Back to Journals » Journal of Experimental Pharmacology » Volume 13
Pathophysiological Mechanisms and Experimental Pharmacotherapy for L-Dopa-Induced Dyskinesia
Authors Fabbrini A, Guerra A
Received 10 February 2021
Accepted for publication 30 March 2021
Published 29 April 2021 Volume 2021:13 Pages 469—485
DOI https://doi.org/10.2147/JEP.S265282
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Andrea Fabbrini,1 Andrea Guerra2
1Department of Human Neurosciences, Sapienza University of Rome, Rome, Italy; 2IRCCS Neuromed, Pozzilli, IS, Italy
Correspondence: Andrea Guerra
IRCCS Neuromed, Sapienza University of Rome, Via Atinense 18, Pozzilli, 86077, IS, Italy
Email [email protected]
Abstract: L-dopa-induced dyskinesia (LID) is the most frequent motor complication associated with chronic L-dopa treatment in Parkinson’s disease (PD). Recent advances in the understanding of the pathophysiological mechanisms underlying LID suggest that abnormalities in multiple neurotransmitter systems, in addition to dopaminergic nigrostriatal denervation and altered dopamine release and reuptake dynamics at the synaptic level, are involved in LID development. Increased knowledge of neurobiological LID substrates has led to the development of several drug candidates to alleviate this motor complication. However, with the exception of amantadine, none of the pharmacological therapies tested in humans have demonstrated clinically relevant beneficial effects. Therefore, LID management is still one of the most challenging problems in the treatment of PD patients. In this review, we first describe the known pathophysiological mechanisms of LID. We then provide an updated report of experimental pharmacotherapies tested in clinical trials of PD patients and drugs currently under study to alleviate LID. Finally, we discuss available pharmacological LID treatment approaches and offer our opinion of possible issues to be clarified and future therapeutic strategies.
Keywords: dyskinesia, L-dopa, Parkinson’s disease, glutamate, serotonin, therapy
Introduction
Parkinson’s disease (PD) is a common progressive neurodegenerative disease characterized by motor symptoms such as bradykinesia, tremor, and rigidity. PD symptoms are mainly the result of the loss of dopaminergic neurons in the substantia nigra.1 Non-motor symptoms, including cognitive, psychiatric, and neurovegetative symptoms, are also common in PD.2 The dopamine precursor levodopa (L-dopa) is currently the most effective symptomatic treatment for PD patients. However, chronic L-dopa treatment is associated with the development of motor fluctuations and L-dopa-induced dyskinesia (LID). More than 50% of patients develop LID after 5 years of L–dopa treatment, with a significant impact on quality of life and a consequent urge in identifying effective treatment strategies for this disabling motor complication.3–5 LID is characterized by choreic and dystonic movements affecting different body parts that occur at different points of the L-dopa cycle (Table 1). Risk factors for LID include an earlier age of PD onset, longer PD duration, longer L-dopa treatment duration, higher L–dopa total exposure, lower body mass index, and female gender.6–10 The role of genetic factors in LID development is still unclear.11
 |
Table 1 Types of Dyskinesia in Parkinson’s Disease |
LID management is one of the most challenging problems in the treatment of PD. The first step in the therapeutic approach to a PD patient with LID is to recognize the clinical pattern of dyskinesia, since OFF period dystonia, peak-dose dyskinesia, and diphasic dyskinesia can be influenced differently by the modification of current antiparkinsonian medication. For instance, in the case of OFF period dystonia, which typically occurs at night or in early morning hours, long-acting formulations of L-dopa can be administered at bedtime.12 On the other hand, while diphasic dyskinesia generally improves by increasing dopaminergic medications, peak-dose dyskinesia management requires the opposite approach. However, L–dopa dose redistribution and overall dopaminergic treatment reduction are sometimes followed by motor symptoms worsening. In these cases, antidyskinetic drugs may be necessary. Amantadine and clozapine are the only available drugs that are recognized to be clinically useful in counteracting LID.13 However, both have efficacy and safety limitations. For this reason, new molecules are needed, and several drugs have been proposed or are currently under study to treat LID. The search for new avenues in LID treatment is difficult due to the complex and incompletely defined pathophysiology of LID. In this review, we first describe the main accredited pathophysiological mechanisms underlying LID in PD patients. We then analyse the experimental pharmacological therapies currently under study for attenuating LID in PD patients. A search was conducted on ClinicalTrials.gov using “dyskinesias” and “Parkinson’s disease” as keywords. Studies from 2015 to the present were included. This search resulted in a list of drugs with diverse pharmacological targets that were studied in Phase I-III clinical trials in the last 5 years (Table 2). Studies on drugs whose mechanisms of action were not supported by strong literature data were excluded from the literature review.
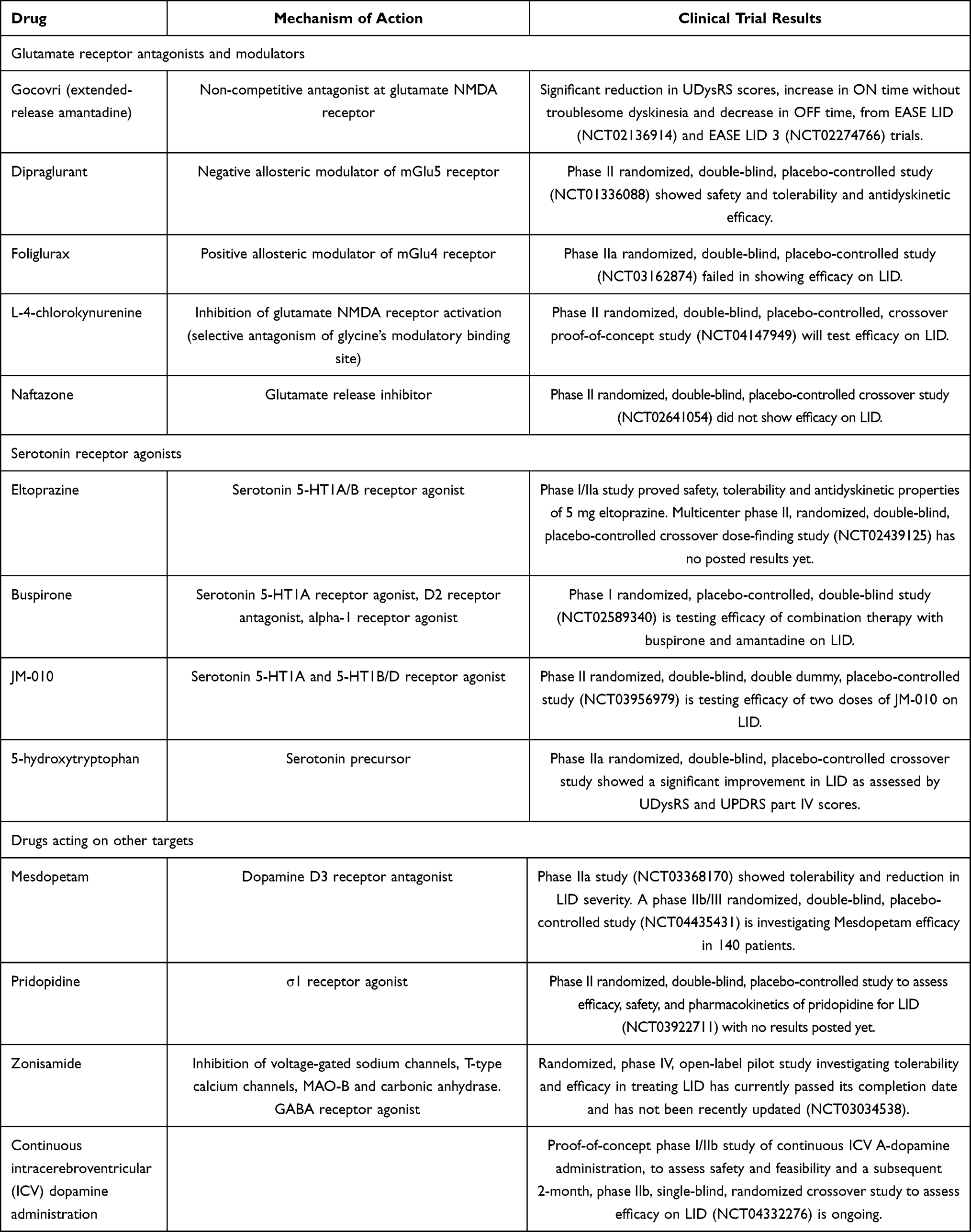 |
Table 2 Investigational Drugs for LID |
Pathophysiological Mechanisms of LID
Dopaminergic Mechanisms
The main factor responsible for LID development in PD is the degree of dopaminergic denervation in the nigrostriatal pathway. Importantly, this factor is independent of the duration of L-dopa exposure. Indeed, experimental studies have demonstrated that administering L–dopa in animals with severe nigrostriatal denervation determines LID emergence from the first doses of the drug.14–19 Similarly, LID appeared very shortly after the initiation of L-dopa therapy in newly diagnosed sub-Saharan PD patients with advanced disease.20 In contrast, chronically administering dopaminergic treatment in conditions characterized by an intact nigrostriatal pathway (eg, healthy animals, patients with restless leg syndrome) did not result in LID.15,17,21,22 However, severe dopaminergic denervation, although essential, is not sufficient per se to cause LID development. A second relevant pathophysiological factor is the introduction of oral short half-life L-dopa, which provides non-physiological pulsatile dopaminergic stimulation in the striatum. This approach makes dopamine (DA) central levels dependent on the pharmacokinetic of exogenous L–dopa.17,18,22–24 This abnormality is indirectly related to progressive neuronal loss of dopaminergic terminals, which leads to altered presynaptic dynamics in DA conversion, release, and reuptake. In fact, in early PD stages, non-degenerated dopaminergic neurons provide for the conversion of L-dopa to DA and for DA storage, ensuring the important fine-tuning of DA release and allowing DA concentration to be maintained at physiological levels in the synaptic cleft. This DA buffering capacity is possible because of D2 autoreceptor and DA transporter (DAT) activity. However, in advanced PD stages, the vast majority of dopaminergic neurons are lost, and DA conversion and release is mainly provided by serotoninergic terminals, which do not have the molecular machinery for feedback control and DA release regulation.25–28 Accordingly, DA concentration cannot be tuned, leading to abnormal swings in extracellular DA following pulsatile oral intake of L–dopa.28–30 A direct evidence of this pathological mechanism has been recently provided in rats, where ectopically expressing the D2 autoreceptor in serotoninergic neurons prevented DA swings and blocked LID development, even using high L-dopa doses.31
The combination of severe dopaminergic denervation and the lack of physiological tonic dopaminergic stimulation in the nigrostriatal circuit causes alterations in DA receptor-mediated signalling and corticostriatal synaptic plasticity. D1 receptor sensitization and D1-related signalling cascade and direct pathway neuron overactivity are postsynaptic abnormalities associated with LID. Molecular biology studies show that excessive intracellular activation in the ERK1/2 and cAMP/PKA signalling of striatal neurons and other signalling pathway changes contribute to D1 hyperactivity in LID.17,18,32 The role of D1 overactivation in LID development is supported by studies demonstrating that D1 receptor agonists and optogenetic D1 stimulation induce dyskinesia, while genetic and pharmacological D1 blockages decrease LID.24,33–35 Although the contribution of the D2 receptor is less clear, recent findings suggest an association between overall inhibition of the indirect pathway and LID. Indeed, dyskinesia determines synaptic modifications in indirect pathway neurons, and the stimulation of these neurons improves LID.18,36–38
Finally, D3 receptor activity changes also play a role in LID. In mice, increased D3 receptor levels in the dorsal striatum of 6-OHDA rats are associated with LID, whereas D3 receptor levels were reduced in a D3 receptor gene knockout model and LID decreased accordingly.39 Increased D3 receptor expression was also observed with PET using the D3 receptor-preferring radioligand [11C]PHNO in the dorsal striatum of PD patients chronically treated with L-dopa and in the globus pallidus of patients with LID.40 Interestingly, D3 overactivation seems to be functionally related to D1 pathway modification,39 and recent evidence suggests that aberrant D1-D3 receptor interactions may also contribute to LID.41
Concerning the alterations in corticostriatal synaptic plasticity, evidence from animal studies suggests that “bidirectional plasticity” impairment may be the electrophysiological hallmark of LID.42,43 Bidirectional plasticity reflects the property to undergo synaptic neurotransmission depression or potentiation with the same plasticity-inducing protocol, depending on the different receptor state. Bidirectional plasticity also encompasses the capacity of potentiated synapses to return to normal activity levels after specific stimulation protocols, a physiological phenomenon known as “depotentiation”. Interestingly, experimental studies have shown that L-dopa can restore long–term potentiation (LTP) plasticity in parkinsonian animals, whereas striatal LTP is not susceptible to depotentiation in animals with LID.42–46 A similar finding has also been described at the motor cortex level in PD patients.47 In summary, synaptic plasticity becomes less “plastic” as LID develops.
