")
Back to Journals » International Journal of Nanomedicine » Volume 14
PD-L1 monoclonal antibody-conjugated nanoparticles enhance drug delivery level and chemotherapy efficacy in gastric cancer cells
Authors Xu S, Cui F, Huang D, Zhang D, Zhu A, Sun X, Cao YM, Ding S, Wang Y, Gao E, Zhang F
Received 25 May 2018
Accepted for publication 29 October 2018
Published 18 December 2018 Volume 2019:14 Pages 17—32
DOI https://doi.org/10.2147/IJN.S175340
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Lei Yang
Shijie Xu,1,* Fangbo Cui,2,3,* Dafu Huang,4,* Dinghu Zhang,5 Anqing Zhu,6 Xia Sun,6 Yiming Cao,7 Sheng Ding,7 Yao Wang,7 Eryun Gao,3 Fenglin Zhang3
1Center for Public Health Research, Medical School, Nanjing University, Nanjing, China; 2Department of Oncology, Wannan Medical College, Wuhu, Anhui, China; 3Department of Oncology, The People’s Hospital of Ma Anshan, Ma Anshan, Anhui, China; 4Department of Oncology, Nanjing Lishui People’s Hospital, Zhongda Hospital Lishui Branch, Southeast University, Nanjing, Jiangsu, China; 5Department of Oncology, Tongde Hospital of Zhejiang Province, Hangzhou, China; 6Department of Oncology, The Affliated Jiangyin Hospital of Southeast University Medical College, Jiangyin, Jiangsu, China; 7Department of General Surgery, The People’s Hospital of Ma Anshan, Ma Anshan, Anhui, China
*These authors contributed equally to this work
Background: Docetaxel (DOC) is widely used as a chemotherapy drug for various tumors, including gastric cancer (GC), but the clinical application of DOC has been limited due to the hydrophobicity of the drug. We aimed to formulate a multifunctional nanoparticle (NP) system to reduce the side effects of the chemotherapy agent, to promote synergistic therapeutic effects, and to achieve targeted delivery of the therapy.
Methods: The polyethylene glycol-poly(ε-caprolactone) NPs (PEG-PCL NPs) were prepared by a ring opening copolymerization technique and were then conjugated with a programmed death-ligand 1 (PD-L1) monoclonal antibody (mAb). The effects of the surface coating on particle size, size distribution, zeta potential, drug encapsulation efficiency, loading capacity, and the drug release kinetics were investigated. By using a panel of PD-L1-expressing human GC cell lines and PD-L1-overexpressing cells, we studied cellular uptake, cytotoxic effects, and cellular apoptosis in the presence of PD-L1 mAb-conjugated NPs.
Results: The characterization of the structure and biological functions of DOC-PEG-PCL-mAb NPs was investigated in vitro. X-ray photoelectron spectroscopy validated the presence of the PD-L1 mAbs on the NP surface. The cellular uptake analysis showed that the antibody-conjugated NPs achieved significantly higher cellular uptake. The results of an in vitro cytotoxicity experiment on three GC lines further proved the targeting effects of the antibody conjugation. In addition, we found that the DOC-PEG-PCL-mAb NPs induced cell apoptosis and enhanced G2-M arrest in cancer cells, indicating the inhibition of microtubule synthesis. When compared with the control groups, DOC-PEG-PCL-mAb NPs are more effective in inhibiting PD-L1 expression in GC cells.
Conclusion: Our results reported here highlight the biological and clinical potential of DOC-PEG-PCL-mAb NPs using PD-L1 mAbs in GC treatment.
Keywords: DOC, gastric carcinoma, PD-L1 monoclonal antibody, nanomedicine, drug delivery
Introduction
Gastric cancer (GC) is the second leading cause of cancer-related death worldwide, with a high incidence in east Asia.1 Along with Japan and Korea, the prevalence of GC in China is the highest in the world.2 Most cases are diagnosed at a later stage, and the choice of treatment options is mostly limited to chemotherapy drugs, which are associated with poor outcomes.3 Currently, cytotoxic chemotherapy, particularly taxoid-based regimens, has been widely used in the treatment of advanced GC.4,5 Despite the fact that monoclonal antibody (mAb) drugs, such as trastuzumab and ramucirumab, have resulted in modest survival improvements in HER2-positive GC patients and in the second-line setting,6,7 there remains a need for effective treatment options for advanced GC.
Drug delivery systems (DDSs) refer to approaches used to deliver drugs at targeted sites inside our body. DDS may specifically deliver a high dose of the therapeutic agent to the cancer cells and employ a sustained drug release with the healthy cells. Compared with the non-specific delivery device, ligand-conjugated nanoparticles (NPs) may be the most prospective among various DDSs. Polymeric NPs can solve the drug hydrophobicity problem by dissolving drugs in a polymeric matrix and providing advantages such as high stability, long bloodstream circulation time, targeted space activation, efficient drug load, and sustained drug release.8,9 The targeted DDS can deliver drugs in an active way, which is achieved via coupling effector molecules to the surface of the NPs, providing DDS for reaching and penetrating into cells overexpressing a given receptor, before releasing the encapsulated drugs into the target cells in a sustained and controlled manner.10 The candidate effector molecules include antibodies, peptides, and affibodies.11–14 Among them, the anti-programmed death-ligand 1 (PD-L1) antibodies have had remarkable clinical benefits in various solid tumors.15–18
PD-1 is a cell surface receptor that plays an important role in downregulating the immune system and promoting self-tolerance by suppressing T cell inflammatory activity. PD-1 binds two ligands, PD-L1 and PD-L2. The binding of PD-L1 to PD-1 transmits an inhibitory signal that reduces the proliferation of antigen-specific T cells in lymph nodes.19,20 PD-L1 expression has been detected in >40% of human GC samples in several studies, suggesting that PD-L1 expression level is significantly upregulated following Helicobacter pylori infection.21–26 The PD-L1 mAb, a promising therapy for advanced GC, is able to specifically bind to the extracellular domain of PD-L1 protein and thus block the interaction between PD-1 and its ligands, being an excellent strategy for drug targeting.
