Back to Journals » Cancer Management and Research » Volume 14
Pediatric Non-Rhabdomyosarcoma Soft Tissue Sarcomas: Standard of Care and Treatment Recommendations from the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG)
Authors Ferrari A ![]() , Brennan B, Casanova M, Corradini N, Berlanga P, Schoot RA, Ramirez-Villar GL
, Brennan B, Casanova M, Corradini N, Berlanga P, Schoot RA, Ramirez-Villar GL ![]() , Safwat A, Guillen Burrieza G, Dall'Igna P
, Safwat A, Guillen Burrieza G, Dall'Igna P ![]() , Alaggio R, Lyngsie Hjalgrim L
, Alaggio R, Lyngsie Hjalgrim L ![]() , Gatz SA, Orbach D, van Noesel MM
, Gatz SA, Orbach D, van Noesel MM
Received 17 July 2022
Accepted for publication 13 September 2022
Published 23 September 2022 Volume 2022:14 Pages 2885—2902
DOI https://doi.org/10.2147/CMAR.S368381
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Andrea Ferrari,1 Bernadette Brennan,2 Michela Casanova,1 Nadege Corradini,3 Pablo Berlanga,4 Reineke A Schoot,5 Gema L Ramirez-Villar,6 Akmal Safwat,7 Gabriela Guillen Burrieza,8 Patrizia Dall’Igna,9 Rita Alaggio,10 Lisa Lyngsie Hjalgrim,11 Susanne Andrea Gatz,12 Daniel Orbach,13 Max M van Noesel5
1Pediatric Oncology Unit, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy; 2Pediatric Oncology, Royal Manchester Children’s Hospital, Manchester, UK; 3Department of Pediatric Oncology, Institut d’Hematologie et d’Oncologie Pédiatrique/Centre, Léon Bérard, Lyon, France; 4Department of Pediatric and Adolescent Oncology, Gustave-Roussy, Cancer Campus, Université Paris-Saclay, Villejuif, France; 5Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands; 6Pediatric Oncology Unit, Hospital Universitario Virgen del Rocío, Seville, Spain; 7Oncology Department and Danish Center for Particle Therapy, Aarhus University Hospital, Aarhus, Denmark; 8Surgical Oncology and Neonatal Surgery, Pediatric Surgery Department, Hospital Infantil Universitari Vall d’Hebron, Barcelona, Spain; 9Department of Emergencies and Organ Transplantation, Pediatric Surgery, University of Bari, Bari, Italy; 10Pathology Department, Ospedale Pediatrico Bambino Gesù IRCCS, Rome, Italy; 11Department of Pediatrics and Adolescent Medicine, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark; 12Cancer Research UK Clinical Trials Unit, Institute of Cancer and Genomic Sciences, University of Birmingham, Birmingham, UK; 13SIREDO Oncology Center, Institut Curie, PSL University, Paris, France
Correspondence: Andrea Ferrari, Pediatric Oncology Unit, Fondazione IRCCS Istituto Nazionale Tumori, Via G. Venezian, 1, Milan, MI, 20133, Italy, Tel +39 02 23902588, Fax +39 02 23902648, Email [email protected]
Abstract: This paper describes the standard of care for patients with non-rhabdomyosarcoma soft tissue sarcomas (NRSTS) and the therapeutic recommendations developed by the European paediatric Soft tissue sarcoma Study Group (EpSSG). NRSTS form a very mixed group of mesenchymal extraskeletal malignancies. Their rarity, heterogeneity, and aggressiveness make the management of children and adolescents with these tumors complex and challenging. The overall cure rate for patients with NRSTS is around 70%, but survival depends on several prognostic variables, such as histotype and tumor grade, extent of disease and stage, tumor size, and tumor site. While surgery remains the mainstay of treatment for most of these tumors, a multimodal therapeutic approach including radiotherapy and chemotherapy is required in many cases. The EpSSG NRSTS 2005 study was the first prospective protocol tailored specifically to NRSTS. Together with the ARST0332 study developed by the North-American Soft Tissue Sarcoma Committee of the Children’s Oncology Group (COG), the EpSSG NRSTS 2005 study currently represents the benchmark for these tumors, establishing risk-adapted standards of care. The EpSSG has developed common treatment recommendations for the large group of adult-type NRSTS (including synovial sarcoma), and specific treatment recommendations for other particular adult-type histologies (ie, alveolar soft-part sarcoma, clear cell sarcoma and dermatofibrosarcoma protuberans); other highly malignant tumors with a biology and clinical behavior differing from those of adult-type NRSTS (ie, rhabdoid tumors and desmoplastic small round cell tumor); and soft tissue tumors of intermediate malignancy (ie desmoid-type fibromatosis, inflammatory myofibroblastic tumors, and infantile fibrosarcoma). New effective drugs are needed for patients whose NRSTS carries the worst prognosis, ie, those with unresectable tumors, metastases at diagnosis, or relapsing disease. Progress in this area relies on our ability to develop international integrated prospective collaborations, both within existing pediatric oncology networks and, importantly, between the communities of specialists treating pediatric and adult sarcoma.
Keywords: non-rhabdomyosarcoma soft tissue sarcomas, NRSTS, EpSSG, treatment, recommendations, pediatric
Introduction
Although soft tissue sarcomas are rare (accounting for less than 1% of all malignant tumors), they amount to about 8% of all malignancies in childhood and adolescence. In this age group, soft tissue sarcomas are the fifth most common type of cancer. About half of the cases are rhabdomyosarcomas, while the remainder are various entities usually grouped by pediatric experts under the label of “non-rhabdomyosarcoma soft tissue sarcomas” (NRSTS).1,2
The term NRSTS describes a very mixed group of mesenchymal extraskeletal malignancies with a clinical behavior varying from relatively benign to highly malignant.3,4 The distribution of the different tumor types varies by age: some histotypes typically occur in small children (eg, infantile fibrosarcoma and rhabdoid tumors); others are generally observed in adolescents and young adults (eg, synovial sarcoma); and many are more common in adults, and rare in children (eg, liposarcoma, leiomyosarcoma and undifferentiated pleomorphic sarcoma).1 Pediatric soft tissue sarcoma experts therefore generally deal with a different pattern of disease from those usually treated by adult sarcoma experts.5,6 For many entities, outcomes also vary by age, with a more favorable prognosis for children than for adults.1
The rarity, heterogeneity, and aggressiveness of NRSTS make the management of children and adolescents with these tumors complex and challenging.7 It is strongly recommended that patients be referred to experienced institutions where multidisciplinary teams can plan an appropriate diagnostic and treatment approach, and enroll patients in any available clinical trials.8,9 While surgery remains the mainstay of treatment for most NRSTS, a multimodal therapeutic approach – including radiotherapy and chemotherapy – is required in many cases.10 The overall cure rate for NRSTS patients is around 70%, but survival is known to depend on several prognostic variables, such as histotype and tumor grade, extent and stage of disease, tumor size, and tumor site.11–14
There has historically been a paucity of data on the natural history and treatment of these tumors.15–18 In the first decade of the new millennium, pediatric cooperative groups developed multimodal risk-adapted trials tailored specifically to NRSTS. In particular, the European paediatric Soft tissue sarcoma Study Group (EpSSG) designed a comprehensive protocol (the EpSSG NRSTS 2005) consisting of two prospective non-randomized studies for patients <21 years old with localized adult-type NRSTS or localized synovial sarcoma (originally distinguished from other adult-type histologies). The study also included specific clinical recommendations for other NRSTS histotypes found in pediatric patients. It was conducted from 2005 to 2016, and involved 100 academic centers and hospitals in 14 different countries.19 In terms of rationale, patient stratification and treatment paradigms (based on both pediatric and adult experiences), the EpSSG NRSTS 2005 study was very similar to the ARST0332 study developed by the North-American Soft Tissue Sarcoma Committee of the Children’s Oncology Group (COG), conducted from 2007 to 2012.20
The EpSSG has already reported on its analyses of various specific histological subgroups,21–32 and on the results of treatment in the whole series of patients enrolled in the prospective non-randomized part of the protocol.19 Table 1 shows the main findings they published. These analyses, together with the results of the COG ARST0332 study,20 currently represent the benchmark for NRSTS treatment in pediatric patients, and establish the risk-adapted standard of care.
 |
Table 1 Different Published Series and Main Results from the EpSSG NRSTS 2005 Study |
Standard of Care for NRSTS
The present paper describes the standard of care for NRSTS and the therapeutic recommendations developed by the EpSSG for newly-diagnosed patients. In particular, we focused on the role of systemic treatment. These recommendations were established in parallel with the development of a non-therapeutic biological study, ie, the MYKIDS study (“Molecular Identification and Characterization of non-Rhabdomyosarcoma Soft Tissue Sarcoma in Kids, Adolescents and Young Adults”). The MYKIDS study is described in more detail below.
NRSTS vary considerably, not only in their biology and clinical behavior, but also in their sensitivity to therapy. To give an example, synovial sarcoma is generally more sensitive to standard chemotherapy than tumor types like alveolar soft-part sarcoma or clear cell sarcoma, which are near-resistant.21,26,33–35 In the light of this heterogeneity, specific therapeutic recommendations should be adopted for specific histotypes.