Glutamatergic Mechanisms
Besides dopaminergic mechanisms, alterations in several non-dopaminergic systems have been linked to LID pathophysiology in the last two decades. First, significantly enhanced glutamatergic neurotransmission has been consistently found at multiple levels within the basal ganglia-thalamo-cortical circuit.48–52 The excitatory effect of glutamate (Glu) is mediated by ionotropic (NMDA - GluN, AMPA - GluA, and kainate - GluK) and metabotropic (mGlu) receptors. Excessive amounts of NMDA and AMPA receptors are present in the striatum of parkinsonian patients and animals with LID.50,53 Moreover, neuroimaging studies have shown increased levels of NMDA-related Glu both in the striatum and motor cortical areas in dyskinetic, but not in non-dyskinetic, PD patients after L–dopa administration, confirming the role of abnormal Glu release in LID.54 NMDA receptors also undergo several molecular structural and functional changes, including altered phosphorylation, trafficking, GluN2A and GluN2B subunit composition rearrangement, and abnormal GluN2A and GluN2B striatal expression (ie, increased and reduced expression of GluN2A and GluN2B, respectively), which eventually enhance glutamatergic excitatory activity in the corticostriatal synapses.48–50,55,56 A confirmation of the link between increased NMDA-related Glu and LID also comes from pharmacological studies using NMDA antagonist drugs, which have been demonstrated to be effective in reducing dyskinesia in animals57–60 and humans (see below). The involvement of mGlu receptors in LID pathophysiology has also been clearly demonstrated, and mGlu receptors have been found to modulate Glu intracellular signalling without affecting Glu excitatory action on synaptic neurotransmission. Three main groups of mGlu receptors exist according to sequence homology and G-protein coupling and ligand-binding profile.51 Group I receptors (mGluR1 and mGluR5) are coupled with phospholipase Cβ and modulate intracellular calcium release through inositol-triphosphate formation and protein kinase C activation, while group II (mGluR2 and mGluR3) and III receptors (mGluR4, mGluR6, mGluR7, and mGluR8) are coupled with inhibitory G proteins and reduce the formation of cyclic adenosine monophosphate (cAMP).51,61 Group I receptors amplify NMDA receptor currents and increase D1-related pathway activity. Animal studies have shown that striatal mGluR5 levels increase in parallel with LID development.62,63 Similarly, mGluR5 is enhanced in the putamen and globus pallidus of dyskinetic PD patients.64 In line with these data, mGluR5 antagonist drugs were shown to be effective in attenuating LID in various animal models of PD.65 In contrast to the effect of group I receptors, group II mGlu stimulation inhibits Glu release in corticostriatal and subthalamonigral terminals,51,66 while group III mGlu activation reduces glutamatergic neurotransmission in the globus pallidus.67–69 Finally, a recent study in patients with PD demonstrated that a specific neurophysiological measure reflecting non-NMDA glutamatergic activity in the primary motor cortex is abnormally enhanced in dyskinetic patients, but can be normalized by using safinamide, a drug that inhibits glutamate release by blocking voltage-gated sodium channels.52,70
Figure 1 shows the main alterations in dopaminergic and glutamatergic synapses in the striatum.
 |
Figure 1 Alterations in glutamatergic and dopaminergic synapses in the striatum. The most relevant abnormalities in glutamatergic (Glu – left) and dopaminergic (DA – right) neurotransmission are illustrated. Increased expression of AMPA, NMDA and mGluR5 receptors, as well as structural and functional changes in NMDA receptors, are associated with overactive Glu neurotransmission. DA neurons have the molecular machinery for feedback control and dopamine release regulation, ie, DA transporter (DAT) and D2 autoreceptors. The latter can decrease dopamine synthesis and release and increase dopamine reuptake through DAT activity modulation. The progressive loss of DA terminals leads to abnormal swings in extracellular dopamine following the L-dopa intake, which are associated with alterations in D1 and D3 receptor-mediated signalling. |
Serotoninergic Mechanisms
While the bulk of evidence supports the role of abnormal glutamatergic activity in LID pathophysiology, some data suggest the involvement of additional non-dopaminergic systems. The involvement of the serotoninergic system in LID is supported by data showing significant striatal hyper-innervation in parkinsonian animals71,72 and enhanced 5-HT transporter (SERT) binding and increased SERT-to–DAT binding ratio in the putamen and globus pallidus of dyskinetic patients.73–76 The contribution of the serotoninergic system is also corroborated by evidence in animal models that 5-HT1A and 5-HT1B receptor agonist drugs normalize serotoninergic neurotransmission, attenuate excessive DA release following L–dopa administration, and reduce LID.77–82 Also, Vilazodone, a selective serotonin reuptake inhibitor and partial 5-HT1A agonist, significantly suppressed developing and established LID without compromising the promotor effects of L-dopa in 6-hydroxydopamine-lesioned hemi-parkinsonian rats.83 Moreover, drugs directly or indirectly modulating SERT, like citalopram, paroxetine and UWA-101/121/122, as well as the combination of SERT- and 5-HT1A-targeting compounds, showed definite antidyskinetic effects in animals.84 In this regard, a recent clinical study conducted in 111 patients demonstrated that the early SSRI exposure in the PD course delays LID onset.85 Increased extracellular DA levels after L-dopa intake have been confirmed in dyskinetic PD patients using positron emission tomography (PET) imaging,86 and buspirone, a partial 5-HT1A agonist, was able to decrease this abnormality.87
Other Mechanisms
For instance, cholinergic neurotransmission mediated by nicotinic receptors has been putatively implicated in dyskinesia in PD.88 Indeed, nicotinic receptors are located close to dopaminergic receptors, are similarly distributed, and their activation modulates DA release.89 In addition, both nicotine and nicotinic receptor agonists were found to prevent or alleviate LID in mice, rat, and primate PD models.90–95 Alpha adrenergic receptors are also located in the striatum and modulate GABA release.96 Alpha receptor stimulation activates the direct pathway, thus possibly contributing to LID generation mechanisms. In further support of this hypothesis, alpha adrenergic receptor antagonists were found to reduce dyskinesia in animal studies.97,98 Alterations in opioid peptide expression and opioid receptor-mediated intracellular signalling are also described in PD patients and animal models of PD with LID.99 The µ opioid receptors (OPRM1) are located in the striatum and their expression is reduced in LID, while OPRM1-related signal transduction is increased in the dyskinetic state.100 Moreover, drugs blocking or downregulating OPRM1 activity attenuated dyskinesia in parkinsonian monkeys.101,102 Finally, the dysregulation of calcium channels has been hypothesized as a mechanism involved in LID. Supporting this idea, in a recent study, a genetic silencing of striatal CaV1.3 voltage‐gated L‐type calcium channels was found to completely prevent LID in severely parkinsonian rats and to ameliorate pre-existing severe LID.103
All the events described in this section (Figure 2) contribute to pathological changes in neuronal firing patterns, which in turn lead to excessive disinhibition of thalamocortical neurons and overactivation of cortical motor areas, and ultimately determine LID development.17,18
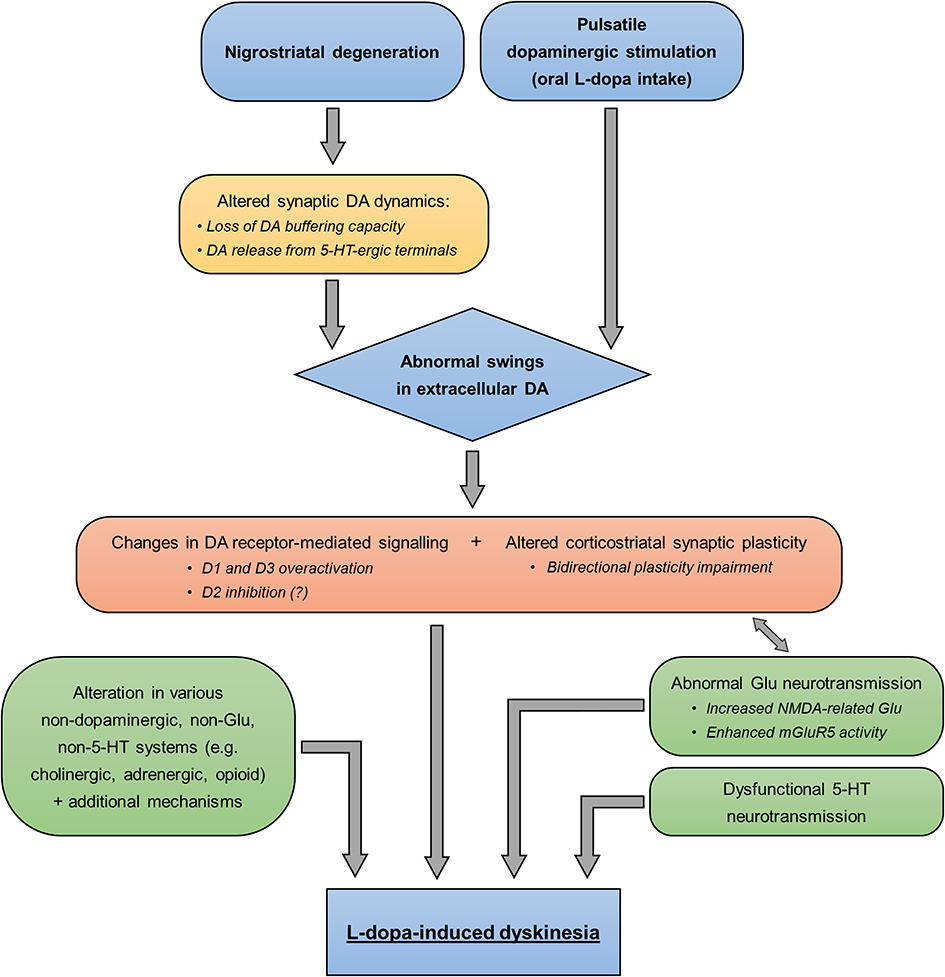 |
Figure 2 Pathophysiological mechanisms of LID. The diagram summarizes the known pathophysiological changes leading to LID development. Pre- and post-synaptic abnormalities are shown in the yellow and orange, respectively. Alterations in non-dopaminergic systems are indicated in the green boxes. Abbreviations: DA, dopamine; Glu, glutamate; 5-HT, serotonin. |