Successful synthesis of functional nanocarriers depends on the matrix materials and surface properties. A candidate nanomaterial that has been identified in the literature is polyethylene glycol-poly(ε-caprolactone) (PEG-PCL) NPs, one of the biodegradable polymers widely applied in the drug delivery realm. The NPs consist of a PEG-coated layer and a PCL core, thus allowing the NPs to achieve passive targeting and providing the reaction site for functional molecular decoration. The amphiphilic NPs contain an inner hydrophobic core containing the poorly soluble drugs and an outer hydrophilic shell, which protects the drugs from inactivation under biological environments.27
In this study, we intended to show the feasibility of the antibody conjugation strategy, in which the PEG-PCL NPs were modified with a PD-L1 mAb and then loaded with docetaxel (DOC). We also have demonstrated this modification’s impact on cellular uptake efficiency, cytotoxicity, and apoptosis in vitro. For the first time in the literature, we have developed DOC-PEG-PCL-mAb NPs and studied their physicochemical characteristics. In addition, we have demonstrated that the surface density of the targeted molecules, believed to be an important property in term of cancer cell inhibition efficiency, showed significant difference between cells transfected with a PD-L1 expression plasmid and untransfected controls. The in vitro cellular uptake of NPs was investigated qualitatively by a confocal cell laser scanning microscope. The in vitro cytotoxicity was evaluated by MTT assay. Apoptosis and cell cycle stage were analyzed by flow cytometry. The overall performance of the designed drug delivery carrier was evaluated in terms of consistency.
Materials and methods
Materials
DOC was purchased from Jiangsu Hengrui Medicine Company (Jiangsu, China). Methoxy polyethylene glycol (MePEG; Mn: 5,000; Sigma-Aldrich Co., St Louis, MO, USA) was dehydrated with toluene, following vacuum drying at 50°C for 12 hours. ε-Caprolactone (ε-CL; Fluka, Shanghai, China) was purified by drying over CaH2 at room temperature (RT) and distillation under reduced pressure. Coumarin 6, stannous octoate, dichloromethane (DCM), chloroform, ethyl ether, sodium borate, PBS (pH 7.4), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDAc), N-hydroxysulfosuccinimide (Sulfo-NHS), trypsin-EDTA solution, propidium iodide (PI), dimethyl sulfoxide (DMSO), and MTT were all provided by Sigma-Aldrich, and polyvinyl alcohol (PVA) was purchased from Aladdin Co. Ltd. (Shanghai, China). Methanol and acetic acid, used as mobile phase in HPLC, were purchased from EMD Millipore (Billerica, MA, USA). Human PD-L1/B7-H1 mAb and negative control mAb (mouse IgG1 isotype control) were supplied by R&D Systems, Inc (Minneapolis, MN, USA). FBS and DMEM were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals were of analytical grade and were used without further purification. MGC803, MKN45, and HGC27 GC cell lines were obtained from the Shanghai Institute of Cell Biology (Shanghai, China).
Synthesis of PEG-PCL block copolymers
The PEG-PCL copolymers were synthesized via ring opening copolymerization as described previously.28 Briefly, a predetermined volume of MePEG or PEG (50 g, 10 mmol) was introduced into a polymerization tube containing ε-CL (1.15 g), and then stannous octoate (0.05 g) was added as a catalyst. The tube was then connected to a vacuum line, evacuated, sealed off, and placed in a thermostat at 130°C for 48 hours. After the polymerization was complete, the resulting complexes were dissolved in DCM and precipitated into a large amount of cold ethyl ether to remove the monomer and oligomer. Then, the precipitates were filtered and washed with water several times before being dried at reduced pressure.
PD-L1 mAb conjugation and ligand density on surface of PEC-PCL copolymers
PD-L1 mAbs were dissolved in borate buffer (pH 8.4) at a concentration of 10 mg/mL. Freeze-dried PEG-PCL NPs powder was re-suspended in borate buffer with a designated volume of mAb solution. The EDAc and Sulfo-NHS were added to conjugate the free primary amine groups on the NPs surface with the carboxylic groups on the antibody molecules.29 Following a 16-hour incubation at RT, the NPs were obtained by centrifugation and washed twice in borate buffer. The pellets were rinsed with ultrapure water and freeze dried for further analysis. The supernatant was collected after each centrifugation and scanned for measurement of the antibody concentration. The antibody amount bound on the NPs surface was obtained by subtracting the amount in the supernatant from the original amount. The negative control for mAb conjugation (PEG-PCL-IgG) NPs was prepared in a same way with the PD-L1 mAb replaced by a mouse IgG1 isotype control mAb.
Preparation of DOC-loaded NPs
The DOC-loaded (DOC-PEG-PCL-mAb and DOC-PEF-PCL-IgG) NPs were formulated by a single-emulsion solvent evaporation technique according to previously published work with slight modifications.30 In brief, 5 mg of DOC was dissolved in 0.5 mL of chloroform in a beaker and mixed with 20 mg of mAb-conjugated PEG-PCL. To generate the emulsion, the mixture was added dropwise to 1.5 mL of a 3% (w/v) PVA solution and then sonicated (XL2000; Misonix Inc., Farmingdale, NY, USA) for 1 minute (32.5 W) to obtain an O/W emulsion. This emulsion was then emulsified in 2.5 mL of an aqueous solution containing 0.5% (w/v) PVA by sonication for 30 seconds (26 W). The O/W emulsion was then gently stirred at RT in a fume hood until all the organic solvent evaporated. The resulting solution was filtered through a 0.45-μm microporous membrane to remove non-incorporated drugs. Blank PEG-PCL NPs were prepared in the same manner, but without adding DOC. The fluorescent NPs were prepared in a same way except that the DOC was replaced with Coumarin 6.
Characterization of NPs
Size, polydispersity, zeta potential, and morphology of NPs
The particle size, polydispersity, and zeta potential of PEG-PCL NPs, DOC-PEC-PCL NPs, and PEG-PCL-mAb NPs were measured by a dynamic light scattering (DLS) detector (Brookhaven Instruments Corporation, Holtsville, NY, USA) and ZetaPlus (Brookhaven Instruments Corporation) at RT in ultrapure water. The values were shown as the average of triplicate measurements for a single sample. The surface morphology of the DOC-PEG-PCL-mAb NPs was investigated by a field emission scanning electron microscopy system (FESEM; JEOL JSM-6700F, Japan). The samples were prepared by dripping a single drop of the NP suspension on a copper grip covered with nitrocellulose membrane and air dried before observation.