Treatment Recommendations Applicable to Adult-Type NRSTS in General
The definition of “adult-type NRSTS” was historically created to identify to identify groups of subtypes which were as homogeneous as possible, ie, “definitely malignant soft tissue sarcomas, typical of adulthood, with morphological features resembling differentiated/mature tissues”.13 This historical definition originally included synovial sarcoma, malignant peripheral nerve sheath tumor (MPNST), adult-type fibrosarcoma, epithelioid sarcoma, alveolar soft part sarcoma (ASPS), clear cell sarcoma (CCS), leiomyosarcoma, liposarcoma, malignant fibrous histiocytoma, hemangiopericytoma, angiosarcoma, and dermatofibrosarcoma protuberans (DFSP). It excluded other histotypes, such as those characteristically occurring in young patients (ie infantile fibrosarcoma [IFS] or rhabdoid tumor), tumors with a primitive morphology (ie, small round cell sarcomas like extraosseous Ewing sarcoma and desmoplastic small round cell tumor [DSRCT]), and borderline tumors (ie, epithelioid hemangioendothelioma, desmoid-type fibromatosis, and inflammatory myofibroblastic tumors [IMT]).13 This original definition of adult-type NRSTS was adopted in the EpSSG NRSTS 2005 protocol.19
In recent years, the understanding of these tumors has improved, and their classification have been modified accordingly for some tumor entities. To give some examples: undifferentiated high-grade pleomorphic sarcoma is the name currently used for what was previously called malignant fibrous histiocytoma3,36; new entities have been recognized (such as BCOR or CIC-rearranged sarcomas);37 other entities have been recognized as clearly distinct in their clinical behavior, making a combination of ifosfamide-doxorubicin clearly inappropriate as the standard of care (eg, ASPS and CCS).26,33–35 In general terms, the definition of adult-type NRSTS may still be of value, however. Although it includes a variety of different tumors (Table 2),38 there is a consensus that most adult-type NRSTS warrant the same treatment approach.
 |
Table 2 A Practical Classification of the Main Pediatric NRSTS Histotypes |
Adult-type NRSTS are usually assumed to be relatively insensitive to chemotherapy, with reported tumor response in the range of 40–50%, or even less (and always much lower than is generally seen in rhabdomyosarcoma).39 Surgery remains the unquestionable keystone of treatment. In principle, radiotherapy has a role in local control after incomplete resections and even after wide excisions in the case of large tumors.40–43 The indications for radiotherapy are usually more limited in children, however, because of the higher risk of severe late effects. The aggressiveness and intensity of surgery and radiotherapy should be discussed and customized for each patient, based on anatomical site, tumor size, patient’s age, and response to initial chemotherapy. The aim is to achieve optimal local control while bearing in mind treatment sequelae, and the preservation of function.
The standard systemic treatment consists of ifosfamide-doxorubicin chemotherapy.19,20 Ifosfamide-doxorubicin was historically derived from adult sarcoma experience.44–49 Peri-operative chemotherapy is generally given in local/locoregional disease, as front-line treatment in patients with locally advanced disease, and/or when surgeons are not sure they can achieve a complete resection at the first attempt. Chemotherapy is preferably given in the neo-adjuvant setting. Neo-adjuvant chemotherapy may have a role not only in converting such cases into conservative complete resections but also for promptly treating any micrometastases.39
There is much debate on whether adjuvant chemotherapy should be administered for adult-type NRSTS to prevent distant recurrences after initial surgery.44–49 Various reports suggest that, after initial tumor resection, a high tumor grade combined with a large tumor size pose a very high risk of metastases, irrespective of the radicality of the initial surgery, so survival might be better in patients given adjuvant chemotherapy.50
In the light of the results of the EpSSG NRSTS 2005 study,19 current EpSSG recommendations divide patients into four treatment groups based on surgical stage according to the Intergroup Rhabdomyosarcoma Study (IRS) classification, tumor size, nodal involvement, and histopathological tumor grade (tumor site is added as a criterion for synovial sarcoma). (Figure 1)
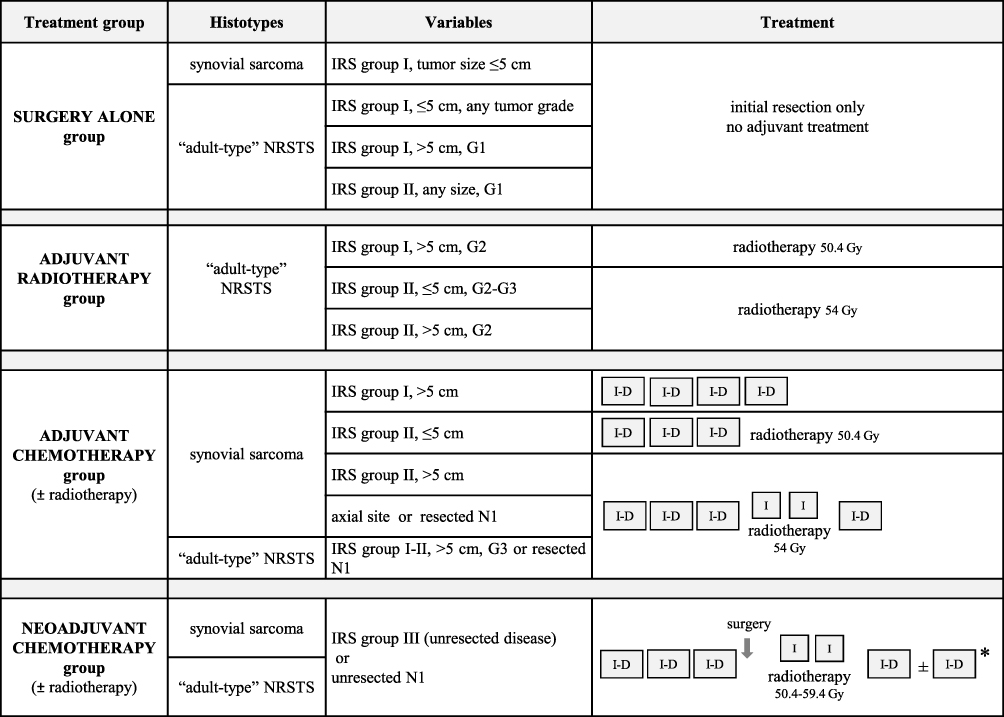 |
Figure 1 EpSSG standard-risk stratification and treatment recommendations for local/locoregional adult-type NRSTS (including synovial sarcoma). Abbreviations: IRS, Intergroup Rhabdomyosarcoma Study; IRS group I, complete resection at first surgery (initial R0 surgery); IRS group II, microscopic residual disease after initial R1 surgery); IRS group III, biopsy or initial macroscopic residual disease after R2 surgery; IRS group IV, metastatic disease at onset; G, tumor grade (according to the Fédération Nationale des Centres de Lutte Contre le Cancer grading system); N1, nodal involvement. Notes: I-D=ifosfamide (3 g/m² per day intravenously, for 3 days) plus doxorubicin (37.5 mg/m² intravenously per day for 2 days). I=ifosfamide (3 g/m² intravenously per day for 2 days). Ifosfamide should be given with hyperhydration and mesna infusion (3 g/m² per day intravenously). Chemotherapy cycles should be administered every 21 days. *Total of 6 courses for synovial sarcoma, 7 courses for the other adult-type NRSTS. |
The group recommended for surgery alone includes low-grade adult-type NRSTS treated with an initial R0-R1 resection (IRS group I–II), and high-grade NRSTS smaller than 5 cm treated with an initial R0 resection. The 5-year event-free survival (EFS) and overall survival (OS) recorded for this category in the EpSSG NRSTS 2005 study (ie, 91.4% and 98.1%, respectively) suggest that adjuvant chemotherapy and radiotherapy can be safely omitted for this low-risk population in an effort to contain short- and long-term treatment-related morbidity.19
Radiotherapy (at a dose of 54.0 Gy) is recommended for high-grade tumors after R1 resections (IRS group II). Based on the NRSTS 2005 study data, the EpSSG also recommends radiotherapy (50.4 Gy) for patients classified as IRS group I, with tumors >5 cm in size, and G2 disease.19
In the adjuvant chemotherapy group, the recommended number of cycles depends on the variables listed in Figure 1. In the EpSSG NRSTS 2005 study, the 5-year EFS and OS were 65.6% and 75.8%, respectively, suggesting that – despite a generally favorable prognosis for grossly resected NRSTS – patients with high-grade and/or large tumors are at high risk of treatment failure. The study struggled to investigate the role of adjuvant chemotherapy in this patient category, however, due to a relatively limited sample size (93 cases).19
For the neoadjuvant chemotherapy group (ie, patients with locally advanced and/or unresectable disease), the EpSSG NRSTS 2005 study reported an overall response rate of 54%, and 5-year EFS and OS of 56.4% and 70.4%, respectively.19 A major open issue remains for this patient category concerning the local treatment to administer after initial chemotherapy, given the diverse clinical situations encountered, relating to patients’ ages, and the tumors’ subtype, size, site, and resectability. The EpSSG recommends planning (and customizing) the “best possible local treatment” with the aim of maximizing the chances of local control, while minimizing radiation-related sequelae and preserving function. To give an example of the different clinical situations that can occur when delayed resection is feasible, patients might be given radiotherapy (50.4 Gy) before surgery, or surgery without any radiotherapy (before or afterwards), or surgery followed by radiotherapy (50.4 Gy after R0 resections, 54.0 Gy after R1, and 59.4 Gy after R2). Decisions should be based on a multidisciplinary discussion, considering a patient’s age (eg, trying to avoid radiotherapy for patients less than 6 years old), or initial tumor size (eg, using radiotherapy for tumors initially over 10 cm in size), or surgical margins at delayed surgery (in principle, postoperative radiotherapy should be recommended after delayed resections other than R0, when re-surgery is not possible or would lead to undesirable anatomic or functional outcomes). Then, there are patients whose tumor cannot be resected: for them, radiotherapy alone is the only local therapy possible, at a recommended dose of 59.4 Gy.
