Back to Journals » OncoTargets and Therapy » Volume 12
Pathophysiologic Mechanisms And Management Of Large Granular Lymphocytic Leukemia Associated Pure Red Cell Aplasia
Authors Qiu ZY, Qin R ![]() , Tian GY, Wang Y, Zhang YQ
, Tian GY, Wang Y, Zhang YQ
Received 8 July 2019
Accepted for publication 13 September 2019
Published 4 October 2019 Volume 2019:12 Pages 8229—8240
DOI https://doi.org/10.2147/OTT.S222378
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Zhi-Yuan Qiu,1 Rong Qin,1 Guang-Yu Tian,1 Yan Wang,1 Ye-Qing Zhang2
1Department of Oncology, The Affiliated People’s Hospital of Jiangsu University, Zhenjiang 212002, Jiangsu, People’s Republic of China; 2Department of Vascular Surgery, The Second Affiliated Hospital of Soochow University, Suzhou 215004, Jiangsu, People’s Republic of China
Correspondence: Yan Wang
Department of Oncology, The Affiliated People’s Hospital of Jiangsu University, 8 Dianli Road, Zhenjiang 212002, Jiangsu, People’s Republic of China
Tel/Fax +86-511-88915061
Email [email protected]
Ye-Qing Zhang
Department of Vascular Surgery, The Second Affiliated Hospital of Soochow University, Suzhou 215004, Jiangsu, People’s Republic of China
Email [email protected]
Abstract: Large granular lymphocytic leukemia (LGLL) is a chronic clonal lymphoproliferative disease of mature T or NK cells, and produces a variety of hematological abnormalities. Pure red cell aplasia (PRCA) is a rare haematological disease and is one of the most common complications of LGLL. LGLL-associated PRCA may represent a relatively indolent type and may be more common than reported, but its natural history and clinical course have not been well described. The ethnic origin of the patients is an important consideration in determining the relationship between PRCA and LGLL. Guidelines and progresses for management of LGLL-associated PRCA rely on accumulation of empirical experiences, integrative analyses of several cases and clinical trials. The purpose of this review is to evaluate occurrence, possible mechanisms, diagnosis, clinical features, treatments and outcomes of LGLL-associated PRCA.
Keywords: large granular lymphocytic leukemia, lymphocytosis, PRCA
Introduction
Large granular lymphocytic leukemia (LGLL) is a chronic clonal lymphoproliferative disease of mature T cells or NK cells, with most patients being asymptomatic and requiring no treatment during the course of the disease.1 These conditions are thought to arise from chronic antigenic stimulation, while the long-term survival of the abnormal LGLs appears to be sustained by impaired survival signaling. LGLL produces a wide variety of hematological and immunological abnormalities1,2 and pure red cell aplasia (PRCA) is one of the most common complications of LGLL in Asian patients.3,4
PRCA is a rare haematological disease, characterized by normocytic anemia, reticulocytopenia, and severe erythroid hypoplasia of bone marrow (BM).5 PRCA includes an idiopathic form or a secondary form. Acquired secondary PRCA may be associated with thymoma, lymphoproliferative disorders, viral infections, solid tumors and a variety of other diseases.6,7 There is a close association between LGLL and PRCA, and LGLL-associated PRCA accounts for a significant portion of secondary PRCA. Because LGLL is frequently underdiagnosed, it is likely that a significant proportion of idiopathic PRCA is secondary to LGLL and LGLL-associated PRCA may be more common than reported. T-LGLL is regarded to surpass all other pathologies as the most common cause of PRCA.8 However, its natural history and clinical course have not been well described because of its rarity. Guidelines and progresses for management of LGLL-associated PRCA rely on accumulation of empirical experiences, integrative analyses of several cases and clinical trials. The purpose of this review is to evaluate occurrence, possible mechanisms, diagnosis, clinical features, treatments and outcomes of LGLL-associated PRCA.
Occurrence
The incidence of LGLL-associated PRCA is 7.3–68.2%, as shown in Table 1. The age of the patients with LGLL-associated PRCA at diagnosis ranges from 18 to 89 years old and the LGL count ranges from 0.3 to 11.9×109/L (Table 1). Oriental LGLL patients tend to present different features from occidental patients. In Asians, PRCA is the most common hematological complication of LGLL, while in western patients, rheumatoid arthritis (RA) and neutropenia are more frequent.6,9–13 These differences might be partly due to ethnicity. So it is intriguing to explore the contributing factors, such as the genetic background.