Surface chemistry analysis
The existence of mAbs on the NPs’ surface was confirmed by X-ray photoelectron spectroscopy (XPS; AXIS His-165 Ultra; Kratos Analytical, Shimadzu Corporation, Kyoto, Japan). The XPS was used to investigate the surface chemistry of the NPs. The NPs surface was further analyzed in terms of the specific binding energy (eV) of the elements. The fixed transmission mode was utilized with a passing energy of 80 eV, and the binding energy spectrum was recorded from 0 to 1,500 eV. The nitrogen element was analyzed under fine mode with 0.5 eV as a step.
Drug encapsulation efficiency (EE) and loading capacity (LC)
EE and LC of DOC-PEG-PCL-mAb NPs were analyzed by HPLC as we previously described.31 Chromatographic separation was achieved using a Zorbax C18 column (150 × 4.6 mm, 5 μm; Agilent Technologies, Santa Clara, CA, USA). The injection volume was 20 μL, and the wavelength was set at 360 nm. The column temperature was 30°C. The mobile phases included a mixture of methanol/water (93:7, v/v) containing 0.1% acetic acid. The EE% and LC% were calculated by equations (1) and (2), respectively:
|
|

|
|

In vitro DOC release from NPs
The in vitro release of DOC from DOC-PEG-PCL-mAb NPs was monitored by dialysis in PBS containing 0.1% Tween-80, which improves the solubility of DOC in PBS to simulate the sink condition. About 0.5 mL of DOC-PEG-PCL-mAb NPs was sealed into a dialysis bag with a 14-kDa molecular weight cutoff. The dialysis bag was immerged in a 5-mL release medium (PBS containing 0.5% v/v Tween-80) with gentle agitation at 37°C for 240 hours. At predetermined time points, a volume of 5 mL of release medium was withdrawn, and an equivalent volume of fresh medium was added. Each sample was centrifuged at 10,000 r/min to remove the possible impurities that were mixed into the release medium during the operating process. The samples were extracted with DCM, vacuum dried at 37°C, and reconstituted with methanol. The DOC was determined by HPLC analysis under the same conditions described previously.
Cellular uptake of NPs by fluorescence microscopy
Cellular uptake studies were performed using Coumarin 6 dye and fluorescence microscope analysis. MKN45 cells were cultivated on 24-well plates for 1 day. After the cells reached 70% confluence, half of the cells were transfected with a PD-L1 plasmid. Next, the culture medium was changed to the suspension of Coumarin 6-loaded mAb-conjugated NPs 48 hours later at the concentration of 0.125 mg/mL for either 1 or 2 hours of incubation. The cells were washed with 0.5 mL PBS three times after incubation and then fixed with 4% cold paraformaldehyde for 20 minutes. The cells were then washed thrice with PBS, followed by the addition of DAPI. Finally, the cells were washed twice with PBS and imaged under a confocal laser scanning microscope (CLSM; Olympus Fluoview FV1000; Olympus, Tokyo, Japan). The pictures were further analyzed using ImageJ software (National Institute of Health, Bethesda, MD, USA).
In vitro cytotoxicity
The cytotoxic effects of NPs on MGC803, MKN45, and HGC27 cells were measured via MTT assay. The cells were layered at a density of 5×103 cells/well (0.1 mL) in 96-well plates. After 24 hours, the cells were divided into two groups, with or without PD-L1 plasmid transfection for 48 hours. After that, the culture medium was replaced with prepared doses of PBS, PEG-PCL, PEG-PCL-mAb, PEG-PCL-IgG, DOC, DOC-PEG-PCL, DOC-PEG-PCL-mAb, or DOC-PEG-PCL-IgG for 48 hours. Next, 20 μL of MTT (5 mg/mL) was added to each well. MTT was removed after incubation for an additional 4 hours, and DMSO was added to dissolve the formazan crystals in the cells. Absorbance was measured at 570 nm using a microplate reader. Cell viability was calculated using the following formula:
|
|

All the results were derived from three independent experiments and tested in triplicate each time.
Apoptosis assay and cell cycle analysis
For the apoptosis assay, the HGC27 cells were plated in six-well culture plates at a density of 1×105 cells/well. After incubating overnight, the cells were treated with blank PEG-PCL, PEG-PCL-IgG, PEG-PCL-mAb, DOC, DOC-PEG-PCL-IgG, and DOC-PEG-PCL-mAb NPs groups for 72 hours. The cells were co-stained with 5 μL Alexa Fluor 488 annexin-V and 1 μL 100 μg/mL PI working solution (Thermo Fisher Scientific) to each 100 μL of cell suspension for 15 minutes, as described in the cell apoptosis kit, and then stained cells were analyzed by flow cytometry with a BD FACS Calibur (BD Biosciences, San Jose, CA, USA) as soon as possible. Green fluorescence represented apoptotic cells, cells stained with both red and green fluorescence represented dead cells, and live cells showed little or no fluorescence.
For the cell cycle analysis, the cells were analyzed using PI/RNase buffer (BD Pharmingen, San Jose, CA, USA) staining according to the instruction book.32 HGC27 cells were plated in six-well plates at a density of 1×105 cells/well overnight. Next, the cells were treated with blank PEG-PCL NPs, PEG-PCL-IgG, PEG-PCL-mAb, DOC, DOC-PEG-PCL-IgG, and DOC-PEG-PCL-mAb for 24 hours. The cells were collected, fixed in 75% ethanol at −20°C for at least 2 hours, washed twice to remove the ethanol by cold PBS, and stained with PI/RNase staining buffer for 15 minutes at RT. Cells were then analyzed via flow cytometry within 1 hour of collection.