Experimental Pharmacological Therapies for LID in PD Patients
Glutamate Receptor Antagonists and Modulators
Amantadine hydrochloride (HCl) is currently considered the most effective drug for the treatment of LID in patients with PD.104 Originally developed as an anti-influenza agent, it acts as an uncompetitive antagonist at the glutamate NMDA receptor and has predictable anticholinergic (eg, dry mouth, urinary retention, constipation) and anti-glutamatergic (eg, hallucinations) adverse effects. Its immediate-release formulation (amantadine IR) has been extensively used over the years for the treatment of LID. Gocovri (ADS-5102) is a capsule containing 137 mg of extended-release amantadine developed by Adamas Pharmaceuticals in order to provide a therapeutic level of amantadine in the blood for a longer period of time than amantadine IR. Gocovri is administered at bedtime (two capsules (274 mg) corresponding to 340 mg of amantadine HCl) and was formulated to achieve a slow increase in amantadine blood levels during sleep, peak concentrations in the morning, and sustained concentrations throughout the day. Two Phase III randomized, double-blind, placebo-controlled studies (EASE LID (ClinicalTrials.gov identifier: NCT02136914) and EASE LID 3 (ClinicalTrials.gov identifier: NCT02274766)) were carried out between 2014 and 2016 to test Gocovri efficacy in treating LID as measured by changes in Unified Dyskinesia Rating Scale (UDysRS) scores.105,106 The trials included 121 and 75 patients, respectively, and had similar designs, apart from an additional timepoint of 24 weeks in the EASE LID trial and other secondary outcomes. Both trials included PD patients with LID (score of at least 2 on question 4.2 of the Unified Parkinson’s Disease Rating Scale - UPDRS), at least two episodes of ON time with troublesome dyskinesia lasting 30 minutes, and stable antiparkinsonian medication during the last 30 days, with L-dopa administered at least 3 times daily. Both trials demonstrated that Gocovri treatment determined a significant reduction in UDysRS scores, an increase in ON time without troublesome dyskinesia, and a decrease in OFF time, while no differences were observed in UPDRS parts I, II, and III scores, suggesting no interaction with motor function. Common recorded adverse effects were visual hallucinations, peripheral edema, dizziness, orthostatic hypotension, dry mouth, and constipation. Based on the results of these trials, Gocovri was approved in 2017 by the Food and Drug Administration for the treatment of LID in PD patients on concomitant L-dopa-based therapy. The drug underwent a final 2-year open-label trial in 223 PD patients with LID and showed long-term safety, tolerability, and efficacy on dyskinesia and OFF time.107 However, the drug is not available outside the US.
ADX48621 or dipraglurant is a negative allosteric modulator of the metabotropic glutamate 5 receptor developed by Addex Therapeutics. Its antidyskinetic properties had already been tested in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) macaque model.108 A Phase II randomized, double-blind, placebo-controlled study on 76 participants testing dose escalation from 50 mg once daily to 100 mg three times a day (ClinicalTrials.gov identifier: NCT01336088) showed that dipraglurant was safe and well tolerated and determined a statistically significant improvement (32% reduction in LID severity with 100 mg three times a day) at day 14 in peak-dose dyskinesia as measured by the modified Abnormal Involuntary Movement Scale (mAIMS). However, a strong placebo response at day 28 resulted in significance being lost.109 A phase IIb/III pivotal clinical trial on 140 PD patients with LID that was postponed due to the Covid-19 pandemic is scheduled to start at the end of 2020 and will use the UDysRS instead of the mAIMS to measure efficacy of the primary endpoint. Concerning these two scales, the mAIMS focuses on anatomical distribution and severity of dyskinesia whereas the UDysRS combines both patient-based and objective evaluation of dyskinesia severity and related disability.110,111 The UDysRS was shown to be more sensitive to treatment effects and less prone to placebo responses than mAIMS.112
With regard to the possible role of group II and III metabotropic glutamate receptors in PD and LID, a new molecule, foliglurax (PXT002331), an mGlu4 receptor positive allosteric modulator, showed improvement in parkinsonian symptoms and dyskinesia in MPTP macaque disease models.113 However, a phase IIa randomized, double-blind, placebo-controlled study involving 157 PD patients experiencing end-of-dose wearing-off and LID (AMBLED; ClinicalTrials.gov identifier: NCT03162874) failed to show a significant reduction in OFF time or dyskinesia after 4 weeks of foliglurax treatment. As a consequence, the foliglurax development program was terminated.
L-4-chlorokynurenine (AV-101) inhibits glutamate NMDA receptor activation by selective antagonism of glycine’s modulatory binding site on this receptor.114 L-4-chlorokynurenine efficacy in mitigating overactive glutamatergic transmission has been investigated in a previous Phase I study in patients suffering from neuropathic pain. The drug showed an excellent safety profile, and was effective in reducing allodynia and mechanical and heat hyperalgesia.115 Due to extensive preclinical and clinical evidence linking excessive glutamatergic transmission to LID pathogenesis, VistaGen Therapeutics has agreed to sponsor a phase II randomized, double-blind, placebo-controlled, crossover proof-of-concept study to test the efficacy and safety of L-4-chlorokynurenine in PD patients with LID (ClinicalTrials.gov identifier: NCT04147949). The study will include 20 participants (30 to 80 years of age) with PD and moderate dyskinesia for at least 25% of the day who will be randomly assigned to L-4-chlorokynurenine or placebo administration for 14 days, with a 1-week washout period before crossover. Primary outcomes are the area under the curve and peak UDysRS part III score at day 14. The secondary outcome is UPDRS part III score. The study is currently not yet recruiting participants and will end in April 2022.
Another molecule, naftazone (CVXL-0107), a glutamate release inhibitor, showed antidyskinetic properties in the MPTP macaque model.116 A subsequent phase II randomized, double-blind, placebo-controlled crossover study (ClinicalTrials.gov identifier: NCT02641054) in 16 PD patients did not show any significant differences in motor symptoms (UPDRS part III) or dyskinesia (mAIMS).117
Serotonin Receptor Agonists
Eltoprazine (originally DU-28853) is a strong 5-HT1A/B receptor agonist initially developed for the treatment of pathological aggression in intellectually disabled patients.118 The antidyskinetic properties of this molecule are due to 5-HT1A- and 5-HT1B-mediated inhibition of serotonin neurons responsible for the uncontrolled stimulation of supersensitized dopamine D1 receptors, which is characteristic of LID state. Acute administration of eltoprazine reduced LID in 6-OHDA lesioned rats and in MPTP monkeys treated with L-dopa.79
Based on the antidyskinetic properties shown in preclinical studies, the drug underwent a phase I/IIa study with three tested doses (2.5 mg, 5 mg, and 7.5 mg) in 22 PD patients with LID to evaluate its safety profile and antidyskinetic properties. Besides proving its safety and tolerability, this study showed that only the 5 mg dose determined a significant improvement in dyskinesia (12% reduction in maximum LID severity) as compared with randomized placebo dosing.119 A multicenter Phase II, randomized, double-blind, placebo-controlled crossover dose-finding study on eltoprazine safety, tolerability, and efficacy in LID (as measured by total UDysRS change) began in 2015 with an estimated enrollment of 60 participants, and was estimated to end in December 2017 (ClinicalTrials.gov identifier: NCT02439125). However, the study’s recruitment status is currently listed as “unknown” and no results have been posted yet.