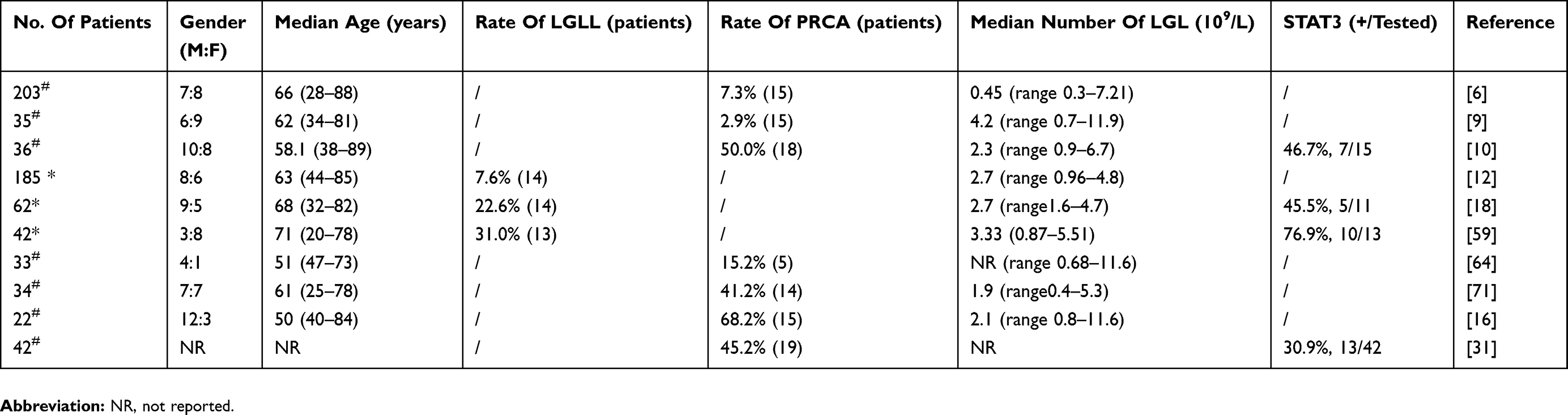 |
Table 1 Summary Of Articles About LGLL-Associated PRCA |
The close association between T-LGLL and PRCA has been well described in both Japanese and Chinese patients. In 2 reports from Japan, 15 patients (42.9%) in one report met the criteria of PRCA among 35 T-LGLL patients and the incidence in the other was 11.0% (14/127).9,14 In China, we reported PRCA was present in 18 (50%) of 36 T-LGLL patients and Zhao X et al reported 19 (67.9%) of 28 T-LGLL patients.10,15 Kwong YL et al also reported PRCA was more frequent in Asians than western LGLL patients (52/110, 47% versus 6/143, 4%).16
In contrast, PRCA was less frequently observed in western LGLL patients. In the reports from western countries, LGLL-associated PRCA appeared in 15 (7%) of 203 patients and 6 patients (7.6%) were found to have PRCA in 201 T-LGLL patients.6,13 In another report, Dhodapkar et al17 identified 5 PRCA patients (7.4%) in 68 T-LGLL patients. In the latest study, Balasubramanian SK et al reported 14 PRCA patients (22%) in 62 LGLL patients.18
The LGLL seems to be triggered and sustained by antigenic drive, and hematopoietic progenitors may be major targets of this process.19 Because antigen presentation depends not only on the peptide processing but also on HLA backgrounds, polarity of HLA distribution could affect the utilization of target molecules by LGLL. Therefore, the difference of HLA distribution between Asian and western populations may partially explain the distinction of the incidence and it would be important to know the ethnic origin of the patients. In oriental patients, an underlying LGLL should be excluded in the diagnostic evaluation of PRCA and different clinicopathologic and epidemiologic factors may be involved.
Possible Mechanisms
Are LGLs causing PRCA, or is their presence more likely to be an epiphenomenon as a consequence of chronic antigen stimulation? The pathophysiological mechanism of LGLL-associated PRCA has not been evaluated and proper distinction of pathogenesis is essential for selecting effective therapeutic modality. Multiple mechanisms have been postulated, including inhibition of erythropoiesis by LGLs, chronic antigenic stimulation due to viral infections, autoimmune diseases, humoral and cytotoxic mechanisms.20,21
Viral Infections
LGLL-associated PRCA may be associated with long-term antigenic stimulation due to viral infections, especially parvovirus B19, which may result in LGL lymphocytosis and may play an etiologic role in some PRCA patients. Parvovirus B19 infection has been reported to be associated with PRCA and there are several reports of T-LGLL in response to parvovirus B19 infection.21–24
Ergas D et al reported parvovirus B19 infection in 3 LGLL-associated PRCA patients, which showed a cross-link between parvovirus B19 infection and the appearance of PRCA.21 These patients had had years of LGLL with a stable hemoglobin level, but PRCA developed after the occurrence of parvovirus B19 infection. Some patients with parvovirus B19-induced PRCA have been reported to have had LGLL.24 LGL expansion could have been initiated after parvovirus B19 infection. There have been other reports of LGLL-associated PRCA that developed after parvovirus B19 infection. In some LGLL-associated PRCA patients, parvovirus B19 was found to be positive after diagnosis.18 Kondo H et al reported a T-LGLL patient developed parvovirus B19-associated PRCA.25 These observations suggest that LGLL patients are at an increasing risk to develop parvovirus B19 infection and underscore the need to screen routinely for this infection. LGLs in the peripheral blood should be carefully examined in patients with parvovirus B19-induced PRCA, and LGLL patients who exhibit anemia should be evaluated for parvovirus B19 infection.
A direct toxic effect of parvovirus B19 on pronormoblasts may result in PRCA. Parvovirus B19 directly infects erythroid progenitors through the red cell surface P antigen. Individuals whose erythroid progenitors do not express P antigen are resistant to parvovirus B19 infection.26–28 Parvovirus B19 has been identified in proliferating CFU-E and serum that contains parvovirus B19 inhibits CFU-E growth in vitro, and this effect can be inhibited by antibody to this virus.29,30 To find out whether parvovirus B19 is related with LGLL, additional studies are warranted to address the role of parvovirus B19 in the pathogenesis of LGLL.