Western blot assay
The protein β-III tubulin in cell extracts was used directly for microtubule dynamics detection. The expression of proapoptotic and antiapoptotic proteins, caspase-3, caspase-8, caspase-9, and Bcl-2, were analyzed to confirm cell death, and cell cycle arrest was evaluated by measuring the cell cycle markers cyclin A and cyclin B in NP-treated cells. The HGC27 cells were seeded in six-well plates (3×105/well) in DMEM with 10% FBS at 37°C with 5% CO2. After 24 hours, the cells were treated with blank PEG-PCL NPs, PEG-PCL-IgG, PEG-PCL-mAb, DOC, DOC-PEG-PCL-IgG, and DOC-PEG-PCL-mAb for 48 hours, and then the cells of each group were collected and lysed using RIPA lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA) on ice for 30 minutes before being centrifuged at 12,000× g for 10 minutes at 4°C. After each sample was separated by SDS-PAGE, the protein was electrotransferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked using blocking buffer for 1 hour and then incubated with primary antibodies for 2 hours at RT or overnight at 4°C. After three washes with PBS-0.1% Tween-20 buffer, the membranes were incubated with a secondary antibody conjugated with horseradish peroxidase (Cell Signaling Technology, Beverly, MA, USA) for 1 hour at RT. After washing three times, the blots were visualized under an enhanced chemiluminescence method (EMD Millipore). Rabbit and mouse monoclonal antibodies against β-III tubulin, caspase-3, caspase-8, caspase-9, and Bcl-2 were purchased from Abcam (Cambridge, UK).
Statistical analyses
The data are represented as the mean±SD. The comparisons of the mean value were performed by two-tailed Student’s t-test. P<0.005, P<0.01, and P<0.05 were considered significant.
Results
NPs conjugated by PD-L1 mAb
Antibody-mediated targeting of GC cells was accomplished by using precision engineering. The method was based on the PEG-PCL NP synthesis as described in the “Materials and methods” section. The NPs were conjugated with antibody by an amidation reaction with EDAc and Sulfo-NHS in aqueous phase (Figure 1A). We also investigated correlation of the amount of antibody conjugated on the NP surface with the ratio of the NPs to PD-L1 mAb. A series of weight ratios (w/w) of NPs/mAb from 10% to 60% were designed to prepare PEG-PCL-mAb copolymers. The final levels of PD-L1 mAb conjugated to the NPs surface are listed in Table 1. Therefore, the 40% wt ratio (w/w) of PEG-PCL/PD-L1 mAb was selected to synthesize PEG-PCL-mAb copolymers for all subsequent experiments.
 | Figure 1 Synthesis of DOC-PEG-PCL-mAb NPs and XPS spectrum results. |
 | Table 1 PD-L1 mAb content of the PEG-PCL-mAb NPs of various NPs/mAb ratios used in the process |
The antibody conjugation of NPs was confirmed by analyzing their surface chemistry using XPS to identify the changes in nitrogen signal according to the specific binding energy. The antibody molecules contain a considerable quantity of nitrogen atoms, which should respond to a stronger signal than that from the amine groups in the PEG-PCL NPs alone. A distinct peak of signals from the orbital of nitrogen (N 1s) qualitatively verifies that the antibody molecules had been conjugated to the polymeric matrix cores in the ‘PEG-PCL + mAb + Sulfo-NHS’ group. Meanwhile, the absence of the N 1s signal in the measurement of the control group without the addition of Sulfo-NHS further confirmed that the PD-L1 mAb had been successfully conjugated on the polymer matrix (Figure 1B).
Structure characterization and surface morphology of the NPs
The particle size and size distribution of three formulations of the NPs were in the range of 170–200 nm with polydispersity of 0.203–0.358 determined by DLS, an acceptably narrow size distribution (Table 2; Figure 2A). As expected, the size of DOC-PEG-PCL-IgG NPs and DOC-PEG-PCL-mAb NPs was slightly larger than that of the PEG-PCL alone due to the addition of DOC. The zeta potentials were negative, ranging from −8.89 to −15.45 mV, and the EE and LC of the DOC-PEG-PCL-IgG NPs were 58.6%±2.5% and 46.6%±0.07% and of the DOC-PEG-PCL-mAb NPs were 48.8%±1.3% and 46.5%±3.4%, respectively. These results suggested that the NPs had high EE and narrow size distribution. In addition, the stability data of particle sizes were determined by DLS every 2 days for 10 days to evaluate stability (Figure 2B). FESEM was employed to image the morphology of the DOC-PEG-PCL-mAb NPs. It was revealed that the NPs are generally spherical in shape and that the particle size observed from these FESEM images was in good agreement with that determined by DLS, which facilitated the enrichment of NPs in the tumor tissue by increasing the enhanced permeability and retention (EPR) effect (Figure 2C).
 | Table 2 Comparison of the characteristics of PEG-PCL, DOC-PEC-PCL-IgG, and DOC-PEG-PCL-mAbs |
 | Figure 2 The structural characterization and surface morphology of the NPs. |
In vitro drug release profile
The drug release profile of the DOC-loaded PEG-PCL NPs after PD-L1 mAb conjugation was detected in vitro over 10 days. DOC presented a burst release from NPs in the first 8 hours, with nearly 35% drug released from the carriers. Afterward, a steady sustained release was observed during the following 10 days and is expected to provide a sustainable drug concentration in an internal environment (Figure 3). These results indicate that DOC-PEG-PCL-mAb NPs could sustain a continued release of DOC for up to 10 days.