Synovial Sarcoma
Among all adult-type NRSTS, synovial sarcoma is the subtype most often occurring in pediatric age.51 Its hallmark is a specific t(X;18)(p11.2;q11.2) chromosomal translocation, and the SYT::SSX fusion transcript (in its various forms). Many pediatric oncologists had long considered synovial sarcoma separately from the other histotypes, given the relatively high rates of response to chemotherapy reported in historical pediatric synovial sarcoma series (approximately 60%, which is higher than that usually reported for adult soft tissue sarcomas).52–57 Up until 2005, most European pediatric oncologists treated synovial sarcoma patients with the protocols designed for rhabdomyosarcoma (which meant, eg, intensive multidrug chemotherapy for at least 6 months, and adjuvant chemotherapy for all patients, even if they had completely excised, small tumors).52–57 This changed with the EpSSG NRSTS 2005 study, the “rhabdomyosarcoma-like” strategy was replaced with a treatment approach that also draws on adult experiences with: chemotherapy given according to patients’ risk stratification (based on tumor size, site and stage); the ifosfamide-doxorubicin regimen; a shorter duration of chemotherapy, even for higher-risk patients; and the omission of chemotherapy for low-risk cases (completely resected limb tumors under 5 cm in size).21,58
The EpSSG NRSTS 2005 study achieved higher survival rates than those published previously by pediatric groups, with 5-year EFS and OS of 80.7% and 90.7%, respectively, for patients with localized synovial sarcoma.21 The study demonstrated the feasibility of a “surgery-alone” strategy for low-risk patients, and this was subsequently confirmed by a joint retrospective EpSSG-COG analysis on 60 cases of small tumors completely resected at diagnosis: there were only 8 local relapses, no metastatic recurrences, and the 3-year EFS and OS were 90% and 100%, respectively.24
Management of Metastatic Disease and Relapsing NRSTS
The EpSSG NRSTS 2005 study did not include patients with adult-type NRSTS who had metastatic disease at diagnosis. Instead, the EpSSG reported on a series of 49 cases of metastatic NRSTS treated between 2008 and 2013 included in the BERNIE protocol. This was an open-label, multicenter, randomized Phase II study assessing the role of bevacizumab in addition to the intensive chemotherapy designed for rhabdomyosarcoma, and including ifosfamide, vincristine, actinomycin-D and doxorubicin (the IVADo regimen), and maintenance therapy with vinorelbine and oral low-dose cyclophosphamide. The 2-year EFS was 27.3% and the 3-year OS was 35.2%. The study showed that adding the anti-angiogenic agent did not prompt any statistically significant improvement in patients’ survival.32
The treatment of metastatic NRSTS remains a big challenge, and a clear standard of care has not yet been defined. Patient management should always be multidisciplinary, taking into account the diverse clinical presentation and histotypes. In principle, patients should be included in specific clinical trials, if available. There is a general agreement to treat metastatic NRSTS patients with systemic therapy and aggressive surgery on the primary tumor and metastatic sites. The ifosfamide-doxorubicin regimen is generally used in children and adolescent with metastatic adult-type NRSTS. In adult experience, there is no formal demonstration that multi-agent chemotherapy is superior to single-agent doxorubicin in terms of OS, though higher response rate and longer progression-free survival were reported.59,60
Similar to patients with metastases at diagnosis, those with relapsing NRSTS also have a very unfavorable prognosis: in a recent study, post-relapse OS was 25.8% at 5 years, with a median survival of 20 months.61 Poor survival rates are also reported for patients with relapsing synovial sarcoma, despite the better outcomes for patients with primary localized disease compared to other adult-type histologies.62
A detailed discussion of the possible treatment options for patients with relapsing NRSTS goes beyond the scope of this article. As a general recommendation, surgery and radiotherapy should be regarded as a key part of the treatment in cases of local relapse. Aggressive resection may be lifesaving, including extensive procedure that might not have been considered acceptable in primary treatment. Surgery and radiotherapy may have a role in cases of distant relapse as well, especially when the lung is the only site, and when the number of metastases is small.
Effective systemic therapies are limited, and no standardized second-line chemotherapy is available. In patients already treated with the ifosfamide–doxorubicin regimen, possible options might be high-dose ifosfamide63–65 (one a particular regimen involves ifosfamide 14 g/m2 given in a continuous 14-day infusion via an external pump)66; regimens such as gemcitabine-docetaxel, or gemcitabine-vinorelbine67,68; or disease-specific drugs, such as paclitaxel in angiosarcoma, eribulin in liposarcoma, and trabectedin in liposarcoma and DSRCT.69–73
Every effort should be made by the pediatric sarcoma community to develop prospective Phase I–II trials for this patient category.74 A stronger cooperation with the adult sarcoma community is also warranted, to enable children and adolescents to benefit from new agents that have already proven effective in adult patients.75,76 A good example concerns the T-cell therapies engineered to target MAGE-A4+ in synovial sarcoma. These modified T-cells are only intended for patients positive for HLA-A*02 whose tumors express MAGE-A4 (expected to be 30% of all patients), but this therapy has shown outstanding disease control rates and objective response rates (85% and 35%, respectively) in adults with synovial sarcoma. Importantly, the SPEARHEAD-1 trial is currently open to patients >10 years old at a few selected European pediatric oncology centers. The EpSSG recommends a (remote) prescreening of all patients with relapsing synovial sarcoma at the time of the first relapse, so that suitable patients can be referred to the centers where the trial is running.77
Treatment Recommendations Specific to Certain Adult-Type NRSTS
ASPS, CCS, and DFSP necessitate specific treatment recommendations, particularly as concerns medical therapies (Table 3).
 |
Table 3 Summary of Treatment Recommendations for Specific Histotypes |
Alveolar Soft Part Sarcoma
ASPS is a highly malignant tumor, carrying a high risk of metastatic dissemination, but conventional chemotherapy has proven ineffective.26,33,34 In cases of localized ASPS, surgery (possibly combined with radiotherapy) is the treatment of choice, and no adjuvant chemotherapy is recommended. For locally advanced/unresectable or metastatic disease, the clinical approach is more debatable.78 Metastatic disease may take an indolent course, and surgery is recommended for its treatment, if feasible. There is a growing body of evidence to indicate that targeted agents like sunitinib, cediranib, pazopanib, tivantinib or bevacizumab may produce a tumor response and prolong survival in cases of ASPS.79,80 Promising findings regarding immune checkpoint inhibitors would also suggest a role for immunotherapy (PD1/PDL1 inhibitors) administered within clinical trials (if available) or on compassionate grounds.81–84
Clear Cell Sarcoma
CCS of tendons and aponeuroses is extremely rare in childhood and little information is available on its clinical behaviour in pediatric age.35 It is generally described as a very aggressive tumor with a very limited responsiveness to standard chemotherapy. CCS exhibits an immunohistochemical similarity to melanoma, so the success of immunotherapy with checkpoint inhibitors in cases of melanoma has prompted studies to assess whether the same clinical benefit can be achieved in CCS as well, with promising initial results.85
Dermatofibrosarcoma Protuberans
DFSP occurring in children is often reportedly a low-grade, small, superficial tumor, completely resected at diagnosis in most cases.31,86 Advanced unresectable or metastatic cases are extremely uncommon in pediatric age, as well as the transformation to high-grade fibrosarcoma. Targeted therapy has proven effective medical treatment in such cases. In most cases, DFSP is caused by a chromosomal translocation resulting in the fusion protein COL1A1::PDGFB, which promotes tumor growth through overproduction of platelet-derived growth factor (PDGF). The multi-tyrosine kinase inhibitor imatinib mesylate has been used successfully to obtain a clinical response in adult patients with advanced and metastatic DFSP, and may be recommended as systemic treatment in such cases. Target therapy can improve the chances of subsequent complete resection even in cases of advanced disease.87–89
Treatment Recommendations Specific to Other Types of NRSTS
Dedicated treatment recommendations are also needed for certain highly malignant tumors with biological features, and a clinical course and response to treatment clearly different from those of adult-type NRSTS (Table 3). Two particularly challenging histotypes are rhabdoid tumors and DSRCT.