LGLs-Mediated Inhibition Of Erythropoiesis
PRCA may be mediated by LGLs against erythroid progenitors.20,23,31–39 The mechanism of inhibition may operate at different levels of erythroid differentiation and even be genetically restricted.34,40 This is likely via LGL-mediated killing of erythroid colony units (CFU-E) and burst forming units (BFU-E), thereby inhibiting erythroid precursor progression to mature erythrocytes.20,31,34,35
The T-LGLs can mediate direct cellular cytotoxicity and inhibit enythroid cell differentiation. It has been demonstrated that LGLs inhibited erythroid hematopoiesis with co-culture techniques in vitro. Hara T et al demonstrated that the LGLs from a LGLL-associated PRCA patient inhibited BFU-E in culture.33 Abkowitz et al demonstrated T-LGLs from PRCA patients mediated inhibition of erythropoiesis in marrow culture systems.34 In another report, T-LGLs inhibited the growth of erythroid colonies in vitro and CFU-E growth increased markedly after removal of the T-LGLs.37 Nagasawa T et al investigated the interaction between CFU-E and the LGLs in vitro.36 The CFU-E was strongly suppressed when the LGLs were cocultured with normal allogeneic BM cells and the LGLs frozen before treatment also suppressed CFU-E of autologous BM cells after the recovery from PRCA. These demonstrated directly that the LGLs were capable of suppressing erythropoiesis. In contrast, erythroid progenitors were significantly increased when LGLs were removed. In vitro, growth of the patient’s erythroid progenitors has been accomplished either after removal of LGLs or after induction of remission.41 These results indicated that the interaction between the T-LGLs and hematopoietic stem cell may be important for the mechanism of erythroid suppression. That T-LGLs are able to suppress erythroid hematopoiesis is consistent with the observations that T-LGLs may be causative in aplastic anemia.42,43 Furthermore, these results obtained from in vitro coculture studies were consistent with the clinical findings.
NK cells might be involved in the physiological regulation of haematopoiesis. A possible pathogenetic impact of hyperactive NK cells should be considered in acquired PRCA. As with T cells, NK cells may have a negative regulation on erythropoiesis and can suppress CFU-E in vitro.38,44 Normal human thymus and BM contain immature hematopoietic cells which are sensitive to NK-cell-mediated cytotoxicity in vitro.45–47 Partanen S et al reported NK cells from PRCA patients inhibited BFU-E and CFU-E in vitro and showed a very high NK activity against the human erythroblastic leukemia cell line K562.38
Human Leukocyte Antigen (HLA) Class I Expression
The reduced expression of HLA class I genes in the erythropoietic lineage accounts for the selective cytotoxicity of LGLs against erythroid progenitors. KIRs (Killer-cell immunoglobulin-like receptors) on the LGLs bind to HLA class I molecules and then prevent LGLs to cause cytolysis.47 HLA class I molecules are expressed by erythroid precursors, whereas mature erythroid precursors have reduced the expression of HLA class I molecules.48,49 LGLs expressing KIRs can destroy erythroid precursors because of the physiologic down-regulation of HLA class I molecules. The reduced expression of HLA class I molecules prevents the transmission of adequate inhibitory signals, which enhances major histocompatibility (MHC)-unrestricted cytotoxicity of LGLs and allows LGLs to cause cytolysis.47 Erythroid precursors become sensitive to MHC-unrestricted cytolysis as the expression of HLA class I molecules is reduced.
NK cells express KIRs and the repertoire of KIRs differs from person to person. Like NK cells, cytotoxic γδ T cells also express KIRs, suggesting that both NK cells and γδ T cells survey the body for missing self HLA class I molecules. Handgretinger R et al reported the γδT-LGLs inhibited erythroid precursors by KIRs in a patient with γδT-LGLL-associated PRCA.50 These γδT-LGLs expressed KIRs and killed erythroid progenitors in an MHC-unrestricted manner. Although most LGLs were of the T cell receptor (TCR) ɑβ-type, some ɑβT-LGLs express the KIRs.
Immune Mechanisms
Immune mechanisms contribute to the pathogenesis of LGLL-associated PRCA as indicated in favourable response to immunosuppressive treatment.5,36 LGLL-associated PRCA develops due to autoantibody-dependent immune mechanisms and has been shown to be associated with immunoglobulins.50 These LGLs have exhibited surface Fc receptors for lgG and suppressed immunoglobulin biosynthesis by B cells.36 IgG antibodies directed against erythroid precursors have been demonstrated in some cases.51,52 Antibodies against erythroid cells or erythropoietin can cause PRCA.53 LGLs activation is dependent on the antibody against erythroid precursors.47
Molecular Mechanism
Genetic changes might lead to the formation of LGLL. STAT3 mutations resulting in persistent proliferation of LGLs are a frequent event in LGLL and aberrant STAT3 signaling underlies the pathogenesis.54–56 LGLL-associated PRCA may also involve activation of the STAT3 pathway. However, studies evaluating this pathway in LGLL-associated PRCA are lacking.
