (<10 or 12 years) comprises 15%–20%.13 The female/male ratio was equal in EOPMS, and it was almost doubled in LOPMS, which may put forward the influential effects of pubertal status rather than the chronological age;1,12,15 which could explain the increase in relapse rate during the peri-menarche period.16 Genetic susceptibility of MS was displayed by several studies; which demonstrated a 7-fold increased risk of MS in relatives of adult MS patients17 and reported a positive family history between 6% and 22% in the POMS according to the degree of kinship, as some studies included first-degree, while others involved more distant relatives.13,18,19 In our study, first and second degree relatives with MS were included, with a reported family history in 8.06%.
Generally, the presenting clinical features in pediatric patients are similar to those observed in adults;1,2 however, polyfocal presentations and ADEM-like onset (encephalopathy) are common in children with MS and tend to be more frequent with younger age of onset.18,20 In our series, polyfocal presentations were reported in 28.49%; most of them had EOPMS; which was in accordance with Huppke et al,20 who found that children younger than 11 years were more likely to manifest with polyfocal features (49% compared with 37% of patients aged 14–16 years). A higher rate of polyfocal onset was recorded in the Turkish pediatric multiple sclerosis database (55%).21 On the contrary, low rates of polyfocal presentation are reported mostly from Canada and the USA,13,15 which could suggest the genetic, geographic, and/or environmental influence.21,22 Additionally, ADEM-like onset could depict a challenge in the diagnosis of POMS; as ADEM followed later by a non-encephalopathic clinical event with new lesions on brain MRI consistent with MS is one of the common clinical scenarios that account for 18%–29% of patients;23 yet, in a prospective study following the IPMSSG definitions on children with ADEM, only 6% developed MS in a 9-year follow up.24 In our series; about 10% of our MS patients developed one encephalopathic and one non-encephalopathic event.
In the current cohort, motor weakness was the presenting symptom in about half of the patients, and was the most commonly experienced symptom during the disease course (81.21%), followed by sensory deficits; both symptoms are more frequent in LOPMS. On the other hand, cerebellar presentation was detected in 34.95%, mainly in EOPMS. The frequency of presenting symptoms in our series are similar to what was reported by Belman et al;3 however, cerebellar and brain stem involvement were the ushering symptoms in about 50% of patients in other studies.21–23,25 The reason for this discrepancy is unclear, but it may be due to the inability of young children to accurately describe symptoms like sensory impairment, vertigo, or diplopia, or due to inconsistent definitions between different studies, some including both clinical and MRI involvement.21
Seizures occur in about 5% of children with MS during the course of disease, and they are much more common in children under the age of 10 years.1 This is consistent with our findings, where seizures occurred in about 6% during the follow-up period of our cohort and they were significantly more detected in EOPMS.
Cognitive deficits, fatigue, and mood disorders are frequently underdiagnosed and undertreated trifecta in POMS.26 In our cohort, cognitive profile was explored in only one-third of our cases (n=53); which showed impairment in about half of them (n=28). Cognitive dysfunction in POMS was reported in about 30%–50% of cases,3 with higher propensity with younger age, longer disease duration, and higher disability.3,27,28 It has not escaped our notice that fatigue and depression remained under-recognized by our patients or their parents; this may be due to difficulties in expressing them by children or their parents, who were minded mainly by the overwhelming outward signs of physical disability.
Patients with POMS typically have relapsing-remitting course with a tendency of having more frequent relapses early in their course, whereas the primary progressive form is exceptional.1,3,21,29–31 In our series, the overall mean ARR was 0.89; however, the mean relapse rate in the first year was significantly higher in EOPMS (1.64) vs LOPMS (1.23); which reflected the pronounced inflammatory pattern. In accordance with previous reports, none of our patients had PPMS; yet, we detected SPMS in 5.38% of our patients; which is comparable with Mikaeloff et al,18 higher than what was reported by the German Centre of Multiple Sclerosis in Childhood and Adolescents (3/166),30 but far lower than some previously reported percentages.19,31 The median duration from symptoms onset to developing a secondary progressive course was 140 months (~12 years); which emphasized that the key determinant of progression to SPMS is disease duration, which is variable in different cohorts and ranged from 4.818 to 28 years.19 This discrepancy could be attributed to sample size and duration of follow-up in each cohort.