Malignant Rhabdoid Tumor
Extracranial malignant rhabdoid tumors are rare, highly aggressive tumors occurring in infants and young children with biallelic mutations of the SMARCB1 (95%) or SMARCA4 (5%) genes encoding respectively for INI1 and BRG1, which are both core subunits of the SWI/SNF chromatin remodeling complex.90 Although an aggressive multimodal strategy is generally adopted to treat young patients with rhabdoid tumors, the outcome remains largely unsatisfactory (due partly to the difficulties of delivering optimal local therapy in patients that are often very young).23 In the absence of dedicated clinical trials, the EpSSG recommends the treatment plan described in the amended version of the EpSSG NRSTS 2005 protocol, ie, dose-intensive chemotherapy every 2 weeks with vincristine-doxorubicin-cyclophosphamide (VDCy), and ifosfamide-etoposide (IE). This treatment proved feasible (with no short-term toxicity of note) in a French series of 35 cases treated between 2014 and 2019, and achieving a 2-year EFS and OS of 47.6% and 42.9%, respectively.91 Further investigations are needed to better clarify the potential role of targeted therapies, such as epidrugs (eg, EZH2 or HDAC inhibitors)92,93 or checkpoint inhibitors.94,95
Desmoplastic Small Round Cell Tumor
Characterized by the recurrent t(11;22)(p13;q12) chromosomal translocation that gives rise to the EWS::WT1 fusion gene, DSRCT generally occurs in the abdominal and pelvic cavity of teenagers or young adults. It presents as a large abdominal mass and is already widely disseminated at the time of diagnosis in most cases, classically with multiple peritoneal lesions and seeding. DSRCT is associated with a very poor prognosis (median survival reportedly ranging from 17 to 25 months).96,97 Attempts to improve the outcome over the past 10–20 years have included aggressive surgery, hyperthermic peritoneal perfusion with cisplatin chemotherapy (HIPEC), high-dose chemotherapy with autologous peripheral stem cell rescue, whole abdomen radiotherapy, and targeted therapy with monoclonal antibodies and multikinase inhibitors.98–106 Whilst awaiting new effective therapies and dedicated trials, the EpSSG recommends an intensive therapeutic approach to DSRCT patients, such as Kushner’s P6 regimen (which includes VDCy and IE chemotherapy),98 the IVADo regimen used for metastatic rhabdomyosarcoma,107 or chemotherapy with irinotecan, ifosfamide, vincristine, actinomycin-D (IrIVA).108 Intensive chemotherapy should be integrated with local control measures, including aggressive surgery (with debulking of large peritoneal tumors and resection of all detectable nodules), and whole abdominopelvic radiotherapy. The addition of prolonged vinorelbine and low-dose oral cyclophosphamide maintenance therapy may also be an option.109
Soft Tissue Sarcomas with BCOR and CIC Rearrangement
Another category of aggressive tumors is the BCOR family of undifferentiated sarcomas, characterized by genetic abnormalities involving BCOR (a transcriptional repressor gene, with a key role in regulating development, hematopoiesis and mesenchymal stem cell differentiation).110 BCOR::CCNB3 sarcomas have a predilection for young males and tend more frequently to involve bones, but also soft tissues and also visceral sites (eg, kidney). BCOR-ITD sarcomas are another BCOR subtype that reveals internal tandem duplications (ITD).110,111
The CIC-rearranged sarcomas (CIC-DUX4 sarcomas) form another group of aggressive tumors112,113 that, like BCOR sarcomas, have been identified relatively recently.
Scant information is available on the clinical course of BCOR and CIC rearranged sarcomas and the best therapeutic approach. These tumors are generally treated using a multimodal approach that includes intensive multi-agent chemotherapy derived from protocols used for Ewing sarcoma. Shared and clear-cut treatment recommendations are lacking, however, and international collaborative studies are being developed to collect more clinical and biological data, compare the available treatments, and hopefully reach an international consensus on how these tumors should be managed.
Treatment Recommendations for Soft Tissue Tumors of Intermediate Malignancy
The vast group of NRSTS also includes three specific entities defined as soft tissue tumors of intermediate malignancy, ie, desmoid-type fibromatosis, IMT and IFS. The approach to the treatment of these three sarcomas has changed to some degree in recent years.
Desmoid-Type Fibromatosis
Desmoid-type fibromatosis (or aggressive fibromatosis) is a deep-seated, musculo-aponeurotic, fibroblastic tumor of borderline malignancy. It is characterized by a marked local aggressiveness and a strong tendency for local recurrence, but no tendency to metastasize (as truly malignant tumors do). Its pathogenesis is multifactorial, and may involve endocrine factors, trauma (including surgical trauma) and genetic predisposition. Desmoid tumors can be sporadic, due mainly to a pathogenic somatic variant in CTNNB1, which encodes beta-catenin; or they can arise in the setting of familial adenomatous polyposis (FAP), in which case they often feature mesenteric lesions with a more aggressive behavior.114,115
In the last decade, the approach to treating desmoid-type fibromatosis has shifted significantly from surgery to a wait-and-see strategy in the case of non-evolving disease.116 This is largely because surgery is rarely a definitive solution (while it can be mutilating and also stimulate fibromatosis), and because desmoids may remain stable for lengthy periods, and even regress spontaneously. Based on the results obtained in the prospective series enrolled in its NRSTS 2005 study, the EpSSG recommends first adopting a wait-and-see strategy to assess the tumor’s rate of growth (or potential spontaneous regression), especially for tumors not at life-threatening sites (Figure 2).22 Treatment should therefore be considered in cases of frank tumor progression, increasing pain/symptoms, or that involve life-threatening sites. The proposed first treatment in such cases is a minimal-morbidity systemic therapy, ie, low-dose intravenous methotrexate plus vinblastine. The therapy should be given at full dose for at least 6 months (the response may take time), possibly followed by another 6 months with wider-spaced doses (ie, doubling the time between injections). The role of surgery after systemic therapy remains an open question, and it is unclear whether delayed resection (if complete and non-mutilating) or a wait-and-see approach would be better after tumor stabilization. The currently preferred approach is to wait and see, without performing any surgery that might stimulate tumor cell growth during wound healing due to the release of local growth factors.
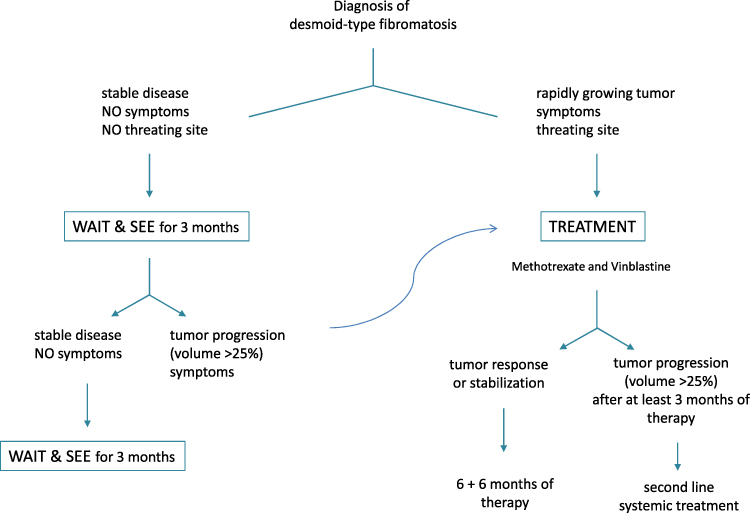 |
Figure 2 EpSSG flow chart for desmoid-type fibromatosis. |
The pharmacological treatment of desmoids can involve various non-cytotoxic and cytotoxic agents, and the response rate is generally in the range of 25–40%. Multi-tyrosine kinase inhibitors are promising (in particular pazopanib and sorafenib), and in adult patients are often consider the first systemic choice.116 However, their role needs to be clarified in prospective pediatric studies.117 Other novel drugs that hold promise, such as gamma-secretase inhibitors (AL10, Nirogacestat) or β-catenin inhibitors (tegavivint), are now under investigation in adults/adolescents and/or in children.116
Due to the potential for long-term cosmetic or functional morbidity in children, radiation therapy may have a role only after several systemic therapies have failed, or in the case of progression despite multiple surgical procedures. Finally, it is worth mentioning the recent interest shown in cryotherapy as a safe and effective treatment modality for extra-abdominal desmoid tumors.118
Inflammatory Myofibroblastic Tumors
IMT is another mesenchymal tumor of intermediate malignancy that occurs mainly in the lungs or abdomen of children and young adults (but also at other sites). It is mainly a monoclonal tyrosine-kinase-driven neoplasm characterized by a rearrangement of the ALK (anaplastic lymphoma kinase) gene with various partner genes (in more than 60% of the cases) or fusions involving ROS1, PDGFRβ, RET, and NTRK.119 Although it generally takes a benign course, IMT can be locally aggressive, and even carry some risk of metastases (in 5% of the cases). Surgery remains the mainstay of treatment and the prognosis is usually good when the tumor is widely resected. In cases of advanced disease, the treatment approach was historically based on corticosteroids or chemotherapy. Various degrees of response were reported, with some authors in favor of low-morbidity regimens like those used for desmoid fibromatosis.30,120,121
The treatment strategy has changed substantially in recent years, however, with emerging evidence of the high levels of activity of ALK inhibitors (crizotinib, ceritinib, alectinib, and brigatinib), and – more recently – ROS1, RET and NTRK inhibitors (for cases with the corresponding molecular abnormality).122,123 The EpSSG recommends no adjuvant therapy after initial R0 or R1 resection, but chemotherapy (vinblastine plus low-dose methotrexate) is still a valid first option in cases of unresectable disease.30 The role of ALK inhibitors (like that of tyrosine kinase inhibitors in general) as a first treatment option still needs to be clarified. That said, tyrosine kinase inhibitors (based on the molecular fusion identified) – if available, and preferably within clinical trials – could be a valuable option in the armamentarium of systemic therapies, given the high likelihood of a response. Things are changing rapidly in the field of tyrosine kinase inhibitors, with several clinical trials currently recruiting IMT patients, some even for upfront treatment if non-mutilating surgery is not feasible. When targeted treatments against ROS1, RET and NTRK translocated IMT become available as well, the approach to this disease can be expected to change.124