STAT3 mutations were frequently present in PRCA patients, irrespective of the etiology. STAT3-mutated T-cell subclones were found in a subset of PRCA, suggesting contributions of STAT3-mutated T cells in this disorder.57 STAT3 mutations were also frequently present in LGLL-associated PRCA patients.31,58 Significant associations between STAT3 mutations and PRCA further imply their roles in PRCA. STAT3 mutations were found in 5 of 15 patients with idiopathic PRCA (33%), and 10 of 13 patients with T-LGLL-associated PRCA (77%).18 Among the PRCA patients, 3 of the 29 PRCA patients without T-LGLL and 6 of the 13 PRCA patients with T-LGLL were positive for STAT3 mutations.59 These results suggested that the STAT3 mutations were limited to PRCA patients with or without T-LGLL. LGLL-associated PRCA displayed a significant association with STAT3 mutation. PRCA with STAT3 mutation was recognized in 13 T-LGLL patients.31 In our research, 7 patients with LGLL-associated PRCA (25%) were found to have STAT3 mutations.10 Balasubramanian et al recently reported the results of targeted sequencing for PRCA patients with and without T-LGLL.18 They detected STAT3 mutations in 5 of 11 T-LGLL-associated PRCA patients but not in any of 17 patients with idiopathic PRCA. But Jerez A et al could not detect STAT3 mutations in patients with LGLL-associated PRCA (0/48) and PRCA were invariably related to STAT3 wild-type patients (6%, 8/122).55 Shi et al also reported that PRCA was seen exclusively in the STAT3 wild-type T-LGLL patients, and patients with STAT3 mutations were more prone to develop RA.60 The absence of STAT3 mutations may be due to the ethnic background of the patients. T-LGLL is known to be frequently accompanied by RA or neutropenia in western countries, whereas PRCA is frequently associated with T-LGLL in East Asian populations.10,12,16
However, it is still unclear whether STAT3-mutated T cells have an inhibitory effect on erythroid cell production or not. Because STAT3 mutations could develop in T cells that were chronically stimulated by inflammatory cytokines or various antigens, the presence of STAT3-mutated T cells may be an epiphenomenon rather than pathogenic.61,62 The mutational process in the STAT3 gene and the functional aspects of STAT3-mutated T cells thus require further investigation.
Cytokines
Cytokines released by LGLs, such as tumor necrosis factor (TNF) which is known as inhibitors of hematopoiesis, may be responsible for the PRCA in LGLL. The patients with untreated PRCA showed high levels of IL-6, IL-8, IL-10, TNFα and sIL-2R, while patients with PRCA treated with immunosuppressive agents showed low levels of TNFα and sIL-2R.63 Interferon-γ production by T-LGLs may have mediated the suppressive effects of erythropoiesis.7
Diagnosis
PRCA was recognized in conjunction with or shortly after LGLL diagnosis. But the diagnosis of LGLL-associated PRCA is still challenging. The currently diagnostic criteria do not distinguish a idiopathic PRCA or a secondary PRCA. It is important to separate the categories of PRCA, because immunosuppression will not only be ineffective in clonal myelopoietic disorders but also may even accelerate transformation.
Based on the published literature, there are several important considerations in making a diagnosis of LGLL-associated PRCA: (1) the lymphocyte count is usually 2-20×109/L and LGL counts > 2×109/L (when LGL counts < 2×109/L diagnosis is confirmed by the detection of clonality); (2) the predominance of LGLs in the peripheral blood smear samples; (3) the clonality of LGL lymphocytosis; (4) the phenotypes of LGLs are usually CD2+CD3+CD8+CD57+ T-cell or surfaceCD3-cCD3epsilon+CD16+ NK-cell; (5) PRCA was defined as isolated anemia (untransfused Hb < 8g/dL) with an absolute number of reticulocyte <2.0×104/L (< 1%) or BM erythroblasts <10% of all nucleated cells;64 (6) Other known causes of PRCA were excluded.
The BFU-E assay can distinguish among MDS, systemic autoimmunity, LGLL and PRCA, which may be an important adjunct to the work-up of the undiagnosed cytopenias.65 Low BFU-E growth (<10 BFU-E/105 marrow mononuclear cells) is more consistent with a diagnosis of MDS and helps to exclude the diagnosis of PRCA, LGLL, or cytopenias from autoimmune diseases. More robust BFU-E growth (≥20 BFU-E/105 marrow mononuclear cells) is usually associated with external suppression hematopoiesis such as occurs in LGLL, PRCA, or autoimmunity. BFU-E growth for patients with LGLL-associated PRCA was higher than that of patients with non-LGLL-associated PRCA. Growth above the normal median (40 BFU-E/105 marrow mononuclear cells) always excluded MDS and was also associated with response to IST.
It is important for hematopathologists and hematologists to be aware of this entity in order to guide treatment strategies. We propose a diagnostic algorithm for the diagnosis of LGLL-associated PRCA (Figure 1).