In our cohort, 29.57% reached an EDSS score of 4 in a median time of 91 months. In long-term follow-up studies, EDSS scores of 4, 6, or 7 are considered milestones in terms of disability and disease progression. Permanent disability (EDSS ≥4.0) was reported by one study in only 15% of patients within 5 years follow-up;21,22 however, the higher percentage in the current study could be explained by the longer follow-up period, that reached up to 20 years in some patients.
In the current series, the frequency of atypical confluent MR brain lesions and the infratentorial location were significantly higher in EOPMS. Altough the imaging behavior of pediatric MS has significant similarities to adult-onset RRMS; yet, younger children often show larger lesions with less well-demarcated margins with frequent infratentorial location, which may completely resolve early in the disease.32 None of our MS patients had initial normal brain MRI; which foregrounded the MRI role in predicting future MS in children after having first demyelinating event.33 In 68.28% of our patients, cervical MRI was positive and it showed typical lesions similar to those with adult onset; the frequency of spinal cord lesions in our series is similar to rates reported by other studies.21,30
CSF analysis was done for only 18.29% of enrolled MS patients. The reason of this low percentage is bi-factorial; driven both by patients and physicians. Patients perceived a lumber puncture (LP) as a frightening and invasive procedure, resulting in high refusal rates. Also, neurologists feel reluctant to proceed with it on a routine basis, as in many patients the diagnosis was established on clinical and radiological grounds, and LP was not a prerequisite according to diagnostic criteria,8 unless alternative diagnoses were suspected.
Current therapeutic strategies in POMS are largely center-specific, with no formal consensus guidelines; however, data accumulating from retrospective and observational studies in children suggested that IFN-b1 and glatiramer acetate have similar safety and efficacy rates as an adult population.21,34 In acute relapses of POMS, IVMP 20–30 mg/kg is recommended, whereas IVIG is considered if there is an incomplete response or in case of a severe attack;1 this was adopted in our series, and the use of IVIG was considered only for poorly responsive relapses.
According to the current recommendations, POMS should be treated with disease-modifying drugs, as early as possible.34 In our series; 72.04% received either immunomodulatory or immunosuppressant treatment throughout their disease course, and about half of them received their treatment within the first year from symptoms onset (early DMT) with better outcome than those who had delayed DMT initiation. About 37% (n=69) of our patients received first-line therapies (IFN-b, GA, or Teriflunamide). Gradual titration of the interferon dosing over 4–6 weeks was a common practice in children, the majority of patients were escalated to full dose, unadjusted for age or body weight.35
In general, injectable DMTs were well-tolerated in our series, with only 4/66 in the IFN-b group who discontinued their treatment.
On the other hand, patients with highly active disease received natalizumab, fingolimod, or other IS. Huppke et al36 confirmed that natalizumab and fingolimod were well tolerated and at least equally effective in POMS highly active disease as in adults. Among the IS used, mitoxantrone was the only Food and Drug Administration (FDA) approved drug for MS in adults; however, vigorous safety-monitoring protocols are required for its use. The safety and efficacy of these powerful immunosuppressive drugs in children with MS requires a collaborative study.
The main strength of the current study is the formulation of results over a relatively large patient cohort pooled from multicenter registries using standardized, unified electronic records that provided remarkable data quality and a high level of homogeneity in the definition of clinical measures.
The main limitations to our study included a potential source of selection bias, as all centers are located in great Cairo, which could have led to an over-representation of aggressive presentations with more motor affection or encephalopathy rather than with optic or other cranial nerves. Other limitations were related to the use of different imaging protocols with different scanners among patients and different lab techniques used in CSF analyses and AQP4-antibody assessment, which made radiographic comparison and laboratory verification a bit difficult. Generally, the need for nationwide unification of patients’ data urged the MS researchers in Egypt to adopt standardized radiological and laboratory protocols among all universities and major medical institutes to allow for more multicenter studies with more homogeneous data.