Infantile Fibrosarcoma
IFS is the most common soft tissue sarcoma occurring in infants under 1 year old. It is considered a tumor with a low malignant potential (rarely metastasizing). It is characterized by the recurrent t(12;15)(p13;q25) translocation with the transcript ETV6::NTRK3, or, in a minority of cases, by fusions involving other genes, such as RAF, RET, or NTRK1.125 IFS is generally located in deep soft tissues of the distal extremities (and less frequently in the trunk). Its clinical behavior may be peculiar, with a rapid initial growth to a huge size, but a good response to preoperative chemotherapy – even with a mild regimen like the vincristine–actinomycin-D (VA) combination – and an excellent prognosis.126,127
The EpSSG recommends conservative tumor resection for localized disease and VA chemotherapy as first-line therapy for patients with unresectable disease (no adjuvant chemotherapy is recommended in cases classified as IRS group I–II).22 More intensive regimens should be considered only in the event of no response to VA. Particular attention should be paid to the dosage of chemotherapy (given patients’ very young age) and to limiting the functional sequelae of surgery. The treatment options for IFS have very recently begun to change, however, with the arrival on the scene of very effective biological agents: NTRK inhibitors like larotrectinib and entrectinib have produced very rapid responses in the vast majority of children with IFS, with a limited acute toxicity.128 An international consensus has proposed front-line treatment with either conventional chemotherapy or NTRK inhibitors for patients with advanced localized disease (at the physician’s and parents’ discretion), while NTRK inhibitors might be the best option for patients with metastatic disease, or to avoid mutilating surgery in cases of insufficient response to chemotherapy.125
Future Studies
The new challenge for the EpSSG is developing a systematic multicenter molecular and epigenetic characterization of cases of NRSTS as a standard approach at the time of their diagnosis.129–133 The EpSSG MYKIDS study is a prospective biological investigation recruiting patients in EpSSG countries, that was ready to start at the time of writing this review. It aims to identify novel translocations and genetic drivers, and to characterize as yet unknown or unclassified sarcomas using whole-exome sequencing (WES), mRNA sequencing (mRNAseq), and DNA methylation profiling. Ultimately, this should lead to an integrated diagnosis, and a comprehensive understanding of the treatment options for a given patient. MYKIDS also aims to identify new, reproducible molecular signatures for predicting outcome and refine risk stratification. In particular, the study will examine the predictive role of chromosomal instability, assessed with a genomic index or a more complex biological signature known as CINSARC (Complexity Index in Sarcoma, a 67-gene signature related to chromosome integrity and genomic complexity).27,134 Other working packages within the MYKIDS study focus on liquid biopsy,127 the development of faithful tumor models such as organoids,132 and post-treatment molecular changes.
New effective drugs are needed for patients with NRSTS that carry a poor prognosis, ie those with unresected tumors, metastases at diagnosis, or relapsing disease. Pediatric sarcoma experts are looking for novel agents that can target the multiple signaling pathways involved in tumorigenesis across NRSTS subtypes, and might enhance the effect of conventional cytotoxic chemotherapy. The impact of new targeted agents for the pediatric population has not paralleled the progress seen in adult sarcoma patients. More international collaboration between pediatric and adult sarcoma communities is crucial, both to facilitate the transfer of potentially effective new agents from adults to children and adolescents, and to improve research programs. The ultimate goal should be to develop shared clinical trials for children, adolescents and adults with the same disease. Whether the results seen in adults can be achieved in pediatric patients, and whether a given soft tissue subtype has the same behavior (and response to treatment) in different age groups are questions that remain to be answered, and deserve to be explored.
The EpSSG is currently working on the development of a study to investigate whether adding regorafenib to the standard ifosfamide-doxorubicin chemotherapy can improve survival in high-risk NRSTS. If it gets underway, this study will compare to the North-American trial on pediatric and adult NRSTS patients, ie, the COG ARST1321 study conducted between 2014 and 2018, in which pazopanib was added to the standard treatment. At a planned interim analysis, the study showed a significant improvement in the pathological response rate after adding pazopanib, which reached a preset threshold so patient enrolment was stopped.135
Conclusions
The current paper describes the state of the art and the therapeutic recommendations developed for NRSTS by the EpSSG. The EpSSG NRSTS 2005 study, together with the COG ARST0332 study, was able to define a risk-adapted standard of care, and demonstrated the importance of a standardized multimodal approach to such rare and diverse tumors as NRSTS.
Improving survival for patients with advanced or metastatic disease remains challenging for a heterogeneous group of tumors with variable or uncertain chemosensitivity. While awaiting new effective drugs with novel mechanisms of action, more standardised or optimised use of available therapy might improve patients’ outcome. The current standard of care for pediatric NRSTS demonstrates how, in recent years, the clinical approaches adopted in the pediatric sarcoma community and in adult setting have converged towards common strategies.
To further refine the multidisciplinary treatment strategy for the different tumor types and risk groups, it is important to develop a uniform approach to the specific challenges of managing these tumors, and devise possible solutions together. It is with this goal in mind that the INternational Soft Tissue SaRcoma ConsorTium (INSTRuCT) was recently established. INSTRuCT was founded by three large cooperative groups – the EpSSG, COG, and Cooperative Weichteilsarkom Studiengruppe (CWS) – with the aim of pooling expertise and resources on a broader international level, developing consensus standards to guide diagnosis and treatment, and comparing clinical data across different groups and studies.7,10,136
Disclosure
Daniel Orbach and Max M van Noesel are co-last authors for this study. Dr Susanne Andrea Gatz reports personal fees from TESARO, BAYER, and EMD Serono, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Ferrari A, Sultan I, Huang TT., et al. Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer. 2011;57(6):943–949. doi:10.1002/pbc.23252
2. Corey RM, Swett K, Ward WG. Epidemiology and survivorship of soft tissue sarcomas in adults: a national cancer database report. Cancer Med. 2014;3:1404–1415. doi:10.1002/cam4.288
3. WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours. Lyon (France): International Agency for Research on Cancer; 2020. (WHO classification of tumours series, 5th ed.; vol.3). ISBN 978-92-832-4502-5. https://publications.iarc.fr/588.
4. Pfister SM, Reyes-Múgica M, Chan JKC, et al. A summary of the inaugural WHO Classification of Pediatric Tumors: transitioning from the optical into the molecular era. Cancer Discov. 2022;12(2):331–355. doi:10.1158/2159-8290.CD-21-1094
5. van der Graaf WTA, Orbach D. Soft tissue sarcomas in adolescents and young adults: a comparison with their paediatric and adult counterparts. Lancet Oncol. 2017;18:e166–e175. doi:10.1016/S1470-2045(17)30099-2
6. Ferrari A, Bleyer A, Patel S, et al. The challenge of the management of adolescents and young adults with soft tissue sarcomas. Pediatr Blood Cancer. 2018;65(7):e27013. doi:10.1002/pbc.27013
7. Ferrari A, Spunt SL, Sparber-Sauer M, et al. Controversies and challenges in the management of pediatric non-rhabdomyosarcoma soft tissue sarcomas. Lancet Child Adolesc Health. 2022;6(4):221–223. doi:10.1016/S2352-4642(22)00036-0
8. Gutierrez JC, Perez EA, Moffat FL, et al. Should soft tissue sarcomas be treated at high-volume centers? An analysis of 4205 patients. Ann Surg. 2007;245(6):952–958. doi:10.1097/01.sla.0000250438.04393.a8
9. Collignon C, Carton M, Brisse HJ, et al. Soft tissue sarcoma in children, adolescents and young adults: outcomes according to compliance with international initial care guidelines. Eur J Surg Oncol. 2020;46(7):1277–1286. doi:10.1016/j.ejso.2019.11.518
10. Ferrari A, Orbach D, Sparber-Sauer M, et al. The treatment approach to pediatric non-rhabdomyosarcoma soft tissue sarcomas: a critical review from the INternational Soft Tissue SaRcoma ConsorTium. Eur J Cancer. 2022;169:10–19. doi:10.1016/j.ejca.2022.03.028
11. Spunt SL, Poquette CA, Hurt YS, et al. Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children’s Research Hospital. J Clin Oncol. 1999;17:3697e705. doi:10.1200/JCO.1999.17.12.3697
12. Spunt SL, Hill DA, Motosue AM, et al. Clinical features and outcome of initially unresected nonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol. 2002;20:3225e35. doi:10.1200/JCO.2002.06.066
13. Ferrari A, Casanova M, Collini P, et al. Adult-type soft tissue sarcomas in pediatric age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol. 2005;23:4021e30. doi:10.1200/JCO.2005.02.053
14. Ferrari A, Miceli R, Casanova M, et al. Adult-type soft tissue sarcomas in pediatric age: a nomogram-based prognostic comparison with adult sarcomas. Eur J Canc. 2007;43:2691e7. doi:10.1016/j.ejca.2007.09.012
15. Pratt CB, Maurer HM, Gieser P, et al. Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol. 1998;30:201–209. doi:10.1002/(SICI)1096-911X(199804)30:4<201::AID-MPO1>3.0.CO;2-K