Buspirone, a 5-HT1A receptor agonist, weak D2 receptor antagonist, and alpha-1 receptor agonist, is an anxiolytic whose activity on serotonergic neurons has been exploited to test its efficacy in treating LID. In a study on 7 patients with LID, buspirone administered at 10 mg twice daily determined a significant reduction in LID severity in 5 patients with more severe dyskinesia, but not in the remaining 2 patients with less severe dyskinesia.120 In a study in 3 patients with OFF-state dyskinesia post transplantation with dopamine-rich fetal mesencephalic tissue who displayed excessive serotonergic innervation of the grafted striatum in neuroimaging analysis, the administration of buspirone significantly attenuated dyskinesia, further supporting the serotonergic hypothesis of LID pathophysiology.121 A randomized, placebo-controlled, double-blind, two–period crossover phase I study (ClinicalTrials.gov identifier: NCT02589340) began in 2015 to test the efficacy of combination therapy with buspirone and amantadine on LID. The study is enrolling participants 18 to 99 years of age with mild dyskinesia treated with amantadine (200–500 mg/day) with insufficient benefit on LID and will randomize them to placebo or buspirone titrated to 30 mg/day over 2 weeks. The primary outcomes are area under the curve measurements for dyskinesia for a 6-hour L-dopa dose cycle, total UDysRS change after 6 weeks, and adverse events monitoring. The study’s estimated primary completion date is December 2020 and is currently listed as “active, not recruiting” on ClinicalTrials.gov.
Building on buspirone’s potential for LID treatment, a new drug, JM-010, was developed by Contera Pharma and combines buspirone’s agonist properties on 5-HT1A receptors with zolmitriptan’s 5-HT1B/5-HT1D agonist properties. JM-010 was shown to be effective in improving dyskinesia in the 6-OHDA rat model. A previous phase IIa/proof-of-concept study in 30 PD patients with LID in South Africa met the criteria for efficacy and safety (ClinicalTrials.gov identifier: NCT02439203). A phase II randomized, double-blind, double dummy, placebo-controlled parallel group study (ASTORIA study) (ClinicalTrials.gov identifier: NCT03956979) sponsored by Contera Pharma is currently ongoing to study the efficacy of two dose levels of JM-010 (4 mg buspirone/0.8 mg zolmitriptan and 8 mg buspirone/0.8 mg zolmitriptan) as compared with placebo in PD patients 18 to 80 years of age on a stable L-dopa regimen experiencing stable peak-dose dyskinesia (at least 1 hour of ON-state LID). The primary outcome is total UDysRS score over 12 weeks. The study is currently listed as “recruiting” and should end in 2022.
Building on the results of a preclinical study in the 6-OHDA lesioned rat,122 Meloni et al carried out a phase IIa randomized, double-blind, placebo-controlled crossover study to assess the efficacy of the serotonin precursor 5-hydroxytryptophan (5-HTP) in treating LID in PD patients.123 The study enrolled 12 participants with LID and motor fluctuations randomized to 50 mg of 5-HTP or placebo for 16 weeks in a crossover design over a period of 4 weeks and showed a significant improvement in dyskinesia as assessed by UDysRS and UPDRS part IV scores, with no modifications in motor performance as measured by UPDRS part III. The authors hypothesized two antidyskinetic mechanisms for 5-HTA: i) competition between increased intracellular 5-HTP-derived serotonin and L-dopa-derived dopamine for storage in serotonin synaptic vesicles, leading to a reduction in uncontrolled dopamine release from serotonergic neurons; ii) activation of serotonin autoreceptors resulting in a reduced serotonin neuron firing rate.
D3 Receptor Antagonists
The potential use of D3 receptor antagonists in LID treatment is supported by studies showing increased D3 receptor expression in the striatum of animals with LID chronically treated with L-dopa (see Pathophysiological Mechanisms of LID).39,124
Mesdopetam (IRL-790), developed by Integrative Research Laboratories, is a preferential dopamine D3 receptor antagonist with psychomotor stabilizing properties.125 A phase Ib study with mesdopetam as an adjunct treatment to regular antiparkinsonian medication in 15 PD patients experiencing peak-dose dyskinesia showed a median UDysRS score reduction of 11.5 points vs placebo.126 In 2019, a randomized, double-blind, placebo-controlled, multicenter phase IIa study on the efficacy and tolerability of mesdopetam was carried out in 74 PD patients with LID on stable antiparkinsonian medication (ClinicalTrials.gov identifier: NCT03368170). The study showed a meaningful reduction in LID severity as assessed by UDysRS, UPDRS part IV, and patient-reported diaries (Hauser diaries), and no side effects were reported. A larger phase IIb/III randomized, double-blind, placebo-controlled study (ClinicalTrials.gov identifier: NCT04435431) began in November 2020 to investigate the efficacy of mesdopetam (three dose groups) vs placebo in 140 PD patients with LID. The primary outcome measure was the change in average daily hours of ON time without troublesome dyskinesia as assessed by 24-hour patient home diaries from baseline to the end of treatment (week 12). The study’s estimated completion date is January 2022.
Other Targets
Pridopidine is a molecule originally developed for the treatment of Huntington’s disease (HD) that has a high affinity for the σ1 receptor and a moderate-to-low affinity for other central nervous system receptors, including D2, D3, σ2, adrenergic α2A and α2C, 5-HT1A, 5-HT2A, and 5-HT7 receptors. In a study in 6-OHDA lesioned mice, pridopidine administration determined neuroprotective effects on dopaminergic cell bodies and increased dopaminergic fiber density in the striatum and striatal upregulation of several growth factors.127 In a further study in the MPTP macaque model of PD with LID, pridopidine determined a significant reduction in dyskinesia (71% reduction), reduced ON time with disabling dyskinesia, and no change in the antiparkinsonian benefit of L-dopa administration.128 Since safety and tolerability were established in previous studies in HD patients,129,130 a phase II randomized, double-blind, placebo-controlled study was carried out to assess the efficacy, safety, and pharmacokinetics of pridopidine (in 2 dose regimens) vs placebo for the treatment of LID in PD patients (ClinicalTrials.gov identifier: NCT03922711). The trial is currently listed as “terminated” due to Covid-19 issues after enrolling 23 of the originally estimated 135 participants, and no results have been posted.
Zonisamide, an antiepileptic drug with inhibitory activity on voltage-gated sodium channels, T-type calcium channels, MAO-B and carbonic anhydrase and with enhancing properties on GABA receptor activity, has previously proved its efficacy in treating wearing-OFF symptoms,131 and is currently approved for the treatment of motor fluctuations in Japan. Zonisamide efficacy in treating LID has been investigated in preclinical studies carried out in 6-OHDA rat and mouse models of PD, showing improving and detrimental effects on LID, respectively.132,133 A randomized, Phase IV, 12-week open-label pilot study investigating zonisamide tolerability and efficacy in treating LID has currently passed its completion date and has not been recently updated (ClinicalTrials.gov Identifier: NCT03034538).
In 2017, Laloux et al carried out a study in chronic MPTP lesioned mice and acute 6–OHDA lesioned rats to assess the feasibility and efficacy of continuous intracerebroventricular (ICV) dopamine administration on motor function. ICV dopamine administration can be performed through an abdominal pump connected by a subcutaneous catheter to the lateral ventricle, close to the lateral striatum. Previous preclinical and human attempts to administer dopamine this way had failed as a result of dopamine oxidation causing excessive oxidative stress or dopamine metabolism and tachyphylaxis.134–136 The study by Laloux used an anaerobic dopamine preparation (A-dopamine) to reduce oxidative stress and programmable pumps to minimize tachyphylaxis. The study showed that A-dopamine effectively restored motor function in preclinical animal models without inducing tachyphylaxis or motor complications, such as dyskinesia, and had a broader therapeutic index than peripheral L-dopa treatment. Furthermore, in the MPTP mouse model, A-dopamine induced a dose-dependent increase in nigrostriatal dopaminergic neurons that was not evident with either O-dopamine (aerobically prepared dopamine) or peripheral L-dopa.137,138 Based on these results, a proof-of-concept phase I/IIb study of continuous ICV A-dopamine administration, with a 1-month phase I trial to assess safety and feasibility and a subsequent 2-month, phase IIb, single-blind, randomized crossover study to assess efficacy on motor fluctuations, including LID, over optimized oral treatment began in September 2020 (ClinicalTrials.gov identifier: NCT04332276). The study will enroll 20 participants and the primary outcome measure is the percentage of time that bradykinesia, as measured by actimetry, exceeds a prespecified target (26). The study is currently listed as “not yet recruiting” and should end in 2023.
Discussion and Concluding Remarks
Pharmacological treatment of LID is a major challenge in PD patients. First, LID is a complex clinical phenomenon characterized by a combination of choreic and dystonic abnormal movements in different body parts, with specific features linked to each L-dopa cycle. A significant limitation in many clinical trials is that LID assessment has often been based on patient self-assessment diaries. However, PD patients may not be completely aware of LID, and therefore data collected through self-assessment questionnaires may be biased.139 An important step to overcome this limitation has been the introduction and validation of the UDysRS, a new LID rating scale that is now increasingly used in clinical trials. UDysRS, indeed, has four parts and part III and IV consist in the objective evaluation by the researcher of LID severity and related disability, thus excluding possible biases due to the patient’s subjective perceptions. Importantly, the objective sections of UDysRS have demonstrated high internal consistency and good inter- and intra-rater reliability.111 However, because different pathophysiological substrates possibly underlie dyskinesia, the drugs tested for LID treatment may be effective for only a specific type of dyskinesia (eg, for peak-dose dyskinesia, not onset- or end-of-dose dyskinesia). Finding the right drug for LID is also problematic since LID pathophysiology is complex and may involve several neurochemical pathways and neurophysiological mechanisms.