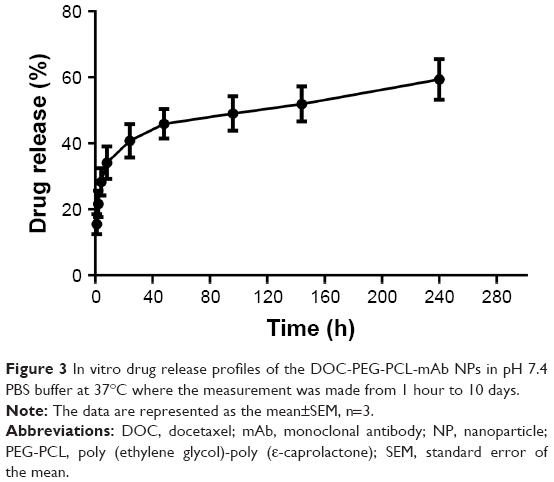 | Figure 3 In vitro drug release profiles of the DOC-PEG-PCL-mAb NPs in pH 7.4 PBS buffer at 37°C where the measurement was made from 1 hour to 10 days. |
Cellular uptake analysis in vitro
To compare the delivery ability of mAb-conjugated NPs, a fluorescent quantitative investigation was conducted by measuring the fluorescence intensity of Coumarin 6 in the cytoplasm of MKN45 cells. The cellular uptake of the Coumarin 6-PEG-PCL-mAb NPs was investigated by CLSM after 1 hour and 2 hours of incubation in MKN45 cells with different levels of PD-L1 ligand expression. The images obtained from the FITC channel, which represents the presence of Coumarin 6, are shown in column “Coumarin 6”; the DAPI channel shows the nuclei in blue fluorescence stained by DAPI; and column “Overlay” displays the images obtained from the merged FITC and DAPI channels. Under the same exciting laser intensity from the same confocal microscope, it can be seen that the fluorescence intensity with the PD-L1 plasmid transfection group in the cytoplasm is significantly higher than that from the untransfected group (Figure 4). This outcome indicates that the recognition between the PD-L1 ligand expressed on the cytomembrane and the PD-L1 mAb on the surface of the Coumarin 6-PEG-PCL-mAb NPs contributes to the enhancement of the uptake of Coumarin 6 encapsulated in the NPs. Over time, the cell uptake of Coumarin 6 increased; however, the nonspecific uptake was also increased.
 | Figure 4 Cellular uptake analyses in MKN45 cells. |
In vitro cytotoxicity
The cell killing efficacy of the NPs formulations is reflected by their cytotoxicity in tumor cells. To simulate tumor microenvironment in vitro, we divided the three kinds of gastric cells into two groups, with and without a PD-L1 plasmid transfected. As shown in Figure 5A, the red curves represent the initial expression level of PD-L1 protein in different GC cells and the orange curves represent the PD-L1 expression level after the plasmid was transfected. The results indicate that we achieved PD-L1 overexpression in our cell lines. Figure 5B illustrates the quantitative analysis on the cytotoxicity of DOC formulated in different NP copolymers, which are of various drug concentrations from 20 to 160 ng/mL. It can be seen that the viability of all three GC cells decreased along drug concentration and expression level of PD-L1 protein increased. Blank and DOC-unloaded NPs had minimal effects on suppression of cell proliferation in the range of our tested concentration. Compared with the isotype IgG control-modified NPs, DOC-PEG-PCL-mAb exhibited significantly increased cytotoxicity in the three GC cell lines despite high drug concentrations before PD-L1 plasmid transfection. These advantages become more obvious after plasmid transfection. The DOC-PEG-PCL-mAb NPs exhibited the highest mortality (ie, the highest cytotoxicity) among all the other formulations with DOC loading, while the DOC-PEG-PCL-mAb NPs exhibited little cell mortality at low drug concentrations without PD-L1 plasmid transfection. This outcome may demonstrate that PD-L1 mAb-conjugated NPs can target cells that specifically express PD-L1 protein and exert a cytotoxic effect on those cells. In addition, HGC27 cells seem more sensitive to drug concentration compared with the others, and this might be due to the relatively high initial PD-L1 expression level in the HGC27 cell line.
 | Figure 5 In vitro viability of three types of gastric cancer cells treated with blank NPs, PEG-PCL, PEG-PCL-IgG, PEG-PCL-mAb, free DOC, DOC-PEG-PCL, DOC-PEG-PCL-IgG, and DOC-PEG-PCL-mAb NPs at different drug concentrations after 48 hours of incubation. |
DOC-PEG-PCL-mAb NPs enhance apoptosis of HGC27 cells
We measured the percentage of apoptotic cells 48 hours after treatment with blank PEG-PCL, PEG-PCL-IgG, PEG-PCL-mAb, DOC, DOC-PEG-PCL-IgG, and DOC-PEG-PCL-mAb NPs. As shown in Figure 6A, cellular incorporation of DOC alone and DOC-PEG-PCL-IgG NPs increased the fraction of cell apoptosis compared with control (16.3%±0.11% and 17.2%±0.13%, respectively). The number of apoptotic cells following treatment with DOC-PEG-PCL-mAb NPs exhibited an obvious increase compared with that of the control (37.4%±0.23%). A comparison between the two antibody-conjugated DOC-loaded groups showed that the difference was significant. Similar trends were also observed in Western blot analysis. The increased expression of proapoptotic proteins, caspase-3, caspase-8, and caspase-9 and the decreased expression of antiapoptotic protein Bcl-2 confirmed apoptosis (Figure 6B). All of these results showed that when combined with PD-L1 mAb, the copolymer NPs induced more apoptosis than that of the other NPs groups.