16. Pratt CB, Pappo AS, Gieser P, et al. Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol. 1999;17:1219.
17. Pappo AS, Rao BN, Jenkins JJ, et al. Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Med Pediatr Oncol. 1999;33:76–82. doi:10.1002/(SICI)1096-911X(199908)33:2<76::AID-MPO3>3.0.CO;2-B
18. Pappo AS, Devidas M, Jenkins J, et al. Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol. 2005;23:4031–4038. doi:10.1200/JCO.2005.03.209
19. Ferrari A, van Noesel MM, Brennan B, et al. Pediatric non-rhabdomyosarcoma soft tissue sarcomas: the prospective NRSTS 2005 study by the European pediatric Soft tissue sarcoma Study Group (EpSSG). Lancet Child Adol Health. 2021;5(8):546–558. doi:10.1016/S2352-4642(21)00159-0
20. Spunt SL, Million L, Chi -Y-Y, et al. A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children’s Oncology Group prospective study. Lancet Oncol. 2020;21(1):145–161. doi:10.1016/S1470-2045(19)30672-2
21. Ferrari A, De Salvo GL, Brennan B, et al. Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol. 2015;26:567–572. doi:10.1093/annonc/mdu562
22. Orbach D, Brennan B, De Paoli A, et al. Conservative strategy in infantile fibrosarcoma is possible: the European pediatric Soft tissue sarcoma Study Group experience. Eur J Cancer. 2016;57:1–9. doi:10.1016/j.ejca.2015.12.028
23. Brennan B, De Salvo GL, Orbach D, et al. Outcome of extracranial malignant rhabdoid tumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study-EpSSG NRSTS 2005. Eur J Cancer. 2016;60:69–82. doi:10.1016/j.ejca.2016.02.027
24. Ferrari A, Chi Y, De Salvo GL, et al. Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: a joint analysis from the European paediatric Soft tissue sarcoma Study Group and the Children’s Oncology Group. Eur J Cancer. 2017;78(1–6). doi:10.1016/j.ejca.2017.03.003.
25. Orbach D, Brennan B, Bisogno G, et al. Desmoid tumors in children: the experience of the European pediatric Soft tissue sarcoma Study Group (EpSSG) – NRSTS 05 study – an international prospective case series. Lancet Child Adolescent Health. 2017;1:284–292. doi:10.1016/S2352-4642(17)30045-7
26. Brennan B, Zanetti I, Orbach D, et al. Alveolar soft part sarcoma in children and adolescents: the European Paediatric Soft Tissue Sarcoma study group prospective trial (EpSSG NRSTS 2005). Pediatr Blood Cancer. 2018;65(4):e26942. doi:10.1002/pbc.26942
27. Orbach D, Mosseri V, Pissaloux D, et al. Genomic complexity in pediatric synovial sarcoma (SYNOBIO study): the European Pediatric Soft Tissue Sarcoma Group (EpSSG NRSTS 2005) experience. Cancer Med. 2018;7(4):1384–1393. doi:10.1002/cam4.1415
28. Spunt SL, Francotte N, De Salvo GL, et al. Clinical features and outcomes of young patients with epithelioid sarcoma: an analysis from the Children’s Oncology Group (COG) and the European paediatric soft tissue Sarcoma Study Group (EpSSG) prospective clinical trials. Eur J Cancer. 2019;112:98–106. doi:10.1016/j.ejca.2019.02.001
29. van Noesel MM, Orbach D, Brennan B, et al. Outcome and prognostic factors in pediatric malignant peripheral nerve sheath tumors: an analysis of the European Pediatric Soft Tissue Sarcoma Group (EpSSG) NRSTS-2005 prospective study. Pediatr Blood Cancer. 2019;66(10):e27833. doi:10.1002/pbc.27833
30. Casanova M, Brennan B, Alaggio R, et al. Inflammatory myofibroblastic tumor: the experience of the European paediatric Soft Tissue Sarcoma Study Group (EpSSG). Eur J Cancer. 2020;127:123–129. doi:10.1016/j.ejca.2019.12.021
31. Brennan B, Zanetti I, De Salvo GL, et al. Dermatofibrosarcoma protuberans in children and adolescents: the European Paediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Pediatr Blood Cancer. 2020;67(10):e28351. doi:10.1002/pbc.28351
32. Ferrari A, Merks JHM, Chisholm JC, et al. Outcomes of metastatic non-rhabdomyosarcoma soft tissue sarcomas (NRSTS) treated within the BERNIE study: a randomised, phase II study evaluating the addition of bevacizumab to chemotherapy. Eur J Cancer. 2020;130:72–80. doi:10.1016/j.ejca.2020.01.029
33. Casanova M, Ferrari A, Bisogno G, et al. Alveolar soft part sarcoma in children and adolescents: a report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol. 2000;11:1445–1449. doi:10.1023/A:1026579623136
34. Orbach D, Brennan B, Casanova M, et al. Paediatric and adolescent alveolar soft part sarcoma: a joint series from European cooperative groups. Pediatr Blood Cancer. 2013;60:1826–1832. doi:10.1002/pbc.24683
35. Ferrari A, Casanova M, Bisogno G, et al. Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer. 2002;94(12):3269–3276. doi:10.1002/cncr.10597
36. Kelleher FC, Viterbo A. Histologic and genetic advances in refining the diagnosis of ”undifferentiated pleomorphic sarcoma”. Cancers. 2013;5(1):218–233. doi:10.3390/cancers5010218
37. Cohen-Gogo S, Cellier C, Coindre JM, et al. Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Societe Francaise des Cancers de L’Enfant. Pediatr Blood Cancer. 2014;61(12):2191–2198. doi:10.1002/pbc.25210
38. Grier HE. Soft tissue sarcoma: apples, oranges, and passion fruit. J Clin Oncol. 1999;17(12):3695–3696. doi:10.1200/JCO.1999.17.12.3695
39. Ferrari A, Miceli R, Rey A, et al. Non-metastatic unresected pediatric nonrhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Canc. 2011;47:724e31. doi:10.1016/j.ejca.2010.11.013
40. Milgrom SA, Million L, Mandeville H, et al. Non-rhabdomyosarcoma soft-tissue sarcoma. Pediatr Blood Cancer. 2021;68(Suppl 2):e28279. doi:10.1002/pbc.28279
41. Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg. 1982;196(3):305–315. doi:10.1097/00000658-198209000-00009
42. Rao AD, Chen Q, Million L, et al. Preoperative intensity modulated radiation therapy compared to three-dimensional conformal radiation therapy for high-grade extremity sarcomas in children: analysis of the Children’s Oncology Group Study ARST0332. Int J Radiat Oncol Biol Phys. 2019;103(1):38–44. doi:10.1016/j.ijrobp.2018.09.005
43. FitzGerald TJ, Followill D, Laurie F, et al. Quality assurance in radiation oncology. Pediatr Blood Cancer. 2021;68(Suppl 2):e28609. doi:10.1002/pbc.28609
44. Frustaci S, Gherlinzoni F, De Paoli A, et al. Adjuvant chemotherapy for adult soft tissue sarcomas of extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol. 2001;19:1238–1247. doi:10.1200/JCO.2001.19.5.1238
45. Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol. 2012;30:850–856. doi:10.1200/JCO.2011.37.7218
46. Gronchi A, Palmerini E, Quagliuolo V, et al. Neoadjuvant chemotherapy in high-risk soft tissue sarcomas: final results of a randomized trial from Italian (ISG), Spanish (GEIS), French (FSG), and Polish (PSG) Sarcoma Groups. J Clin Oncol. 2020;38(19):2178–2186. doi:10.1200/JCO.19.03289
47. Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet. 1997;350:1647–1654. doi:10.1016/S0140-6736(97)08165-8
48. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008;113:573–581. doi:10.1002/cncr.23592
49. Pasquali S, Pizzamiglio S. The impact of chemotherapy on survival of patients with extremity and trunk wall soft tissue sarcoma: revisiting the results of the EORTC-STBSG 62931 randomised trial. Eur J Cancer. 2019;109:51–60. doi:10.1016/j.ejca.2018.12.009
50. Ferrari A, Brecht IB, Koscielniak E, et al. The role of adjuvant chemotherapy in children and adolescents with surgically-resected high-risk adult-type soft tissue sarcomas. Ped Blood Cancer. 2005;45(2):128–134. doi:10.1002/pbc.20376
51. Sultan I, Rodriguez-Galindo C, Saab R, et al. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer. 2009;115(15):3537–3547. doi:10.1002/cncr.24424
52. Okcu MF, Munsell M, Treuner J, et al. Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol. 2003;21:1602–1611. doi:10.1200/JCO.2003.07.008
53. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101:627–634. doi:10.1002/cncr.20386
54. Brecht IB, Ferrari A, Int-Veen C, et al. Grossly-resected synovial sarcoma treated by the German and Italian pediatric soft tissue sarcoma cooperative group: discussion on the role of adjuvant therapies. Pediatr Blood Cancer. 2006;46:11–17. doi:10.1002/pbc.20502
55. Ferrari A, Bisogno G, Alaggio G, et al. Synovial sarcoma of children and adolescents: the prognostic role of axial sites. Eur J Cancer. 2008;44:1202–1209. doi:10.1016/j.ejca.2008.03.016