To date, pharmacological possibilities to reduce LID in PD patients include a few indirect and only two direct approaches. Indirect approaches are based on the idea that replacing pulsatile exogenous administration of L-dopa by strategies possibly mimicking physiological tonic dopaminergic stimulation of nigrostriatal pathways prevents the synaptic alterations and abnormalities in multiple neurotransmitter systems that contribute to LID pathophysiology.140,141 The administration of extended-release dopamine agonists, and particularly the early use of systems allowing continuous dopaminergic drug delivery, including subcutaneous apomorphine and intrajejunal L-dopa infusion, has demonstrated to be effective in attenuating blood L–dopa peaks and produced relatively good results on LID.142–146 Multicenter trials using intrajejunal L–dopa infusion, for instance, showed a significant increase in the ON time without troublesome dyskinesia (2–4 hours) and decrease in the ON time with troublesome dyskinesia (≈2 hours) at both short term and long term.144,147,148 Future approaches for continuous L-dopa delivery in the brain may include the use of viral vectors. In a recent proof-of-principle study, an adenovirus-associated vector was applied to deliver two L-dopa synthesizing enzymes in the striatum of parkinsonian monkeys, and this treatment determined a significant improvement in motor functions without pro-dyskinetic effects.149 Based on clinical data showing a direct correlation between the risk of LID development and the total amount of L-dopa intake,150 another strategy consists in reducing the daily L-dopa dosage using L-dopa-sparing pharmacological (ie, combination of low L-dopa doses with non-dopaminergic therapies) and non–pharmacological interventions (ie, deep brain stimulation).13,22,151–153
Regarding direct approaches, the only drugs currently approved for LID treatment are amantadine and clozapine.13 Amantadine demonstrated moderate antidyskinetic effects in clinical studies,104,154 but achieving LID improvement usually requires high doses, which can induce relevant side effects (eg, confusion, hallucinations, peripheral edema, livedo reticularis, and dizziness).154 Clozapine is an atypical neuroleptic with anti-dopaminergic, anti-serotonergic, anti-muscarinic, and anti-adrenergic properties.155 However, despite the fact that a double-blind placebo-controlled study conducted in 50 patients demonstrated positive results,156 clozapine’s safety profile is affected by the risk of serious adverse events, such as agranulocytosis.
LID treatment therefore remains an unmet need in PD patients. Based on the experimental data reported in this review, promising drug candidates are mainly directed at modulating altered glutamatergic and serotoninergic neurotransmission. However, although pharmacological studies using allosteric modulators of metabotropic glutamate receptors and 5-HT1 agonists produced positive results in animals, the majority of recent clinical trials in patients showed no or minimal LID improvement. Moreover, regarding compounds with 5-HT1 agonist properties, the possibility for an additional worsening of motor function ascribable to a global reduction in dopamine availability should be considered.157 Notably, none of the therapies currently under investigation (apart from extended-release amantadine) reached a phase III clinical trial.
We believe that further research is needed to clarify neurophysiological and neurobiological LID substrates, including whether and to what extent abnormalities in different neurotransmitter systems interact with each other. This would possibly determine the discovery of new altered mechanisms to target. However, beyond researching new potential targets for attenuating LID, we believe that future studies should clarify the reason for the discrepancy between the positive results in animals and negative results in human clinical trials. One possibility could be that placebo effects in PD patients may mask the efficacy of drug candidates due to the strong beneficial effects observed in placebo groups.158 In addition, most clinical trials reviewed in Experimental Pharmacological Therapies for LID in PD Patients involved a relatively small number of participants, and weak, though significant, effects may have been lost. Finally, since LID pathophysiology includes abnormalities in multiple dopaminergic and non-dopaminergic pathways, normalizing alterations in a single system could be insufficient to determine clinically relevant effects on LID in humans. In this regard, an interesting study in MPTP-treated monkeys showed that eltoprazine and amantadine act synergistically to counteract LID, and the combination of these drugs determined a marked increase in ON time without LID.79 Future clinical trials may test whether using a combination of (potentially effective) drugs acting on different neurotransmitter systems determines a strong and reliable LID reduction in PD patients.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896–912. doi:10.1016/S0140-6736(14)61393-3
2. Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci. 2017;18(7):435–450. doi:10.1038/nrn.2017.62
3. Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa–induced dyskinesias. Mov Disord. 2007;22(10):
4. Wu J, Lim E-C, Nadkarni NV, Tan E-K, Kumar PM. The impact of levodopa therapy-induced complications on quality of life in Parkinson’s disease patients in Singapore. Sci Rep. 2019;9(1):9248. doi:10.1038/s41598-019-45110-5
5. Grandas F, Galiano ML, Tabernero C. Risk factors for levodopa-induced dyskinesias in Parkinson’s disease. J Neurol. 1999;246(12):1127–1133. doi:10.1007/s004150050530
6. Bjornestad A, Forsaa EB, Pedersen KF, Tysnes O-B, Larsen JP, Alves G. Risk and course of motor complications in a population-based incident Parkinson’s disease cohort. Parkinsonism Relat Disord. 2016;22:48–53. doi:10.1016/j.parkreldis.2015.11.007
7. Stocchi F, Olanow CW. Continuous dopaminergic stimulation in early and advanced Parkinson’s disease. Neurology. 2004;62(1 Suppl 1):S56–S63. doi:10.1212/WNL.62.1_suppl_1.S56
8. Gilgun-Sherki Y, Djaldetti R, Melamed E, Offen D. Polymorphism in candidate genes: implications for the risk and treatment of idiopathic Parkinson’s disease. Pharmacogenomics J. 2004;4(5):291–306. doi:10.1038/sj.tpj.6500260
9. Zappia M, Annesi G, Nicoletti G, et al. Sex differences in clinical and genetic determinants of levodopa peak-dose dyskinesias in Parkinson disease: an exploratory study. Arch Neurol. 2005;62(4):601–605. doi:10.1001/archneur.62.4.601
10. Fahn S. Parkinson disease, the effect of levodopa, and the ELLDOPA trial. Earlier vs Later L-DOPA. Arch Neurol. 1999;56(5):529–535. doi:10.1001/archneur.56.5.529
11. Falla M, Di Fonzo A, Hicks AA, Pramstaller PP, Fabbrini G. Genetic variants in levodopa-induced dyskinesia (LID): a systematic review and meta-analysis. Parkinsonism Relat Disord. 2021;84:52–60. doi:10.1016/j.parkreldis.2021.01.020
12. Vijayakumar D, Jankovic J. Drug-induced dyskinesia, part 1: treatment of levodopa-induced dyskinesia. Drugs. 2016;76(7):759–777. doi:10.1007/s40265-016-0566-3
13. Fox SH, Katzenschlager R, Lim S-Y, et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2018;33(8):1248–1266. doi:10.1002/mds.27372
14. Winkler C, Kirik D, Björklund A, Cenci MA. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson’s disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2002;10(2):165–186. doi:10.1006/nbdi.2002.0499
15. Di Monte DA, McCormack A, Petzinger G, Janson AM, Quik M, Langston WJ. Relationship among nigrostriatal denervation, parkinsonism, and dyskinesias in the MPTP primate model. Mov Disord. 2000;15(3):459–466. doi:10.1002/1531-8257(200005)15:3<459::AID-MDS1006>3.0.CO;2-3
16. Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9(9):665–677. doi:10.1038/nrn2471
17. Bastide MF, Meissner WG, Picconi B, et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog Neurobiol. 2015;132:96–168.
18. Cenci MA, Jörntell H, Petersson P. On the neuronal circuitry mediating l-DOPA-induced dyskinesia. J Neural Transm. 2018;125(8):1157–1169. doi:10.1007/s00702-018-1886-0
19. Cenci MA, Crossman AR. Animal models of l-dopa-induced dyskinesia in Parkinson’s disease. Mov Disord. 2018;33(6):889–899. doi:10.1002/mds.27337
20. Cilia R, Akpalu A, Sarfo FS, et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain. 2014;137(10):2731–2742. doi:10.1093/brain/awu195
21. Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol. 2009;8(9):844–856. doi:10.1016/S1474-4422(09)70183-8
22. Leta V, Jenner P, Chaudhuri KR, Antonini A. Can therapeutic strategies prevent and manage dyskinesia in Parkinson’s disease? An update. Expert Opin Drug Saf. 2019;18(12):1203–1218. doi:10.1080/14740338.2019.1681966
23. Cerri S, Siani F, Blandini F. Investigational drugs in phase I and phase II for Levodopa-induced dyskinesias. Expert Opin Investig Drugs. 2017;26(7):777–791. doi:10.1080/13543784.2017.1333598
24. Chen X, Wang Y, Wu H, Cheng C, Le W. Research advances on L-DOPA-induced dyskinesia: from animal models to human disease. Neurol Sci. 2020;41(8):2055–2065. doi:10.1007/s10072-020-04333-5
25. Navailles S, De Deurwaerdère P. Imbalanced dopaminergic transmission mediated by serotonergic neurons in L-DOPA-induced dyskinesia. Parkinsons Dis. 2012;2012:323686. doi:10.1155/2012/323686