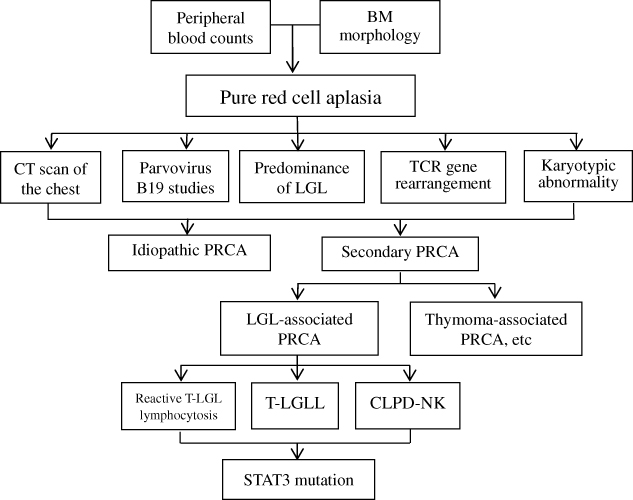 |
Figure 1 Diagnostic algorithm for the differential diagnosis of LGLL-associated PRCA. Abbreviations: BM, Bone marrow; PRCA, pure red cell aplasia; LGL, Large granular lymphocyte; TCR, T-cell receptor. |
Clinical Presentation
There is no specific clinical presentation of LGLL-associated PRCA and the signs and symptoms are those associated with anemia, such as fatigue, pallor, palpitations, peripheral neuropathy and shortness of breath on exertion.64,66 Because PRCA is a pure underproduction anemia, the gradual decline in hemoglobin concentration allows some degree of adaptation, and symptoms may be fewer than would be expected for the degree of anemia.
Comparing STAT3 mutation(+) LGLL-associated PRCA patients with STAT3 mutation(-) patients, the STAT3 mutation(+) patients had more lymphocytes, more CD8+ T cells59 and showed a trend towards a younger age (median age, 52.5 years old and 77.5 years old) than the STAT3 mutation(-) patients.31 LGLL-associated PRCA patients showed a higher frequency of T-cell clonality and STAT3 mutations than PRCA patients without T-LGLL.59
Some other diseases have been associated with LGLL-associated PRCA: thymoma,6,67 polyglandular autoimmune syndrome,36,68 chronic lymphocytic leukemia, non-Hodgkin lymphoma and amegakaryocytic thrombocytopenicpurpura,68,69 but it is notable that RA and neutropenia were infrequently associated.
Types Of LGL Lymphocytosis
PRCA-associated LGL lymphocytosis typically comprises monoclonal or oligoclonal expansion of cytotoxic T- or NK-cells. Reactive LGL lymphocytosis is usually polyclonal, transient, and asymptomatic. Neoplastic clonal LGL expansions are classified as T-LGLL and CLPD-NK.
Reactive LGL Lymphocytosis
Some PRCA patients have elevated levels of LGLs but do not fulfill the diagnostic criteria for LGLL.23 In these patients, the LGL lymphocytosis may be polyclonal and may rarely evolve into LGLL.22,23,70
T-LGLL
T-LGLL is most frequent in LGLL-associated PRCA, and has been reported in case report or case series.6,9,10,12,16,18,59,64,71 These LGL expansions have been defined as neoplastic processes because of their clonal nature. Most T-LGLL is of the αβ type, but some cases of T-LGLL were of the γδ type.72,73 γδT-LGLL is rare, and its association with PRCA is also rare. γδT-LGLL-associated PRCA was first described by Oshimi K et al.74 Tanaka et al reported a case of γδT-LGLL complicated with PRCA at presentation, and later with Epstein-Barr virus (EBV) infection.75 The disease was first diagnosed as PRCA, but γδT-LGLL developed later, and PRCA was retrospectively considered to be caused by γδT-LGLL. Handgretinger R et al also described a patient with γδT-LGLL-associated PRCA.50 In Japan, αβT-LGLL and γδT-LGLL were frequently associated with PRCA.3,76
Although several theories have been proposed, the mechanism by which PRCA develops in αβT-LGLL and γδT-LGLL is not well understood. γδT-LGLL is strongly suspected when LGLs show CD3+CD4-CD8- phenotype. However, the incidence of PRCA (29%) in CD4-/CD8- γδT-LGLL was much higher than that reported in CD8+ γδT-LGLL (6%),77,78 and the incidence of PRCA in γδT-LGLL (29%) was also higher than that reported in αβT-LGLL (7%).17 However, the higher incidences of PRCA need to be confirmed by accumulation of data from a larger number of cases of CD4-/CD8- γδT-LGLL. The higher incidences of PRCA observed in CD4-/CD8- γδT-LGLL may be related to the higher cytotoxic activity observed in the CD5- subset of γδT cells than that in the CD5+ subset.79
CLPD-NK
CLPD-NK is a rare type in secondary PRCA and has been reported in case report.3,38,39,50 Tefferi A et al reported a case of CLPD-NK-associated PRCA and the tumour cells expressed features consistent with NK cells, but EBV was absent from the tumour cells.39 In 1994, Tefferi A et al reported another case of CLPD-NK-associated PRCA.80 Kurt H et al also reported a patient with PRCA in 11 CLPD-NK patients.81
Clonality And PRCA
PRCA is mediated by a spectrum of T cell responses ranging from polyclonal to oligo and monoclonal. The presence of STAT3 mutations may support this notion,23 as STAT3 mutation(+) LGLs are usually a sub fraction of clonal cytotoxic T lymphocytes (CTL). Reactive T lymphocytosis can evolve from polyclonal to clonal and be further fixed by STAT3 mutations.13
High frequencies of clonal TCR gene rearrangements were observed in PRCA patients. Balasubramanian SK et al reported 8 (25%) of the 32 patients were positive for clonal TCR gene rearrangement but did not fulfill the criteria for LGLL.18 Sivakumaran et al reported two PRCA patients had clonal TCR gene rearrangements.82 LGLL-associated PRCA patients had clonal TCR gene rearrangements more often than the PRCA patients (65% vs. 33%, respectively).15 In a report from oriental patients, 19% PRCA patients with TCR gene rearrangement were diagnosed as T-LGLL and the actual frequency might be higher as only some of the patients had been investigated.20 The significance of this observation is that in oriental patients, and perhaps also in occidental patients, an underlying T-LGLL should be excluded in the diagnostic evaluation of PRCA. Motoji et al reported the TCR gene rearrangement could not be detected after a LGLL-associated PRCA patient was treated successfully.83 The significance of this observation is that disappearance of TCR gene rearrangement is an effective indicator of treatment.