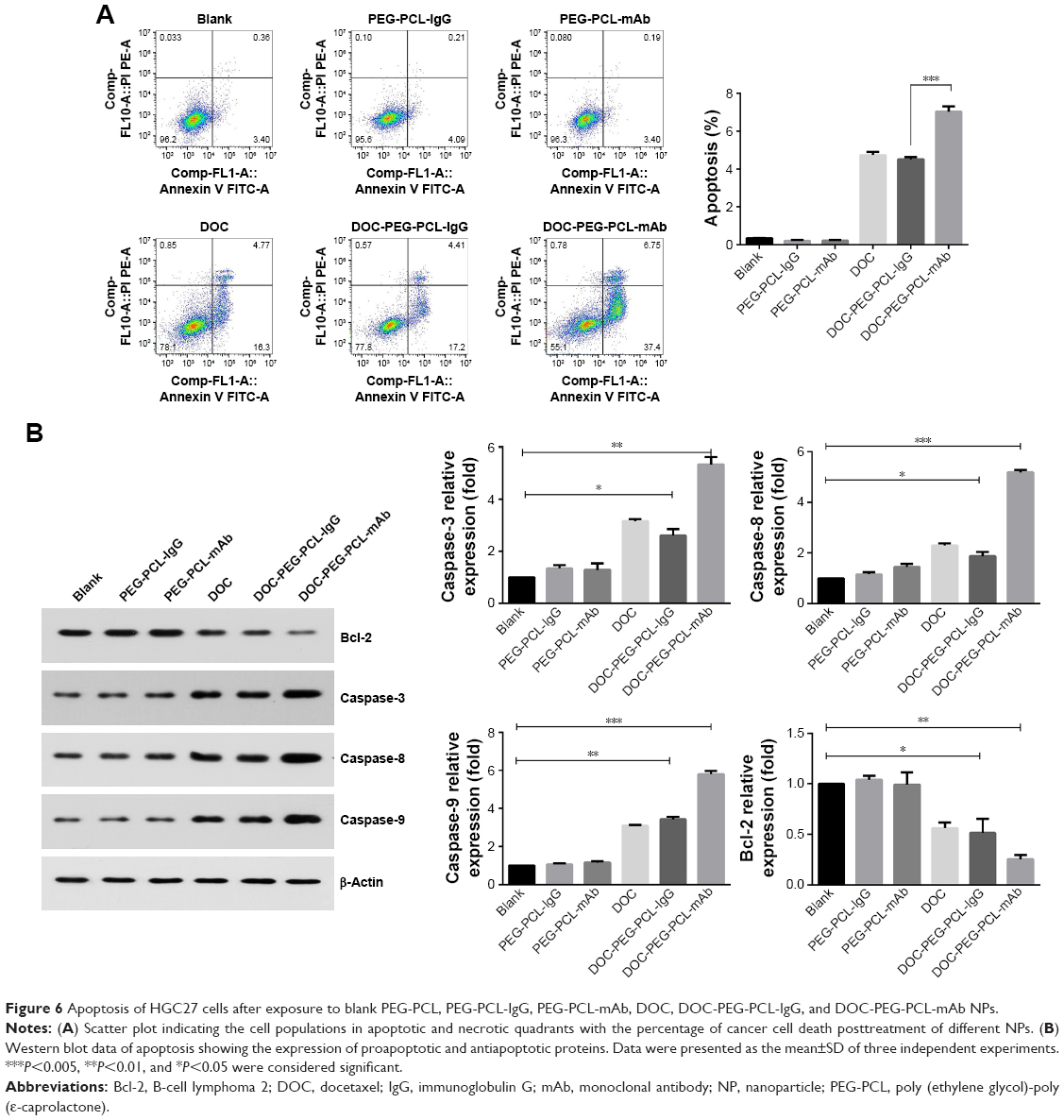 | Figure 6 Apoptosis of HGC27 cells after exposure to blank PEG-PCL, PEG-PCL-IgG, PEG-PCL-mAb, DOC, DOC-PEG-PCL-IgG, and DOC-PEG-PCL-mAb NPs. |
DOC-PEG-PCL-mAb NPs enhanced G2-M arrest
Compared with the control group, treatment with DOC containing groups induced cell cycle arrest at the G2/M phase. The cells treated with PD-L1 mAb-conjugated NPs significantly increased cells arrested at the G2/M phase (Figure 7A). There were significant differences between DOC-PEG-PCL-mAb NPs group and the other DOC containing groups (P<0.001). We investigated the cell cycle markers cyclin A and cyclin B in different NP-treated cells. We found that cyclin A and B proteins were increased in the DOC-PEG-PCL-mAb group, which confirmed the cell cycle arrested at G2/M phase (Figure 7B). Cells arrested in G2/M phase make them more sensitive to the damaging effects of cytotoxic agents. The results demonstrated that DOC-PEG-PCL-mAb NPs arrested more cells at the G2/M phase, which will allow them to be more easily killed by radiation.
 | Figure 7 DOC-PEG-PCL-mAb NPs enhance G2/M arrest. |
DOC-PEG-PCL-mAb NPs influence microtubule dynamics
TUBB3 in cell extracts was used directly for microtubule dynamics detection. We evaluated the TUBB3 protein expression in HGC27 after 48 h cell culture with PEG-PCL, PEG-PCL-mAb, PEG-PCL-IgG, DOC, DOC-PEG-PCL-mAb, and DOC-PEG-PCL-IgG NPs by Western blot. As shown in Figure 8, an evident decrease of TUBB3 expression was observed in both PD-L1 plasmid-transfected and untransfected cells, suggesting that DOC-PEG-PCL-mAb NPs significantly influenced the dynamics of microtubules. No differences between the PD-L1 high expression cells and low expression cells were exhibited.
 | Figure 8 DOC-PEG-PCL-mAb NPs influence microtubule dynamics. |
Discussion
In this study, we have developed PD-L1 antibody-mediated NPs (PEG-PCL-mAb NPs) synergized with DOC to target GC cells, enabling higher drug uptake, excellent sensitivity, widespread cell apoptosis, and microtubule network disruption. According to our knowledge, this is the first report of chemotherapy NPs targeted to the PD-L1 immune checkpoint as a promising agent for antitumor therapy.
DOC has demonstrated promising cytotoxic activity in GC, both as monotherapy33 and in combination with other agents.34 However, due to the extreme hydrophobicity and degradation characteristics exhibited by this compound in vivo, the use of DOC is limited. Furthermore, the poor water solubility of DOC limits the delivery characteristics of the drug to the tumor site at an effective concentration. PD-L1-targeted monoclonal antibodies have been approved by the FDA for the treatments of different cancers. However, only part of the cancer patients respond to the antibodies therapy due to its reliance on high expression of PD-L1 on cancer cells.35 Meanwhile, both transcriptional and posttranscriptional mechanisms have been investigated that the agent paclitaxel (PTX) induced the expression of PD-L1 immunosuppressive molecules via the mitogen-activated protein kinase (MAPK) pathway.36 This evidence suggests a synergy if the agent PTX could colocalize with the PD-L1 mAb. PD-L1 expression is a valuable biomarker for therapies targeting immune checkpoints, which are expressed on various tumor cells.37 However, there is a need for tumor-targeted delivery systems for securing both efficacy and safety since PD-L1 is also expressed on the normal tissues and cells.38 Above all, a NP system may be a key technology to address above issues by offering targeted delivery of various types of immunotherapeutics, resulting in significant improvements in the tumor immunotherapy.