26. Carta M, Bezard E. Contribution of pre-synaptic mechanisms to L-DOPA-induced dyskinesia. Neuroscience. 2011;198:245–251. doi:10.1016/j.neuroscience.2011.07.070
27. Navailles S, Bioulac B, Gross C, De Deurwaerdère P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson’s disease. Neurobiol Dis. 2010;38(1):136–143. doi:10.1016/j.nbd.2010.01.012
28. Carta M, Björklund A. The serotonergic system in L-DOPA-induced dyskinesia: pre-clinical evidence and clinical perspective. J Neural Transm (Vienna). 2018;125(8):1195–1202. doi:10.1007/s00702-018-1865-5
29. Carta M, Carlsson T, Kirik D, Björklund A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain. 2007;130(7):1819–1833. doi:10.1093/brain/awm082
30. Mosharov EV, Borgkvist A, Sulzer D. Presynaptic effects of levodopa and their possible role in dyskinesia. Mov Disord. 2015;30(1):45–53. doi:10.1002/mds.26103
31. Sellnow RC, Newman JH, Chambers N, et al. Regulation of dopamine neurotransmission from serotonergic neurons by ectopic expression of the dopamine D2 autoreceptor blocks levodopa-induced dyskinesia. Acta Neuropathol Commun. 2019;7(1):8. doi:10.1186/s40478-018-0653-7
32. Heiman M, Heilbut A, Francardo V, et al. Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc Natl Acad Sci U S A. 2014;111(12):4578–4583. doi:10.1073/pnas.1401819111
33. Hernández LF, Castela I, Ruiz-DeDiego I, Obeso JA, Moratalla R. Striatal activation by optogenetics induces dyskinesias in the 6-hydroxydopamine rat model of Parkinson disease. Mov Disord. 2017;32(4):530–537. doi:10.1002/mds.26947
34. Perez XA, Zhang D, Bordia T, Quik M. Striatal D1 medium spiny neuron activation induces dyskinesias in parkinsonian mice. Mov Disord. 2017;32(4):538–548. doi:10.1002/mds.26955
35. Girasole AE, Lum MY, Nathaniel D, et al. A subpopulation of striatal neurons mediates levodopa–induced dyskinesia. Neuron. 2018;97(4):787–795.e6. doi:10.1016/j.neuron.2018.01.017
36. Fieblinger T, Graves SM, Sebel LE, et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Commun. 2014;5:5316. doi:10.1038/ncomms6316
37. Suarez LM, Solis O, Aguado C, Lujan R, Moratalla R. L-DOPA oppositely regulates synaptic strength and spine morphology in D1 and D2 striatal projection neurons in dyskinesia. Cereb Cortex. 2016;26(11):4253–4264. doi:10.1093/cercor/bhw263
38. Suárez LM, Solís O, Caramés JM, et al. L-DOPA treatment selectively restores spine density in dopamine receptor D2-expressing projection neurons in dyskinetic mice. Biol Psychiatry. 2014;75(9):711–722. doi:10.1016/j.biopsych.2013.05.006
39. Solís O, Garcia-Montes JR, González-Granillo A, Xu M, Moratalla R. Dopamine D3 receptor modulates l-DOPA-induced dyskinesia by targeting D1 receptor-mediated striatal signaling. Cereb Cortex. 2017;27(1):435–446. doi:10.1093/cercor/bhv231
40. Payer DE, Guttman M, Kish SJ, et al. D3 dopamine receptor-preferring [11C]PHNO PET imaging in Parkinson patients with dyskinesia. Neurology. 2016;86(3):224–230. doi:10.1212/WNL.0000000000002285
41. Lanza K, Centner A, Coyle M, Del Priore I, Manfredsson FP, Bishop C. Genetic suppression of the dopamine D3 receptor in striatal D1 cells reduces the development of L-DOPA-induced dyskinesia. Exp Neurol. 2021;336:113534. doi:10.1016/j.expneurol.2020.113534
42. Picconi B, Hernández LF, Obeso JA, Calabresi P. Motor complications in Parkinson’s disease: striatal molecular and electrophysiological mechanisms of dyskinesias. Mov Disord. 2018;33(6):867–876. doi:10.1002/mds.27261
43. Picconi B, De Leonibus E, Calabresi P. Synaptic plasticity and levodopa-induced dyskinesia: electrophysiological and structural abnormalities. J Neural Transm (Vienna). 2018;125(8):1263–1271. doi:10.1007/s00702-018-1864-6
44. Picconi B, Centonze D, Håkansson K, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6(5):501–506. doi:10.1038/nn1040
45. Prescott IA, Liu LD, Dostrovsky JO, Hodaie M, Lozano AM, Hutchison WD. Lack of depotentiation at basal ganglia output neurons in PD patients with levodopa-induced dyskinesia. Neurobiol Dis. 2014;71:24–33. doi:10.1016/j.nbd.2014.08.002
46. Calabresi P, Pisani A, Rothwell J, Ghiglieri V, Obeso JA, Picconi B. Hyperkinetic disorders and loss of synaptic downscaling. Nat Neurosci. 2016;19(7):868–875. doi:10.1038/nn.4306
47. Huang Y-Z, Rothwell JC, Lu C-S, Chuang W-L, Chen R-S. Abnormal bidirectional plasticity-like effects in Parkinson’s disease. Brain. 2011;134(8):2312–2320. doi:10.1093/brain/awr158
48. Chase TN, Oh JD. Striatal dopamine- and glutamate-mediated dysregulation in experimental parkinsonism. Trends Neurosci. 2000;23(10):S86–S91. doi:10.1016/S1471-1931(00)00018-5
49. Hallett PJ, Dunah AW, Ravenscroft P, et al. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology. 2005;48(4):503–516. doi:10.1016/j.neuropharm.2004.11.008
50. Sgambato-Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol. 2012;96(1):69–86.
51. Sebastianutto I, Cenci MA. mGlu receptors in the treatment of Parkinson’s disease and L-DOPA-induced dyskinesia. Curr Opin Pharmacol. 2018;38:81–89. doi:10.1016/j.coph.2018.03.003
52. Guerra A, Suppa A, D’Onofrio V, et al. Abnormal cortical facilitation and L-dopa-induced dyskinesia in Parkinson’s disease. Brain Stimul. 2019;12(6):1517–1525. doi:10.1016/j.brs.2019.06.012
53. Calon F, Rajput AH, Hornykiewicz O, Bédard PJ, Di Paolo T. Levodopa-induced motor complications are associated with alterations of glutamate receptors in Parkinson’s disease. Neurobiol Dis. 2003;14(3):404–416. doi:10.1016/j.nbd.2003.07.003
54. Ahmed I, Bose SK, Pavese N, et al. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain. 2011;134(4):979–986. doi:10.1093/brain/awr028
55. Gardoni F, Picconi B, Ghiglieri V, et al. A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. J Neurosci. 2006;26(11):2914–2922. doi:10.1523/JNEUROSCI.5326-05.2006
56. Mellone M, Stanic J, Hernandez LF, et al. NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: from experimental models to patients. Front Cell Neurosci. 2015;9:245. doi:10.3389/fncel.2015.00245
57. Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur J Neurosci. 2002;15(1):120–132. doi:10.1046/j.0953-816x.2001.01843.x
58. Bibbiani F, Oh JD, Kielaite A, Collins MA, Smith C, Chase TN. Combined blockade of AMPA and NMDA glutamate receptors reduces levodopa-induced motor complications in animal models of PD. Exp Neurol. 2005;196(2):422–429. doi:10.1016/j.expneurol.2005.08.017
59. Dekundy A, Lundblad M, Danysz W, Cenci MA. Modulation of L-DOPA-induced abnormal involuntary movements by clinically tested compounds: further validation of the rat dyskinesia model. Behav Brain Res. 2007;179(1):76–89. doi:10.1016/j.bbr.2007.01.013
60. Morin N, Di Paolo T. Pharmacological treatments inhibiting levodopa-induced dyskinesias in MPTP-lesioned monkeys: brain glutamate biochemical correlates. Front Neurol. 2014;5:144. doi:10.3389/fneur.2014.00144
61. Rondard P, Pin J-P. Dynamics and modulation of metabotropic glutamate receptors. Curr Opin Pharmacol. 2015;20:95–101. doi:10.1016/j.coph.2014.12.001
62. Samadi P, Grégoire L, Morissette M, et al. mGluR5 metabotropic glutamate receptors and dyskinesias in MPTP monkeys. Neurobiol Aging. 2008;29(7):1040–1051. doi:10.1016/j.neurobiolaging.2007.02.005
63. Sanchez-Pernaute R, Wang J-Q, Kuruppu D, et al. Enhanced binding of metabotropic glutamate receptor type 5 (mGluR5) PET tracers in the brain of parkinsonian primates. Neuroimage. 2008;42(1):248–251. doi:10.1016/j.neuroimage.2008.04.170
64. Ouattara B, Grégoire L, Morissette M, et al. Metabotropic glutamate receptor type 5 in levodopa-induced motor complications. Neurobiol Aging. 2011;32(7):1286–1295. doi:10.1016/j.neurobiolaging.2009.07.014
65. Rascol O, Fox S, Gasparini F, Kenney C, Di Paolo T, Gomez-Mancilla B. Use of metabotropic glutamate 5-receptor antagonists for treatment of levodopa-induced dyskinesias. Parkinsonism Relat Disord. 2014;20(9):947–956. doi:10.1016/j.parkreldis.2014.05.003
66. Conn PJ, Battaglia G, Marino MJ, Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci. 2005;6(10):787–798. doi:10.1038/nrn1763
67. Greco B, Lopez S, van der Putten H, Flor PJ, Amalric M. Metabotropic glutamate 7 receptor subtype modulates motor symptoms in rodent models of Parkinson’s disease. J Pharmacol Exp Ther. 2010;332(3):1064–1071. doi:10.1124/jpet.109.162115
68. Broadstock M, Austin PJ, Betts MJ, Duty S. Antiparkinsonian potential of targeting group III metabotropic glutamate receptor subtypes in the rodent substantia nigra pars reticulata. Br J Pharmacol. 2012;165(4b):1034–1045. doi:10.1111/j.1476-5381.2011.01515.x
69. Konieczny J, Lenda T. Contribution of the mGluR7 receptor to antiparkinsonian-like effects in rats: a behavioral study with the selective agonist AMN082. Pharmacol Rep. 2013;65(5):1194–1203. doi:10.1016/S1734-1140(13)71477-4
70. Gardoni F, Morari M, Kulisevsky J, et al. Safinamide modulates striatal glutamatergic signaling in a rat model of levodopa-induced dyskinesia. J Pharmacol Exp Ther. 2018;367(3):442–451. doi:10.1124/jpet.118.251645
71. Bédard C, Wallman M-J, Pourcher E, Gould PV, Parent A, Parent M. Serotonin and dopamine striatal innervation in Parkinson’s disease and Huntington’s chorea. Parkinsonism Relat Disord. 2011;17(8):593–598. doi:10.1016/j.parkreldis.2011.05.012
72. Gagnon D, Gregoire L, Di Paolo T, Parent M. Serotonin hyperinnervation of the striatum with high synaptic incidence in parkinsonian monkeys. Brain Struct Funct. 2016;221(7):3675–3691. doi:10.1007/s00429-015-1125-5