Treatments
LGLL-assosicated PRCA is an orphan disease without rationally established standard treatments and optimal treatment is controversial. Treatment is mainly based on retrospective case series, as well as on case reports, however, responses are unpredictable, and a sizable proportion of patients may be refractory and transfusion dependent. Several agents, such as methotrexate (MTX), cyclosporine (CsA), cyclophosphamide (CTX), prednisone, hematopoietic growth factors and nucleoside analogs have been used with variable success,60,61 and alemtuzumab and antithymocyte globulin have been proposed for refractory disease.62,63 The goal of treatment is to attain a normal hemoglobin concentration or transfusion independence with clinically acceptable hemoglobin concentration.
Methotrexate (MTX)
Based on the data previously discussed, first-line treatment includes low-dose MTX with or without steroids. MTX is usually administered orally at a weekly dose of 5–13.5 mg/m2. A trial of at least 3 months is necessary before MTX therapy is characterized as ineffective. The efficacy of MTX as first-line treatment was evaluated on 10 LGLL-assosicated PRCA patients.18 The overall CRR (complete response rate) and ORR (overall response rate) was 20% (2/10) and 30% (3/10), respectively. Our series of Chinese T-LGLL-assosicated PRCA patients responded well to MTX.10 In the 18 patients with T-LGLL-assosicated PRCA there were 6 complete and 11 partial responses with an ORR of 94%. Median time-to-response was 3 months (range, 1–5 months). Response rate in PRCA patients was not significantly different from that in all other patients.
Treatment should be continued indefinitely in responders. Given the potential marrow-suppressive effect of MTX, its use should be monitored in patients with hepatic and renal function and avoided in patients with impaired hepatic or renal function. MTX interferes with the cellular utilization of folate, and folate depletion is the cause of most of the side effects. So low-dose oral folate (1–5 mg daily) should be given to prevent side effects. But folate may reduce the efficacy of MTX.
Cyclophosphamide (CTX)
CTX at doses of 50–100 mg orally daily, is effective for inducing remission and preventing relapse in LGLL-associated PRCA.6,12,14,84 There were 4 case series that reported LGLL-associated PRCA showed better outcomes with CTX with response rates of 60–100% when compared with CsA or steroids.6,12,84,85 As an initial therapy, CTX produced a better ORR than prednisone alone (80% versus 43%) and was associated with a longer duration of response, 60 months (range, 22–117 months) versus 7.5 months (range, 5–43 months).6 Five patients received CTX (4 CR, 1 NR), the remission duration ranged from 22 to 120 months.6 Fujishima et al have demonstrated that the ORR to CTX is 75% (2 CR and 4 PR) and the median relapse-free survival was 53 months in LGLL-associated PRCA.12 CTX seemed to have a better activity in LGLL-associated PRCA than CsA, but this was not statistically significant.12 The median duration of CTX therapy was 24 months with a range of 10–124 months.12 The ORR to CTX was 47% (14/30) and there was no difference in ORR between the LGLL and LGLL-associated PRCA group.18 The ORR to the initial CTX therapy was 66% and the median duration of response was 32 months.85 In 7 patients with T-LGLL-associated PRCA, all of them were successfully treated with CTX monotherapy.84 Therapeutic responses began after 8 weeks, and clinical CRs were obtained after 6 months. Clinical remission was associated with the disappearance of TCR gene rearrangement, which suggested that the disappearance of TCR gene rearrangement might be an indicator for the discontinuation of CTX. Hansen RM et al reported a LGLL-associated PRCA patient who experienced a partial and then a complete clinical response to splenectomy and CTX.41 CTX was cytotoxic not only for T-LGLL, but also the polyclonal proliferation of T-LGLs. Rie Tabata et al showed that CTX was effective when PRCA was caused by increased T-LGLs.86 The treatment had to be maintained to achieve continued remission, but CTX was of limited value as a maintenance agent due to its late toxicity.
The precise underlying mechanism is still unknown, and one possible explanation is that CTX is effective to reduce the CTLs, which damaged antibody-bound erythroblasts directly. On the other hand, CTL-mediated damage to hematopoietic progenitors could cause MDS, which lead to anemia, and reduction of CTLs by CTX could avoid this phenomenon.