Conclusion
The results of the present study conducted with a relatively large patient cohort indicate that the characteristics of POMS in Egyptians are comparable with other registries. Monofocal onset with long tract affection is more detected in LOPMS; whereas, polyfocality with cerebellar involvement and encephalopathy are more common in younger ages. The MRI appearance of POMS may show initial diffuse, large white matter lesions with ill-defined borders and a more infratentorial location; yet, resolution of such lesions can occur later. Early initiation of disease-modifying treatment carries a better outcome along the disease course.
Acknowledgments
The authors declare that they have not received any grants for this work. The project pediatric MS database is a university project that is not funded by any medical or private company.
Disclosure
The authors report no conflicts of interest in this work.
References
Banwell B, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M. Multiple sclerosis in children: clinical diagnosis, therapeutic strategies, and future directions. Lancet Neurol. 2007;6(10):887–902. | ||
Waldman A, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M, Banwell B. Multiple sclerosis in children: an update on clinical diagnosis, therapeutic strategies, and research. Lancet Neurol. 2014;13(9):936–948. | ||
Belman AL, Krupp LB, Olsen CS, et al. Characteristics of children and adolescents with multiple sclerosis. Pediatrics. 2016;138(1):e20160120. | ||
Zakaria M, Zamzam DA, Abdel Hafeez MA, et al. Clinical characteristics of patients with multiple sclerosis enrolled in a new registry in Egypt. Mult Scler Relat Disord. 2016;10:30–35. | ||
Hamdy SM, Abdel-Naseer M, Shalaby NM, et al. Characteristics and predictors of progression in an Egyptian multiple sclerosis cohort: a multicenter registry study. Neuropsychiatr Dis Treat. 2017;13:1895–1903. | ||
Krupp LB, Tardieu M, Amato MP, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler. 2013;19(10):1261–1267. | ||
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. | ||
Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–286. | ||
Tedeholm H, Lycke J, Skoog B, et al. Time to secondary progression in patients with multiple sclerosis who were treated with first generation immunomodulating drugs. Mult Scler. 2013;19(6):765–777. | ||
Wingerchuk DM, Banwell B, Bennett JL, et al. International Panel for NMO Diagnosis. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177–189. | ||
Saida T, Tashiro K, Itoyama Y, Sato T, Ohashi Y, Zhao Z; Interferon Beta-1b Multiple Sclerosis Study Group of Japan. Interferon beta-1b is effective in Japanese RRMS patients: a randomized, multicenter study. Neurology. 2005;64(4):621–630. | ||
Ness JM, Chabas D, Sadovnick AD, Pohl D, Banwell B, Weinstock-Guttman B; International Pediatric MS Study Group. Clinical features of children and adolescents with multiple sclerosis. Neurology. 2007;68(16 Suppl 2):S37–S45. | ||
Chitnis T, Glanz B, Jaffin S, Healy B. Demographics of pediatric-onset multiple sclerosis in an MS center population from the Northeastern United States. Mult Scler. 2009;15(5):627–631. | ||
Correia A, Augusto L, Meireles J, Pinto J, Sousa AP. Pediatric multiple sclerosis in Portugal: a multicentre sudy. Acta Med Port. 2016;29(7–8):425–431. | ||
Langille MM, Islam T, Burnett M, Amezcua L. Clinical characteristics of pediatric-onset and adult-onset multiple sclerosis in Hispanic Americans. J Child Neurol. 2016;31(8):1068–1073. | ||