56. Brennan B, Stevens M, Kelsey A, et al. Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children’s Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer. 2010;55:85–90. doi:10.1002/pbc.22453
57. Orbach D, Dowell HM, Rey A, et al. Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) working group. Pediatr Blood Cancer. 2011;57(7):1130–1136. doi:10.1002/pbc.23138
58. Venkatramani R, Xue W, Randall RL, et al. Synovial Sarcoma in Children, Adolescents, and Young Adults: a Report From the Children’s Oncology Group ARST0332 Study. J Clin Oncol. 2021;39(35):3927–3937. doi:10.1200/JCO.21.01628
59. Antman K, Crowley J, Balcerzak SP, et al. An intergroup Phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol. 1993;11(7):1276–1285. doi:10.1200/JCO.1993.11.7.1276
60. Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled Phase 3 trial. Lancet Oncol. 2014;15(4):415–423. doi:10.1016/S1470-2045(14)70063-4
61. Chiaravalli S, Bergamaschi L, Livellara V, et al. Adult-type non-rhabdomyosarcoma soft tissue sarcomas in pediatric age: salvage rates and prognostic factors after relapse. Eur J Cancer. 2022;169:179–187. doi:10.1016/j.ejca.2022.03.037
62. Ferrari A, De Salvo GL, Dall’Igna P, et al. Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer. 2012;48:3448–3455. doi:10.1016/j.ejca.2012.06.017
63. Le Cesne A, Antoine E, Spielmann M, et al. High-dose ifosfamide: circumvention of resistance to standard-dose ifosfamide in advanced soft tissue sarcomas. J Clin Oncol. 1995;13(7):1600–1608. doi:10.1200/JCO.1995.13.7.1600
64. Nielsen OS, Judson I, van Hoesel Q, et al. Effect of high-dose ifosfamide in advanced soft tissue sarcomas. A multicentre phase II study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2000;36(1):61–67. doi:10.1016/S0959-8049(99)00240-3
65. Palumbo R, Palmeri S, Antimi M, et al. Phase II study of continuous-infusion high-dose ifosfamide in advanced and/or metastatic pretreated soft tissue sarcomas. Ann Oncol. 1997;8(11):1159–1162. doi:10.1023/A:1008279426654
66. Meazza C, Casanova M, Luksch R, et al. Prolonged 14-Day Continuous Infusion of High-Dose Ifosfamide With an External Portable Pump: feasibility and Efficacy in Refractory Pediatric Sarcoma. Pediatr Blood Cancer. 2010;55:617–620. doi:10.1002/pbc.22596
67. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002. J Clin Oncol. 2007;25:2755–2763. doi:10.1200/JCO.2006.10.4117
68. Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer. 2007;109(9):1863–1869. doi:10.1002/cncr.22609
69. Fata F, O’Reilly E, Ilson D, et al. Paclitaxel in the treatment of patients with angiosarcoma of the scalp or face. Cancer. 1999;86:2034–2037. doi:10.1002/(SICI)1097-0142(19991115)86:10<2034::AID-CNCR21>3.0.CO;2-P
70. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387:1629–1637. doi:10.1016/S0140-6736(15)01283-0
71. Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol. 2007;8(7):595–602. doi:10.1016/S1470-2045(07)70175-4
72. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol. 2013;24:1703–1709. doi:10.1093/annonc/mds659
73. Frezza AM, Whelan JS, Dileo P. Trabectedin for desmoplastic small round cell tumours: a possible treatment option? Clin Sarcoma Res. 2014;4:3. doi:10.1186/2045-3329-4-3
74. Baldi GG, Orbach D, Bertulli R, et al. Standard treatment and emerging drugs for managing synovial sarcoma: adult’s and pediatric oncologist perspective. Expert Opin Emerg Drugs. 2019;24(1):43–53. doi:10.1080/14728214.2019.1591367
75. van der Graaf WTA, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo controlled phase 3 trial. Lancet. 2012;379:1879–1886. doi:10.1016/S0140-6736(12)60651-5
76. Mir O, Brodowicz T, Italiano A, et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, Phase 2 trial. Lancet Oncol. 2016;17(12):1732–1742. doi:10.1016/S1470-2045(16)30507-1
77. D’Angelo SP, Attia S, Blay JY, et al. Identification of response stratification factors from pooled efficacy analyses afamitresgene autoleucel (“Afami-cel” [Formerly ADP-A2M4]) in metastatic synovial sarcoma and myxoid/round cell liposarcoma phase and phase 2 trials. J Clin Oncol. 2022;40(16):11562. doi:10.1200/JCO.2022.40.16_suppl.11562
78. Bisogno G, Ferrari A, Alaggio R, et al. Treatment options for alveolar soft part sarcoma in paediatric patients. Exp Opin Orphan Drugs. 2014;2(6):579–589. doi:10.1517/21678707.2014.896191
79. Stacchiotti S, Negri T, Zaffaroni N, et al. Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol. 2011;22:1682–1690. doi:10.1093/annonc/mdq644
80. Kummar S, Allen D, Monks A, et al. Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol. 2013;31:2296–2302. doi:10.1200/JCO.2012.47.4288
81. Kerrison WGJ, Lee ATJ, Thway K, et al. Current Status and Future Directions of Immunotherapies in Soft Tissue Sarcomas. Biomedicines. 2022;10(3):573. doi:10.3390/biomedicines10030573
82. Shi Y, Cai Q, Jiang Y, et al. Activity and Safety of Geptanolimab (GB226) for Patients with Unresectable, Recurrent, or Metastatic Alveolar Soft Part Sarcoma: a Phase II, Single-arm Study. Clin Cancer Res. 2020;26:6445–6452. doi:10.1158/1078-0432.CCR-20-2819
83. Naqash AR, Coyne GHO, Moore N, et al. Phase II study of atezolizumab in advanced alveolar soft part sarcoma (ASPS). J Clin Oncol. 2021;39:11519. doi:10.1200/JCO.2021.39.15_suppl.11519
84. Blay JY, Penel N, Ray-Coquard IL, et al. High clinical activity of pembrolizumab in chordoma, alveolar soft part sarcoma (ASPS) and other rare sarcoma histotypes: the French AcSé pembrolizumab study from Unicancer. J Clin Oncol. 2021;39:11520. doi:10.1200/JCO.2021.39.15_suppl.11520
85. Jones AL, Joon A, Haydu LE, et al. Outcomes of melanoma soft parts/clear cell sarcoma (MSP/CCS) patients (pts) with immune and targeted therapies. J Clin Oncol. 2019;37:e21046. doi:10.1200/JCO.2019.37.15_suppl.e21046
86. Krewer J, Rolle U, Koscielniak E, et al. Dermatofibrosarcoma protuberans in children and adolescents: primary and relapsed disease - experience of the Cooperative Weichteilsarkomstudiengruppe (CWS). J Surg Oncol. 2020;122(2):263–272. doi:10.1002/jso.25943
87. McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23(4):866–873. doi:10.1200/JCO.2005.07.088
88. Rutkowski P, Klimczak A, Lugowska I, et al. Long-term results of treatment of advanced dermatofibrosarcoma protuberans (DFSP) with imatinib mesylate - The impact of fibrosarcomatous transformation. Eur J Surg Oncol. 2017;43:1134–1141. doi:10.1016/j.ejso.2017.03.011
89. Navarrete-Dechent C, Mori S, Barker CA, et al. Imatinib treatment for locally advanced or metastatic dermatofibrosarcoma protuberans: a systematic review. JAMA Dermatol. 2019;155(3):361–369. doi:10.1001/jamadermatol.2018.4940
90. Lee RS, Stewart C, Carter SL, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest. 2012;122:2983e8. doi:10.1172/JCI64400
91. Enault M, Minard-Colin V, Corradini N, et al. Extracranial rhabdoid tumours: results of a SFCE series of patients treated with a dose compression strategy according to European Paediatric Soft tissue sarcoma Study Group recommendations. Eur J Cancer. 2021;161:64–78. doi:10.1016/j.ejca.2021.10.025
92. Italiano A, Soria J-C, Toulmonde M, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, Phase 1 study. Lancet Oncol. 2018;19:649–659. doi:10.1016/S1470-2045(18)30145-1
93. Kerl K, Ries D, Unland R, et al. The histone deacetylase inhibitor SAHA acts in synergism with fenretinide and doxorubicin to control growth of rhabdoid tumor cells. BMC Cancer;2013. 13. doi:10.1186/1471-2407-13-13
94. Geoerger B, Zwaan CM, Marshall LV, et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): a multicentre phase 1-2 study. Lancet Oncol. 2020;21:134–144. doi:10.1016/S1470-2045(19)30693-X
95. Geoerger B, Kang HJ, Yalon-Oren M, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020;21:121–133. doi:10.1016/S1470-2045(19)30671-0
96. Kushner BH, LaQuaglia MP, Wollner N, et al. Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol. 1996;14:1526–1531. doi:10.1200/JCO.1996.14.5.1526
97. Scheer M, Vokuhl C, Blank B, et al. Desmoplastic small round cell tumors: multimodality treatment and new risk factors. Cancer Med. 2019;8:527–542. doi:10.1002/cam4.1940
98. Bisogno G, Ferrari A, Rosolen A, et al. Sequential intensified chemotherapy with stem cell rescue for children and adolescents with desmoplastic small round-cell tumor. Bone Marrow Transplant. 2010;45(5):907–911. doi:10.1038/bmt.2009.248