Cyclosporine (CsA)
LGLL-associated PRCA also showed a good response to CsA, which may be proposed as first line therapy.14,15,87,88 In cases not responding to MTX, CsA may be administered as a second-line therapy. A reasonable starting dose is 5–10 mg/kg per day orally in two divided doses for at least 3 months. Fujishima N et al reported CsA produced remissions in 1/4 patients. 5 patients were maintained on CsA and 2 patients relapsed during maintenance therapy.12 In a report, response rate for T-LGLL with or without PRCA was analyzed.15 The 19 patients with T-LGLL-associated PRCA achieved an ORR of 78.9% (11 CR, 4 PR), while 9 patients without PRCA achieved an ORR of 88.9% (5CR, 3PR). In a study on 25 LGLL-assosicated PRCA patients, the ORR was 56% (28% achieved CR).89 ORR was better in non-LGLL vs LGLL-assosicated PRCA (84% vs. 55%, P=0.01).18 Kawakami T et al reported that the response rate to CsA in the STAT3 mutation(+) PRCA patients was significantly lower than that in the STAT3 mutation(-) patients (46% [6 of 13] vs 100% [8 of 8]).59 Two STAT3 mutation(+) patients acquired resistance to CsA, whereas 8 STAT3 mutation(-) patients remained responsive to CsA.
CsA is an immunosuppressive agent which seems to alter T-cell function by inhibiting the production of IL-2 from the helper T cells. One of the advantages of CsA is its rapid effect; the disadvantages of CsA are renal toxicity, hepatotoxicity, gastrointestinal complication, hirsutism, hypertension and so on, but these effects are dose-dependent and reversible. CsA levels should be monitored with target levels of 150 to 250 ng/mL.87 After achieving remission, CsA can be tapered slowly to the lowest effective maintenance dose.
Steroids
Prednisone is usually administered at a dose of 40–60 mg/day and appears to be useful when used in combination with immunosuppressive agents. Single-agent steroid therapy was of limited value and may be associated with unwanted toxicity. Some patients may have a response, but most of them were not durable responses. Go RS et al reported 7 patients were treated with prednisone (3 CR, 4 NR), but all prednisone responders relapsed within 5 to 10 months of treatment.6
Alemtuzumab
The humanized anti-CD52 antibody alemtuzumab (Campath-1H) is capable of selectively killing CD52-expressing cells and might be suitable for LGLL-assosicated PRCA because it is effective in not only T-cell lymphoproliferative diseases,90,91 but also PRCA.87 Alemtuzumab should be used with caution in selected cases that are refractory to other treatments. Alemtuzumab is given at a dose of 10 mg/week for 4–6 weeks, and response is assessed with careful monitoring of the blood counts. Successful outcomes with alemtuzumab in the salvage setting have been described.89,92,93 Alemtuzumab showed promising results in LGLL-assosicated PRCA with an ORR of 75% and overall CRR of 63%.18 It was highly efficacious for 2 patients with LGLL-associated PRCA.94 The mean response time was 17 days compared with a response time of at least 61 days on standard first-line therapy. Wing-Yan Au et al reported treatment with alemtuzumab led to a complete remission of the PRCA in a T-LGLL patient.93 A patient with thymoma, PRCA and T-LGLL was treated with alemtuzumab and there was an increase in haemoglobin but leukaemic remission did not occur.95 Therefore, this observation suggested that alemtuzumab might not be curative for T-LGLL. This response of PRCA to alemtuzumab independent of the leukaemia was reminiscent of that observed when CsA was used to treat T-LGLL,96 where improvement of cytopenias occurred despite persistence of the leukaemia. Schützinger C et al reported successful treatment with alemtuzumab in a patient with γδT-LGLL-associated PRCA refractory to CsA and MTX.92 This case showed that alemtuzumab was highly effective for treatment-resistant γδT-LGLL and led to a rapid and sustained elimination of PRCA.
Infection is an important complication of alemtuzumab. Virustesting should be carried out before treatment. In a report, 56% patients developed opportunistic infections, and herpesvirus infections were the most common.97 Other infections included upper respiratory tract infections, tuberculosis, sepsis and/or bacteraemia.97
Antithymocyte Globulin
Antithymocyte globulin has a 50% response rate in primary autoimmune PRCA, but has relatively low response rate in refractory PRCA. LGLs inhibited normal BM CFU-E growth, and this effect could be abolished by treatment with antithymocyte globulin.42 A T-LGLL-associated PRCA patient was refractory to treatment, but responded to antithymocyte globulin and became transfusion independent.64 In two patients with T-LGLL-associated PRCA, administration with antithymocyte globulin resulted in CR in one patient, whereas the other patient was refractory to all types of treatment and remained transfusion-dependent.21
Notably, there should be a therapeutic trial for at least 2 to 3 months before a specific agent is deemed ineffective. Although CTX, MTX and CsA are effective treatment, they have also been shown to increase the incidence of secondary malignancies. This is thought to be related to the immunosuppressive effect. In some cases, molecular remission was achieved, although this was not a prerequisite for long-term clinical remission. Remissions can be achieved in most cases with sequential immunosuppressive treatment. In about 5% cases, there was no response to several lines of treatment and chronic red cell transfusion was necessary. More intensive cytotoxic treatment such as combination chemotherapy is usually unnecessary. The cumulative dose of the medication and the duration of the maintenance treatment is one of the major concerns considering the late toxicity. Therefore, strategies that reduce the duration of exposure can minimize the long-term risks.