The drug carrier copolymers produced in this study are a core shell system in which PCL chains possess a core matrix, while PEG chains from PEG-PCL copolymers stretch on the PCL core as the shielding shell layer, which can enhance the hydrophilicity and assist the particle in escaping from phagocytosis and opsonization. We conjugated PD-L1 monoclonal antibodies to the surface of PEG-PCL copolymers via EDC/NHS coupling agents and not only improved the delivery efficiency of drug but also reduced the complexity by controlling the combination of administration time, administration site, and dosage. NPs can increase the solubility of hydrophobic therapeutic and offer the proper size and surface properties to prolong blood circulation, allowing for their selective accumulation in tumors via the EPR effect.39 Tumor accumulation may be further improved by modifying the particle surface with cancer targeting ligands.40
Nonspecific interaction between nanocarriers and non-target cells is a critical challenge that limits the therapeutic efficacy resulting in adverse effects.41 By using Coumarin 6 dye-encapsulated PEG-PCL-mAb NPs, we monitored the specific intracellular uptake of NPs using fluorescence microscopy. Compared with the group not transfected with the PD-L1 plasmid, the Coumarin 6 dye within PEG-PCL-mAb NPs was visualized in the cytoplasm of GC cells after plasmid transfection. The PD-L1 targeting ligand in the formulation contributed to improve cellular uptake and enhanced drug transfection efficiency in MKN45 cells (Figure 4) via recognition of conjugated antibody on the surface of NPs. Furthermore, we evaluated the anticancer cell function of different nanoformulations. The results indicate that the PD-L1 protein, a potent biomarker, increases the tumor-targeting effects. The successful treatment of GC partly depends on the sensitivity of cancer cells to conventional chemotherapy. At the cellular level, we found that DOC increases the expression of PD-L1 in cancer cells, making target cells more sensitive to PD-L1 mAbs. This outcome results in the DOC-PEG-PCL-mAb NPs effectively delivering DOC to tumor cells, causing a mortality rate up to 65% (Figure 5B), increasing cell apoptosis (Figure 6), and causing a quantity of cells to arrest in G2/M phase (Figure 7). On the other hand, tumor cells may also be more susceptible to immune attack after exposure to chemotherapy.42,43 Chemotherapy can change the tumor microenvironment to facilitate increased immune activity or recognition, including the formation of additional neo-antigens, the infiltration of cytotoxic T cells, and the reduction in inhibitory T-regulatory cells.44,45
PD-L1 mAb conjugated with PEG-PCL NPs showed TUBB3 expression inhibition similar to the DOC-PEG-PCL-mAb NPs in HGC27 cells, which suggests that the antibody activity was retained even after NP synthesis, as shown in Figure 8. The cellular uptake results indicate that the drug can be ingested to a certain extent even in cells with low PD-L1 expression. Low concentration of NPs will contribute to TUBB3 expression inhibition. This also indicates that the linked antibodies have good biological activity. In addition, according to our research, the inhibition might be caused by the PD-L1 mAb and thus further increases the drug sensitivity of DOC. The synergy between PD-L1 mAb and DOC has been reported in the literature,46,47 and the mechanism may be that the PD-L1 antibody leads to TUBB3 expression inhibition. The advantages of our DOC-PEG-PCL-mAb NPs in terms of cytotoxicity might be mainly due to cellular uptake superiority and synergy between the PD-L1 mAb and DOC. The efficacy of DOC-PEG-PCL-mAb NPs in targeting gastric tumors in vivo will be investigated in future studies.
Before the advent of immune checkpoint antibodies, many clinical studies have shown that the combination of immunotherapy drugs and traditional chemotherapy drugs reveal better therapeutic effects.48,49 Nevertheless, the addition of the mAb on the NPs loaded with DOC is a better solution for anticancer efficacy, as well as the reduction in the side effects. Moreover, this system can be applied to not only GC cells but also to other PD-L1 expressing cancer cells. We believe that the development of novel theranostic NPs and their facile surface chemistries will provide tremendous opportunities for controlling antitumor immunity and improving immunotherapy efficacy.
Conclusion
This research reconstructed NPs (PEG-PCL NPs) for surface coating by PD-L1 monoclonal antibodies for targeted delivery of anticancer drugs for GC treatment. Such a NP system for drug delivery is multifunctional, providing a way to formulate anticancer drugs with no use of harmful adjuvants, reducing the side effects of the formulated anticancer drug, promoting synergistic therapeutic effects with cancer immune checkpoints, and achieving targeted delivery of the therapy. The PD-L1 mAb-conjugated DOC-PEG-PCL NPs have the potential to be applied for targeted chemotherapy for PD-L1-positive cancer. These results should be further confirmed by in vivo experiments.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81502029 and 81672398); the 5th group of the “333 Talent Project” of Jiangsu Province (level 3); and the 14th group of Six Talent Peak Project of the Jiangsu Province (YY-068).
Disclosure
The authors report no conflicts of interest in this work.