73. Rylander D, Parent M, O’Sullivan SS, et al. Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann Neurol. 2010;68(5):619–628. doi:10.1002/ana.22097
74. Smith R, Wu K, Hart T, et al. The role of pallidal serotonergic function in Parkinson’s disease dyskinesias: a positron emission tomography study. Neurobiol Aging. 2015;36(4):1736–1742. doi:10.1016/j.neurobiolaging.2014.12.037
75. Lee J-Y, Seo S, Lee JS, Kim H-J, Kim YK, Jeon BS. Putaminal serotonergic innervation: monitoring dyskinesia risk in Parkinson disease. Neurology. 2015;85(10):853–860. doi:10.1212/WNL.0000000000001909
76. Roussakis -A-A, Politis M, Towey D, Piccini P. Serotonin-to-dopamine transporter ratios in Parkinson disease: relevance for dyskinesias. Neurology. 2016;86(12):1152–1158. doi:10.1212/WNL.0000000000002494
77. Dupre KB, Eskow KL, Barnum CJ, Bishop C. Striatal 5-HT1A receptor stimulation reduces D1 receptor-induced dyskinesia and improves movement in the hemiparkinsonian rat. Neuropharmacology. 2008;55(8):1321–1328. doi:10.1016/j.neuropharm.2008.08.031
78. Bishop C, Krolewski DM, Eskow KL, et al. Contribution of the striatum to the effects of 5-HT1A receptor stimulation in L-DOPA-treated hemiparkinsonian rats. J Neurosci Res. 2009;87(7):1645–1658. doi:10.1002/jnr.21978
79. Bezard E, Tronci E, Pioli EY, et al. Study of the antidyskinetic effect of eltoprazine in animal models of levodopa-induced dyskinesia. Mov Disord. 2013;28(8):1088–1096. doi:10.1002/mds.25366
80. Iderberg H, McCreary AC, Varney MA, Cenci MA, Newman-Tancredi A. Activity of serotonin 5-HT(1A) receptor “biased agonists” in rat models of Parkinson’s disease and L-DOPA-induced dyskinesia. Neuropharmacology. 2015;93:52–67. doi:10.1016/j.neuropharm.2015.01.012
81. Paolone G, Brugnoli A, Arcuri L, Mercatelli D, Morari M. Eltoprazine prevents levodopa-induced dyskinesias by reducing striatal glutamate and direct pathway activity. Mov Disord. 2015;30(13):1728–1738. doi:10.1002/mds.26326
82. Ghiglieri V, Mineo D, Vannelli A, et al. Modulation of serotonergic transmission by eltoprazine in L-DOPA-induced dyskinesia: behavioral, molecular, and synaptic mechanisms. Neurobiol Dis. 2016;86:140–153. doi:10.1016/j.nbd.2015.11.022
83. Meadows SM, Conti MM, Gross L, et al. Diverse serotonin actions of vilazodone reduce l-3,4-dihidroxyphenylalanine-induced dyskinesia in hemi-parkinsonian rats. Mov Disord. 2018;33(11):1740–1749. doi:10.1002/mds.100
84. Lanza K, Bishop C. Serotonergic targets for the treatment of L-DOPA-induced dyskinesia. J Neural Transm (Vienna). 2018;125(8):1203–1216. doi:10.1007/s00702-017-1837-1
85. Mazzucchi S, Frosini D, Ripoli A, et al. Serotonergic antidepressant drugs and L-dopa-induced dyskinesias in Parkinson’s disease. Acta Neurol Scand. 2015;131(3):191–195. doi:10.1111/ane.12314
86. de la Fuente-fernández R, Sossi V, Huang Z, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127(12):2747–2754. doi:10.1093/brain/awh290
87. Politis M, Wu K, Loane C, et al. Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J Clin Invest. 2014;124(3):1340–1349. doi:10.1172/JCI71640
88. Perez XA, Bordia T, Quik M. The striatal cholinergic system in L-dopa-induced dyskinesias. J Neural Transm (Vienna). 2018;125(8):1251–1262. doi:10.1007/s00702-018-1845-9
89. Kosillo P, Zhang Y-F, Threlfell S, Cragg SJ. Cortical control of striatal dopamine transmission via striatal cholinergic interneurons. Cereb Cortex. 2016;26(11):4160–4169. doi:10.1093/cercor/bhw252
90. Quik M, Cox H, Parameswaran N, O’Leary K, Langston JW, Di Monte D. Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys. Ann Neurol. 2007;62(6):588–596. doi:10.1002/ana.21203
91. Quik M, Mallela A, Ly J, Zhang D. Nicotine reduces established levodopa-induced dyskinesias in a monkey model of Parkinson’s disease. Mov Disord. 2013;28(10):1398–1406. doi:10.1002/mds.25594
92. Bordia T, Campos C, Huang L, Quik M. Continuous and intermittent nicotine treatment reduces L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesias in a rat model of Parkinson’s disease. J Pharmacol Exp Ther. 2008;327(1):239–247. doi:10.1124/jpet.108.140897
93. Bordia T, Campos C, McIntosh JM, Quik M. Nicotinic receptor-mediated reduction in L-DOPA-induced dyskinesias may occur via desensitization. J Pharmacol Exp Ther. 2010;333(3):929–938. doi:10.1124/jpet.109.162396
94. Zhang D, Mallela A, Sohn D, et al. Nicotinic receptor agonists reduce L-DOPA-induced dyskinesias in a monkey model of Parkinson’s disease. J Pharmacol Exp Ther. 2013;347(1):225–234. doi:10.1124/jpet.113.207639
95. Zhang D, Bordia T, McGregor M, McIntosh JM, Decker MW, Quik M. ABT-089 and ABT-894 reduce levodopa-induced dyskinesias in a monkey model of Parkinson’s disease. Mov Disord. 2014;29(4):508–517. doi:10.1002/mds.25817
96. Zhang W, Ordway GA. The alpha2C-adrenoceptor modulates GABA release in mouse striatum. Brain Res Mol Brain Res. 2003;112(1–2):24–32. doi:10.1016/S0169-328X(03)00026-3
97. Henry B, Fox SH, Peggs D, Crossman AR, Brotchie JM. The alpha2-adrenergic receptor antagonist idazoxan reduces dyskinesia and enhances anti-parkinsonian actions of L-dopa in the MPTP-lesioned primate model of Parkinson’s disease. Mov Disord. 1999;14(5):744–753. doi:10.1002/1531-8257(199909)14:5<744::AID-MDS1006>3.0.CO;2-7
98. Barnum CJ, Bhide N, Lindenbach D, et al. Effects of noradrenergic denervation on L-DOPA-induced dyskinesia and its treatment by α- and β-adrenergic receptor antagonists in hemiparkinsonian rats. Pharmacol Biochem Behav. 2012;100(3):607–615. doi:10.1016/j.pbb.2011.09.009
99. Pan J, Cai H. Opioid system in L-DOPA-induced dyskinesia. Transl Neurodegener. 2017;6:1. doi:10.1186/s40035-017-0071-y
100. Hanrieder J, Ljungdahl A, Fälth M, Mammo SE, Bergquist J, Andersson M. L-DOPA-induced dyskinesia is associated with regional increase of striatal dynorphin peptides as elucidated by imaging mass spectrometry. Mol Cell Proteomics. 2011;10(10):
101. Henry B, Fox SH, Crossman AR, Brotchie JM. Mu- and delta-opioid receptor antagonists reduce levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Exp Neurol. 2001;171(1):139–146. doi:10.1006/exnr.2001.7727
102. Koprich JB, Fox SH, Johnston TH, et al. The selective mu-opioid receptor antagonist ADL5510 reduces levodopa-induced dyskinesia without affecting antiparkinsonian action in MPTP-lesioned macaque model of Parkinson’s disease. Mov Disord. 2011;26(7):1225–1233. doi:10.1002/mds.23631
103. Steece-Collier K, Stancati JA, Collier NJ, et al. Genetic silencing of striatal CaV1.3 prevents and ameliorates levodopa dyskinesia. Mov Disord. 2019;34(5):697–707. doi:10.1002/mds.27695
104. da Silva-júnior FP, Braga-Neto P, Sueli Monte F, de Bruin VMS. Amantadine reduces the duration of levodopa-induced dyskinesia: a randomized, double-blind, placebo-controlled study. Parkinsonism Relat Disord. 2005;11(7):449–452. doi:10.1016/j.parkreldis.2005.05.008
105. Pahwa R, Tanner CM, Hauser RA, et al. ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson disease (EASE LID Study): a randomized clinical trial. JAMA Neurol. 2017;74(8):941–949. doi:10.1001/jamaneurol.2017.0943
106. Oertel W, Eggert K, Pahwa R, et al. Randomized, placebo-controlled trial of ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 3). Mov Disord. 2017;32(12):1701–1709. doi:10.1002/mds.27131
107. Tanner CM, Pahwa R, Hauser RA, et al. EASE LID 2: a 2-year open-label trial of gocovri (amantadine) extended release for dyskinesia in Parkinson’s disease. J Parkinsons Dis. 2020;10(2):543–558. doi:10.3233/JPD-191841
108. Bezard E, Pioli EY, Li Q, et al. The mGluR5 negative allosteric modulator dipraglurant reduces dyskinesia in the MPTP macaque model. Mov Disorders. 2014;29(8):1074–1079. doi:10.1002/mds.25920
109. Tison F, Keywood C, Wakefield M, et al. A phase 2A trial of the novel mGluR5-negative allosteric modulator dipraglurant for levodopa-induced dyskinesia in Parkinson’s disease. Mov Disord. 2016;31(9):1373–1380. doi:10.1002/mds.26659
110. Guy W. AIMS: ECDEU Assessment Manual for Psychopharmacology. Washington, DC: Government Printing Office; 1976.
111. Goetz CG, Nutt JG, Stebbins GT. The unified dyskinesia rating scale: presentation and clinimetric profile. Mov Disord. 2008;23(16):2398–2403. doi:10.1002/mds.22341
112. Goetz CG, Stebbins GT, Chung KA, et al. Which dyskinesia scale best detects treatment response? Mov Disord. 2013;28(3):341–346. doi:10.1002/mds.25321
113. Charvin D, Di Paolo T, Bezard E, et al. An mGlu4-positive allosteric modulator alleviates parkinsonism in primates. Mov Disord. 2018;33(10):1619–1631. doi:10.1002/mds.27462
114. Kemp JA, Foster AC, Leeson PD, et al. 7-chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-D-aspartate receptor complex. Proc Natl Acad Sci U S A. 1988;85(17):6547–6550. doi:10.1073/pnas.85.17.6547
115. Wallace M, White A, Grako KA, Lane R, Cato AJ, Snodgrass HR. Randomized, double-blind, placebo-controlled, dose-escalation study: investigation of the safety, pharmacokinetics, and antihyperalgesic activity of L-4-chlorokynurenine in healthy volunteers. Scand J Pain. 2017;17(1):243–251. doi:10.1016/j.sjpain.2017.05.004
116. Brotchie JM, Johnston SH, Fox SH, Zerr P, Tiberghien F. FP0011 extends the duration of the anti-parkinsonian actions of L-DOPA and reduces L-DOPA-induced dyskinesia in the MPTP-lesiones macaque model of Parkinson’s disease. Mov Disord. 2007;22:S96.