Lulu S, Graves J, Waubant E. Menarche increases relapse risk in pediatric multiple sclerosis. Mult Scler. 2016;22(2):193–200. | ||
Nielsen NM, Westergaard T, Rostgaard K, et al. Familial risk of multiple sclerosis: a nationwide cohort study. Am J Epidemiol. 2005;162(8):774–778. | ||
Mikaeloff Y, Caridade G, Assi S, Suissa S, Tardieu M. Prognostic factors for early severity in a childhood multiple sclerosis cohort. Pediatrics. 2006;118(3):1133–1139. | ||
Renoux C, Vukusic S, Mikaeloff Y, et al. Natural history of multiple sclerosis with childhood onset. N Engl J Med. 2007;356(25):2603–2613. | ||
Huppke B, Ellenberger D, Rosewich H, Friede T, Gärtner J, Huppke P. Clinical presentation of pediatric multiple sclerosis before puberty. Eur J Neurol. 2014;21(3):441–446. | ||
Yilmaz Ü, Anlar B, Gücüyener K; The Turkish Pediatric Multiple Sclerosis Study Group. Characteristics of pediatric multiple sclerosis: the Turkish pediatric multiple sclerosis database. Eur J Paediatr Neurol. 2017;21(6):864–887. | ||
Banwell B, Krupp L, Kennedy J, et al. Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol. 2007;6(9):773–781. | ||
Mikaeloff Y, Suissa S, Vallée L, et al. First episode of acute CNS inflammatory demyelination in childhood: prognostic factors for multiple sclerosis and disability. J Pediatr. 2004;144(2):246–252. | ||
Mar S, Lenox J, Benzinger T, Brown S, Noetzel M. Long-term prognosis of pediatric patients with relapsing acute disseminated encephalomyelitis. J Child Neurol. 2010;25(6):681–688. | ||
Reinhardt K, Weiss S, Rosenbauer J, Gärtner J, von Kries R. Multiple sclerosis in children and adolescents: incidence and clinical picture – new insights from the nationwide German surveillance (2009–2011). Eur J Neurol. 2014;21(4):654–659. | ||
Suppiej A, Cainelli E. Cognitive dysfunction in pediatric multiple sclerosis. Neuropsychiatr Dis Treat. 2014;10:1385–1392. | ||
Amato MP, Krupp LB, Charvet LE, Penner I, Till C. Pediatric multiple sclerosis: Cognition and mood. Neurology. 2016;87(9 Supp 2):S82–S87. | ||
Amato MP, Goretti B, Ghezzi A, et al. Cognitive and psychosocial features of childhood and juvenile MS; two-year follow-up. Neurology. 2010;75(13):1134–1140. | ||
Gorman MP, Healy BC, Polgar-Turcsanyi M, Chitnis T. Increased relapse rate in pediatric-onset compared with adult-onset multiple sclerosis. Arch Neurol. 2009;66(1):54–59. | ||
Stark W, Huppke P, Gärtner J. Paediatric multiple sclerosis. The experience of the German Centre for Multiple Sclerosis in Childhood and Adolescence. J Neurol. 2008;255(Suppl 6):119–122. | ||
Deryck O, Ketelaer P, Dubois B. Clinical characteristics and long term prognosis in early onset multiple sclerosis. J Neurol. 2006;253:720–723. | ||
Banwell B, Arnold D, Tillema JM, et al. MRI in the evaluation of pediatric multiple sclerosis. Neurology. 2016;87(9 Supp 2):S88–S96. | ||
Tintore M, Rovira A, Martinez MJ, et al. Isolated demyelinating syndromes: comparison of different MR imaging criteria to predict conversion to clinically definite multiple sclerosis. AJNR Am J Neuroradiol. 2000;21:702–706. | ||
Tenembaum SN, Banwell B, Pohl D, et al. Subcutaneous interferon Beta-1a in pediatric multiple sclerosis: a retrospective study. J Child Neurol. 2013;28(7):849–856. | ||
Pena JA, Lotze TE. Pediatric multiple sclerosis: current concepts and consensus definitions. Autoimmune Dis. 2013;2013:673947. | ||
Huppke P, Huppke B, Ellenberger D, et al. Therapy of highly active pediatric multiple sclerosis. Mult Scler. Epub 2017 Sep 1. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.