99. Hayes-Jordan A, Green HL, Lin H, et al. Complete cytoreduction and HIPEC improves survival in desmoplastic small round cell tumor. Ann Surg Oncol. 2014;21:220–224. doi:10.1245/s10434-013-3269-y
100. Desai NB, Stein NF, LaQuaglia MP, et al. Reduced toxicity with intensity modulated radiation therapy (IMRT) for desmoplastic small round cell tumor (DSRCT): an update on the whole abdominopelvic radiation therapy (WAP-RT) experience. Int J Radiat Oncol Biol Phys. 2013;85(1):e67–e72. doi:10.1016/j.ijrobp.2012.09.005
101. Honoré C, Delhorme JB, Nassif E, et al. Can we cure patients with abdominal Desmoplastic Small Round Cell Tumor? Results of a retrospective multicentric study on 100 patients. Surg Oncol. 2019;29:107–112. doi:10.1016/j.suronc.2019.04.002
102. Honoré C, Atallah V, Mir O, et al. Abdominal desmoplastic small round cell tumor without extra peritoneal metastases: is there a benefit for HIPEC after macroscopically complete cytoreductive surgery? PLoS One. 2017;12(2):e0171639. doi:10.1371/journal.pone.0171639
103. Atallah V, Honore C, Orbach D, et al. Role of adjuvant radiation therapy after surgery for abdominal desmoplastic small round cell tumors. Int J Radiat Oncol Biol Phys. 2016;95(4):1244–1253. doi:10.1016/j.ijrobp.2016.02.046
104. Olivier-Gougenheim L, Orbach D, Atallah V, et al. Desmoplastic small round cell tumors with EWS-WT1 transcript expression: should we consider children and adult patients differently? J Pediatr Hematol Oncol. 2022;44(3):e637–e642. doi:10.1097/MPH.0000000000002252
105. Liu KX, Collins NB, Greenzang KA, et al. The use of interval-compressed chemotherapy with the addition of vincristine, irinotecan, and temozolomide for pediatric patients with newly diagnosed desmoplastic small round cell tumor. Pediatr Blood Cancer. 2020;67(10):e28559. doi:10.1002/pbc.28559
106. Italiano A, Kind M, Cioffi A, et al. Clinical activity of sunitinib in patients with advanced desmoplastic round cell tumor: a case series. Target Oncol. 2013;8(3):211–213. doi:10.1007/s11523-012-0251-8
107. Schoot RA, Chisholm JC, Casanova M, et al. Metastatic rhabdomyosarcoma: results of the European paediatric Soft tissue sarcoma Study Group MTS 2008 study and pooled analysis with the concurrent BERNIE study. J Clin Oncol. 2022. doi:10.1200/JCO.21.02981
108. Ferrari A, Bergamaschi L, Chiaravalli S, et al. Multiagent chemotherapy including IrIVA regimen and maintenance therapy in the treatment of desmoplastic small round cell tumor. Tumori. 2022;108(1):93–97. doi:10.1177/0300891621995250
109. Ferrari A, Grosso F, Stacchiotti S, et al. Response to vinorelbine and low-dose cyclophosphamide chemotherapy in two patients with desmoplastic small round cell tumor. Ped Blood Cancer. 2007;49(6):864–866. doi:10.1002/pbc.20682
110. Kao YC, Owosho AA, Sung YS, et al. BCOR-CCNB3 Fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol. 2018;42(5):604–615. doi:10.1097/PAS.0000000000000965
111. Cohen-Gogo S, Cellier C, Coindre JM, et al. Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Société Française des Cancers de L’Enfant. Pediatr Blood Cancer. 2014;61(12):2191–2198.
112. Brčić I, Brodowicz T, Cerroni L, et al. round cell sarcomas with CIC-DUX4 gene fusion: expanding the clinical spectrum. Pathology. 2020;52(2):236–242. doi:10.1016/j.pathol.2019.09.015
113. Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol. 2017;41(7):941–949. doi:10.1097/PAS.0000000000000846
114. Meazza C, Bisogno G, Gronchi A, et al. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer. 2010;116(1):233–240. doi:10.1002/cncr.24679
115. Sparber-Sauer M, Seitz G, von Kalle T, et al. Systemic therapy of aggressive fibromatosis in children and adolescents: report of the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer. 2018;65(5):e26943. doi:10.1002/pbc.26943
116. Desmoid Tumor Working Group. The management of desmoid tumors: a joint global consensus-based guideline approach for adult and pediatric patients. Eur J Cancer. 2020;127:96–107. doi:10.1016/j.ejca.2019.11.013
117. Sparber-Sauer M, Orbach D, Navid F, et al. Rationale for the use of tyrosine kinase inhibitors in the treatment of pediatric desmoid-type fibromatosis. Br J Cancer. 2021;124(10):1637–1646. doi:10.1038/s41416-021-01320-1
118. Vora BMK, Munk PL, Somasundaram N, et al. Cryotherapy in extra-abdominal desmoid tumors: a systematic review and meta-analysis. PLoS One. 2021;16(12):e0261657. doi:10.1371/journal.pone.0261657
119. Antonescu CR, Suurmeijer AJ, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol. 2015;39(7):957e67. doi:10.1097/PAS.0000000000000404
120. Kube S, Vokuhl C, Dantonello T, et al. Inflammatory myofibroblastic tumors - A retrospective analysis of the Cooperative Weichteilsarkom Studiengruppe. Pediatr Blood Cancer. 2018;65(6):e27012. doi:10.1002/pbc.27012
121. Pire A, Orbach D, Galmiche L, et al. Clinical, pathologic, and molecular features of inflammatory myofibroblastic tumors in children and adolescents. Pediatr Blood Cancer. 2022;69(5):e29460. doi:10.1002/pbc.29460
122. Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–1733. doi:10.1056/NEJMoa1007056
123. Mosse´ YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a Children’s Oncology Group Study. J Clin Oncol. 2017;35:3215e21. doi:10.1200/JCO.2017.73.4830
124. Mahajan P, Casanova M, Ferrari A, et al. Inflammatory myofibroblastic tumor: molecular landscape, targeted therapeutics, and remaining challenges. Curr Probl Cancer. 2021;45(4):100768. doi:10.1016/j.currproblcancer.2021.100768
125. Orbach D, Sparber-Sauer M, Laetsch TW, et al. Spotlight on the treatment of infantile fibrosarcoma in the era of neurotrophic tropomyosin receptor kinase inhibitors: international consensus and remaining controversies. Eur J Cancer. 2020;137:183–192. doi:10.1016/j.ejca.2020.06.028
126. Orbach D, Rey A, Cecchetto G, et al. Infantile fibrosarcoma: place of chemotherapy and treatment proposals based on the European experience. J Clin Oncol. 2010;28(2):318–323. doi:10.1200/JCO.2009.21.9972
127. Sparber-Sauer M, Vokuhl C, Seitz G, et al. The impact of local control in the treatment of children with advanced infantile and adult-type fibrosarcoma: experience of the cooperative weichteilsarkom studiengruppe (CWS). J Pediatr Surg. 2020;55(9):1740–1747. doi:10.1016/j.jpedsurg.2019.10.051
128. Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumors harbouring NTRK gene fusions: a multicentre, open-label, phase 1 study. Lancet Oncol. 2018;19(5):705–714. doi:10.1016/S1470-2045(18)30119-0
129. Hingorani P, Janeway K, Crompton BD, et al. Current state of pediatric sarcoma biology and opportunities for future discovery: a report from the sarcoma translational research workshop. Cancer Genet. 2016;209(5):182–194. doi:10.1016/j.cancergen.2016.03.004
130. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12(1):498. doi:10.1038/s41467-020-20603-4
131. Wei J, Liu X, Li T, et al. The new horizon of liquid biopsy in sarcoma: the potential utility of circulating tumor nucleic acids. J Cancer. 2020;11(18):5293–5308. doi:10.7150/jca.42816
132. Colella G, Fazioli F, Gallo M, et al. Sarcoma spheroids and organoids - promising tools in the era of personalized medicine. Int J Mol Sci. 2018;19(2):615. doi:10.3390/ijms19020615
133. van Tilburg CM, Pfaff E, Pajtler KW, et al. The Pediatric Precision Oncology INFORM Registry: clinical outcome and benefit for patients with very high-evidence targets. Cancer Discov. 2021;11(11):2764–2779. doi:10.1158/2159-8290.CD-21-0094
134. Lagarde P, Przybyl J, Brulard C, et al. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol. 2013;31:608–615. doi:10.1200/JCO.2012.46.0147
135. Weiss AR, Chen YL, Scharschmidt TJ, et al. Pathological response in children and adults with large unresected intermediate-grade or high-grade soft tissue sarcoma receiving preoperative chemoradiotherapy with or without pazopanib (ARST1321): a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol. 2020;21(8):1110–1122. doi:10.1016/S1470-2045(20)30325-9
136. Hawkins DS, Bisogno G, Koscielniak E. Introducing INSTRuCT: an international effort to promote cooperation and data sharing. Pediatr Blood Cancer. 2020;e28701. doi:10.1002/pbc.28701
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.