References
Xu W, Chen Q, Wang Q, et al. JWA reverses cisplatin resistance via the CK2-XRCC1 pathway in human gastric cancer cells. Cell Death Dis. 2014;5(12):e1551. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Catalano V, Labianca R, Beretta GD, Gatta G, de Braud F, van Cutsem E. Gastric cancer. Crit Rev Oncol Hematol. 2009;71(2):127–164. | ||
Chabner BA, Roberts TG. Timeline: chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5(1):65–72. | ||
Kim B, Lee KW, Kim MJ. A multicenter randomized Phase II study of Docetaxel vs. Docetaxel Plus Cisplatin vs. Docetaxel Plus S-1 as second-line chemotherapy in metastatic gastric cancer patients who had progressed after Cisplatin Plus Either S-1 or Capecitabine. Eur J Cancer. 2017;49(3):706–716. | ||
Bang YJ, van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697. | ||
Wilke H, Muro K, van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15(11):1224–1235. | ||
Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. | ||
Feng S-S, Zhao L, Zhang Z, et al. Chemotherapeutic engineering: Vitamin E TPGS-emulsified nanoparticles of biodegradable polymers realized sustainable paclitaxel chemotherapy for 168 h in vivo. Chem Eng Sci. 2007;62(23):6641–6648. | ||
Wang X, Yang L, Chen ZG, Shin DM. Application of nanotechnology in cancer therapy and imaging. CA Cancer J Clin. 2008;58(2):97–110. | ||
Yoo HS, Park TG. Folate-receptor-targeted delivery of doxorubicin nano-aggregates stabilized by doxorubicin-PEG-folate conjugate. J Control Release. 2004;100(2):247–256. | ||
Brown KC. Peptidic tumor targeting agents: the road from phage display peptide selections to clinical applications. Curr Pharm Des. 2010;16(9):1040–1054. | ||
Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23(9):1147–1157. | ||
Ahlgren S, Wållberg H, Tran TA, et al. Targeting of HER2-expressing tumors with a site-specifically 99mTc-labeled recombinant affibody molecule, ZHER2:2395, with C-terminally engineered cysteine. J Nucl Med. 2009;50(5):781–789. | ||
Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. | ||
Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. | ||
Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. | ||
Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24(2):207–212. | ||
Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylor infection. J Immunol. 2006;176(5):3000–3009. | ||
Kim JW, Nam KH, Ahn SH, et al. Prognostic implications of immunosuppressive protein expression in tumors as well as immune cell infiltration within the tumor microenvironment in gastric cancer. Gastric Cancer. 2016;19(1):42–52. | ||
Qing Y, Li Q, Ren T, et al. Upregulation of PD-L1 and APE1 is associated with tumorigenesis and poor prognosis of gastric cancer. Drug Des Devel Ther. 2015;9:901–909. | ||
Wu C, Zhu Y, Jiang J, Zhao J, Zhang XG, Xu N. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem. 2006;108(1):19–24. | ||
Hou J, Yu Z, Xiang R, et al. Correlation between infiltration of FOXP3+ regulatory T cells and expression of B7-H1 in the tumor tissues of gastric cancer. Exp Mol Pathol. 2014;96(3):284–291. | ||
Geng Y, Wang H, Lu C, et al. Expression of costimulatory molecules B7-H1, B7-H4 and Foxp3+ Tregs in gastric cancer and its clinical significance. Int J Clin Oncol. 2015;20(2):273–281. | ||
Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther. 2006;112(3):630–648. | ||
Li R, Li X, Xie L, et al. Preparation and evaluation of PEG-PCL nanoparticles for local tetradrine delivery. Int J Pharm. 2009;379(1):158–166. | ||
Liu Y, Li K, Liu B, Feng SS. A strategy for precision engineering of nanoparticles of biodegradable copolymers for quantitative control of targeted drug delivery. Biomaterials. 2010;31(35):9145–9155. | ||
Wang Y, Gao S, Ye WH, Yoon HS, Yang YY. Co-delivery of drugs and DNA from cationic core-shell nanoparticles self-assembled from a biodegradable copolymer. Nat Mater. 2006;5(10):791–796. | ||
Zhang D, Zou Z, Ren W, et al. Gambogic acid-loaded PEG-PCL nanoparticles act as an effective antitumor agent against gastric cancer. Pharm Dev Technol. 2018;23(1):33–40. | ||
Jung J, Park SJ, Chung HK, et al. Polymeric nanoparticles containing taxanes enhance chemoradiotherapeutic efficacy in non-small cell lung cancer. Int J Radiat Oncol Biol Phys. 2012;84(1):e77–e83. | ||
Einzig AI, Neuberg D, Remick SC, et al. Phase II trial of docetaxel (Taxotere) in patients with adenocarcinoma of the upper gastrointestinal tract previously untreated with cytotoxic chemotherapy: the Eastern Cooperative Oncology Group (ECOG) results of protocol E1293. Med Oncol. 1996;13(2):87–93. | ||
Thuss-Patience PC, Kretzschmar A, Repp M, et al. Docetaxel and continuous-infusion fluorouracil versus epirubicin, cisplatin, and fluorouracil for advanced gastric adenocarcinoma: a randomized phase II study. J Clin Oncol. 2005;23(3):494–501. | ||
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. | ||
Gong W, Song Q, Lu X, et al. Paclitaxel induced B7-H1 expression in cancer cells via the MAPK pathway. J Chemother. 2011;23(5):295–299. | ||
Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in Cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–856. | ||
Liang SC, Latchman YE, Buhlmann JE, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33(10):2706–2716. | ||
Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63(63):185–198. | ||
Friedman AD, Claypool SE, Liu R. The smart targeting of nanoparticles. Curr Pharm Des. 2013;19(35):6315–6329. | ||
Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. | ||
Ramakrishnan R, Assudani D, Nagaraj S, et al. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest. 2010;120(4):1111–1124. | ||
van der Most RG, Currie AJ, Cleaver AL, et al. Cyclophosphamide chemotherapy sensitizes tumor cells to TRAIL-dependent CD8 T cell-mediated immune attack resulting in suppression of tumor growth. PLoS One. 2009;4(9):e6982. | ||
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714. | ||
Ménard C, Martin F, Apetoh L, Bouyer F, Ghiringhelli F. Cancer chemotherapy: not only a direct cytotoxic effect, but also an adjuvant for antitumor immunity. Cancer Immunol Immunother. 2008;57(11):1579–1587. | ||
Gadgeel S, Stevenson J, Langer CJ, et al. Pembrolizumab (pembro) plus chemotherapy as front-line therapy for advanced NSCLC: KEYNOTE-021 cohorts A-C. J Clin Oncol. 2016;suppl 15:9016–9016. | ||
Martino EC, Misso G, Pastina P, et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov. 2016;2:16025. | ||
Atzpodien J, Kirchner H, Jonas U, et al. Interleukin-2- and interferon alfa-2a-based immunochemotherapy in advanced renal cell carcinoma: a prospectively randomized trial of the German cooperative renal carcinoma chemoimmunotherapy group (DGCIN). J Clin Oncol. 2004;22(7):1188–1194. | ||
Parra HS, Tixi L, Latteri F, et al. Combined regimen of cisplatin, doxorubicin, and alpha-2b interferon in the treatment of advanced malignant pleural mesothelioma: a Phase II multicenter trial of the Italian Group on Rare Tumors (GITR) and the Italian Lung Cancer Task Force (FONICAP). Cancer. 2001;92(3):650–656. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.