117. Corvol J-C, Durif F, Meissner WG, et al. Naftazone in advanced Parkinson’s disease: an acute L-DOPA challenge randomized controlled trial. Parkinsonism Relat Disord. 2019;60:51–56. doi:10.1016/j.parkreldis.2018.10.005
118. Wigal SB, Duong S. Pharmacokinetic evaluation of eltoprazine. Expert Opin Drug Metab Toxicol. 2011;7(6):775–781. doi:10.1517/17425255.2011.580275
119. Svenningsson P, Rosenblad C, Af Edholm Arvidsson K, et al. Eltoprazine counteracts l-DOPA-induced dyskinesias in Parkinson’s disease: a dose-finding study. Brain. 2015;138(4):963–973. doi:10.1093/brain/awu409
120. Bonifati V, Fabrizio E, Cipriani R, Vanacore N, Meco G. Buspirone in levodopa-induced dyskinesias. Clin Neuropharmacol. 1994;17(1):73–82. doi:10.1097/00002826-199402000-00008
121. Politis M, Oertel WH, Wu K, et al. Graft-induced dyskinesias in Parkinson’s disease: high striatal serotonin/dopamine transporter ratio. Mov Disord. 2011;26(11):1997–2003. doi:10.1002/mds.23743
122. Tronci E, Lisci C, Stancampiano R, et al. 5-Hydroxy-Tryptophan for the treatment of L-DOPA-induced dyskinesia in the rat Parkinson’s disease model. Neurobiol Dis. 2013;60:108–114. doi:10.1016/j.nbd.2013.08.014
123. Meloni M, Puligheddu M, Sanna F, et al. Efficacy and safety of 5-Hydroxytryptophan on levodopa-induced motor complications in Parkinson’s disease: a preliminary finding. J Neurol Sci. 2020;415:415. doi:10.1016/j.jns.2020.116869
124. Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff P, Schwartz JC. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Natl Acad Sci U S A. 1997;94(7):3363–3367. doi:10.1073/pnas.94.7.3363
125. Waters S, Sonesson C, Svensson P, et al. Preclinical pharmacology of [2-(3-fluoro-5-methanesulfonyl-phenoxy)ethyl](propyl)amine (IRL790), a novel dopamine transmission modulator for the treatment of motor and psychiatric complications in Parkinson disease. J Pharmacol Exp Ther. 2020;374(1):113–125. doi:10.1124/jpet.119.264226
126. Svenningsson P, Johansson A, Nyholm D, et al. Safety and tolerability of IRL790 in Parkinson’s disease with levodopa-induced dyskinesia—a phase 1b trial. NPJ Parkinsons Dis. 2018;4(1):35. doi:10.1038/s41531-018-0071-3
127. Francardo V, Geva M, Bez F, et al. Pridopidine induces functional neurorestoration via the sigma-1 receptor in a mouse model of Parkinson’s disease. Neurotherapeutics. 2019;16(2):465–479. doi:10.1007/s13311-018-00699-9
128. Johnston TH, Geva M, Steiner L, et al. Pridopidine, a clinic-ready compound, reduces 3,4-dihydroxyphenylalanine-induced dyskinesia in Parkinsonian macaques. Mov Disord. 2019;34(5):708–716. doi:10.1002/mds.27565
129. de Yebenes JG, Landwehrmeyer B, Squitieri F, et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011;10(12):1049–1057. doi:10.1016/S1474-4422(11)70233-2
130. Reilmann R, McGarry A, Grachev ID, et al. Safety and efficacy of pridopidine in patients with Huntington’s disease (PRIDE-HD): a phase 2, randomised, placebo-controlled, multicentre, dose-ranging study. Lancet Neurol. 2019;18(2):165–176. doi:10.1016/S1474-4422(18)30391-0
131. Murata M, Hasegawa K, Kanazawa I, et al. Zonisamide improves wearing-off in Parkinson’s disease: a randomized, double-blind study. Mov Disord. 2015;30(10):1343–1350. doi:10.1002/mds.26286
132. Oki M, Kaneko S, Morise S, et al. Zonisamide ameliorates levodopa-induced dyskinesia and reduces expression of striatal genes in Parkinson model rats. Neurosci Res. 2017;122:45–50. doi:10.1016/j.neures.2017.04.003
133. Sano H, Nambu A. The effects of zonisamide on L-DOPA–induced dyskinesia in Parkinson’s disease model mice. Neurochem Int. 2019;124:171–180. doi:10.1016/j.neuint.2019.01.011
134. Yebenes JGD, Fahn S, Lovelle S, et al. Continuous intracerebroventricular infusion of dopamine and dopamine agonists through a totally implanted drug delivery system in animal models of Parkinson’s disease. Mov Disorders. 1987;2(3):143–158. doi:10.1002/mds.870020302
135. Venna N, Sabin TD, Ordia JI, Vernon H, Mark MD. Treatment of severe Parkinson’s disease by intraventricular injection of dopamine. SFN. 1984;47(1–2):62–64.
136. Horne MK, Butler EG, Gilligan BS, Wodak J, Stark RJ, Brazenor GA. Intraventricular infusion of dopamine in Parkinson’s disease. Ann Neurol. 1989;26(6):792–794. doi:10.1002/ana.410260620
137. Laloux C, Gouel F, Lachaud C, et al. Continuous cerebroventricular administration of dopamine: a new treatment for severe dyskinesia in Parkinson’s disease? Neurobiol Dis. 2017;103:24–31. doi:10.1016/j.nbd.2017.03.013
138. Devos D, Devedjian J-C, Moreau C. Intracerebroventricular dopamine for Parkinson’s disease. Oncotarget. 2017;8:45034–45035. doi:10.18632/oncotarget.17596
139. Pietracupa S, Fasano A, Fabbrini G, et al. Poor self-awareness of levodopa-induced dyskinesias in Parkinson’s disease: clinical features and mechanisms. Parkinsonism Relat Disord. 2013;19(11):1004–1008. doi:10.1016/j.parkreldis.2013.07.002
140. Jenner P, McCreary AC, Scheller DKA. Continuous drug delivery in early- and late-stage Parkinson’s disease as a strategy for avoiding dyskinesia induction and expression. J Neural Transm (Vienna). 2011;118(12):1691–1702. doi:10.1007/s00702-011-0703-9
141. Fox SH, Brotchie JM. Viewpoint: developing drugs for levodopa-induced dyskinesia in PD: lessons learnt, what does the future hold? Eur J Neurosci. 2019;49(3):399–409. doi:10.1111/ejn.14173
142. Giladi N, Ghys L, Surmann E, Boroojerdi B, Jankovic J. Effects of long-term treatment with rotigotine transdermal system on dyskinesia in patients with early-stage Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(12):1345–1351. doi:10.1016/j.parkreldis.2014.09.016
143. Antonini A, Chaudhuri KR, Martinez-Martin P, Odin P. Oral and infusion levodopa-based strategies for managing motor complications in patients with Parkinson’s disease. CNS Drugs. 2010;24(2):119–129. doi:10.2165/11310940-000000000-00000
144. Antonini A, Fung VSC, Boyd JT, et al. Effect of levodopa-carbidopa intestinal gel on dyskinesia in advanced Parkinson’s disease patients. Mov Disord. 2016;31(4):530–537. doi:10.1002/mds.26528
145. Timpka J, Mundt-Petersen U, Odin P. Continuous dopaminergic stimulation therapy for Parkinson’s disease - recent advances. Curr Opin Neurol. 2016;29(4):474–479. doi:10.1097/WCO.0000000000000354
146. Dafsari HS, Martinez-Martin P, Rizos A, et al. EuroInf 2: subthalamic stimulation, apomorphine, and levodopa infusion in Parkinson’s disease. Mov Disord. 2019;34(3):353–365. doi:10.1002/mds.27626
147. Olanow CW, Kieburtz K, Odin P, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014;13(2):141–149. doi:10.1016/S1474-4422(13)70293-X
148. Lopiano L, Modugno N, Marano P, et al. Motor and non-motor outcomes in patients with advanced Parkinson’s disease treated with levodopa/carbidopa intestinal gel: final results of the GREENFIELD observational study. J Neurol. 2019;266(9):2164–2176. doi:10.1007/s00415-019-09337-6
149. Rosenblad C, Li Q, Pioli EY, et al. Vector-mediated l-3,4-dihydroxyphenylalanine delivery reverses motor impairments in a primate model of Parkinson’s disease. Brain. 2019;142(8):2402–2416. doi:10.1093/brain/awz176
150. Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351(24):2498–2508.
151. Müller T. Current and investigational non-dopaminergic agents for management of motor symptoms (including motor complications) in Parkinson’s disease. Expert Opin Pharmacother. 2017;18(14):1457–1465. doi:10.1080/14656566.2017.1373089
152. Limousin P, Foltynie T. Long-term outcomes of deep brain stimulation in Parkinson disease. Nat Rev Neurol. 2019;15(4):234–242. doi:10.1038/s41582-019-0145-9
153. Gonzalez-Latapi P, Bhowmick SS, Saranza G, Fox SH. Non-dopaminergic treatments for motor control in Parkinson’s disease: an update. CNS Drugs. 2020;34(10):1025–1044. doi:10.1007/s40263-020-00754-0
154. Hubsher G, Haider M, Okun MS. Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology. 2012;78(14):1096–1099. doi:10.1212/WNL.0b013e31824e8f0d
155. Jann MW, Grimsley SR, Gray EC, Chang WH. Pharmacokinetics and pharmacodynamics of clozapine. Clin Pharmacokinet. 1993;24(2):161–176. doi:10.2165/00003088-199324020-00005
156. Durif F, Debilly B, Galitzky M, et al. Clozapine improves dyskinesias in Parkinson disease: a double-blind, placebo-controlled study. Neurology. 2004;62(3):381–388. doi:10.1212/01.WNL.0000110317.52453.6C
157. Lindenbach D, Palumbo N, Ostock CY, Vilceus N, Conti MM, Bishop C. Side effect profile of 5-HT treatments for Parkinson’s disease and L-DOPA-induced dyskinesia in rats. Br J Pharmacol. 2015;172(1):119–130. doi:10.1111/bph.12894
158. Witek N, Stebbins GT, Goetz CG. What influences placebo and nocebo responses in Parkinson’s disease? Mov Disord. 2018;33(8):1204–1212. doi:10.1002/mds.27416
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.