In refractory cases, study of newer immunosuppressive agents may provide new treatment options. Gene signature and mutational profiling may be an effective tool in determining what an appropriate treatment is. Patients with STAT3 mutations also tended to need more treatments.59 In LGLL-associated PRCA patients with STAT3 mutation, STAT3 activation led to multiple gene deregulations and treatment with STAT3 inhibitors induced the apoptosis of leukemic cells.56 As a result, specific STAT3 inhibitors have been evaluated as a novel therapeutic regimen for LGLL-associated PRCA. These observations were not limited to the STAT3 mutation(+) patients with LGLL-associated PRCA; similar findings were also recognized in STAT3 mutation(-) patients with LGLL-associated PRCA, but not in normal controls.98,99 The mechanisms of proliferative and/or anti-apoptotic characteristics of LGLs in STAT3 mutation(+) patients might be similar to those in STAT3 mutation(-) patients through alternative changes other than STAT3 SH2 domain mutations.
Prognosis
LGLL is the disease commonly associated with PRCA. This association predicts superior response to immunosuppressive treatment, but is not correlated with improved survival. The patients with LGLL-associated PRCA had a better response than those with idiopathic PRCA (88% v 56%).20 However, this result did not reach statistical significance, and the survival time was not significantly different between the two groups.20 The median overall survival of idiopathic PRCA was 212.6 months, and the median overall survival of LGLL-associated PRCA was 147.8 months.14 Survival time was not significantly different between the idiopathic PRCA and LGLL-associated PRCA. Response of patients with relapsing PRCA to immunosuppressive treatment was inferior to those of untreated PRCA patients (65.8% vs 80%).14 We sought to identify potential prognostic factors and to review response rates to various treatment regimens. Patients with STAT3 mutation had briefer treatment-free survival than patients with wild-type STAT3 (5 months [range, 0.5–13 months] vs 19 months [range, 3–97 months]). Time-to-response and response-duration were also briefer in STAT3 mutation patients but these differences were not significant.58 Infection and organ failure, but not progression of LGLL, were the principal causes of death. Refractoriness to induction immunosuppressive treatment and relapse of anaemia may be risk factors for death in LGLL-associated PRCA. Effective maintenance treatment, treatment of post-transfusion iron overload and the management of infectious complications are crucial for improving the prognosis of LGLL-associated PRCA.
Conclusion
LGLL remains an underdiagnosed disorder. A significant proportion of idiopathic PRCA may be secondary to LGLL. T-LGLL is the disease most commonly associated with PRCA in Asian patients. Immunosuppressive treatments have efficacy in LGLL-associated PRCA and distinction of pathogenetic mechanisms is essential for selecting the most effective therapeutic modality. As the pathogenetic mechanisms of LGLL-associated PRCA become elucidated, more targeted and less toxic treatments are expected to be developed. Nevertheless, comparison of these studies is limited by unavoidable heterogeneity in terminology, diagnostic criteria and patient populations.
Acknowledgments
This research was funded by “Liu Ge Yi Gong Cheng”of Jiangsu Province (Grant No. LGY2017022), Youth Foundation of Jiangsu Province (Grant No. QNRC2016448) and Natural Science Foundation of Jiangsu Province (BK20161354) and Social Development Foundation of Zhenjiang (SH2019069).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sokol L, Loughran TP
2. Kondo H, Watanabe J, Iwasaki H. T-large granular lymphocyte leukemia accompanied by an increase of natural killer cells (CD3-) and associated with ulcerative colitis and autoimmune hepatitis. Leukemia Lymphoma. 2001;41:207–212. doi:10.3109/10428190109057973
3. Oshimi K, Yamada O, Kaneko T, et al. Laboratory findings and clinical course of 33 patients with granular lymphocyte proliferative disorders. Leukemia. 1993;7:782–788.
4. Kondo H, Narita K, Iwasaki H, Watanabe J. Effectiveness of cyclosporine A in a patient with pure red cell aplasia associated with T cell-lineage granular lymphocyte proliferative disorders resistant to cyclophosphamide. Eur J Haematol. 2000;64:206–207. doi:10.1034/j.1600-0609.2000.9l097.x
5. Means RT
6. Go RS, Li CY, Tefferi A, Phyliky RL. Acquired pure red cell aplasia associated with lymphoproliferative disease of granular T lymphocytes. Blood. 2001;98:483–485. doi:10.1182/blood.v98.2.483
7. Lai DW, Loughran TP
8. Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist. 2004;9:247–258. doi:10.1634/theoncologist.9-3-247
9. Kawahara S, Sasaki M, Isobe Y, et al. Clinical analysis of 52 patients with granular lymphocyte proliferative disorder (GLPD) showed frequent anemia in indolent T-cell GLPD in Japan. Eur J Haematol. 2009;82:308–314. doi:10.1111/j.1600-0609.2009.01213.x
10. Qiu ZY, Fan L, Wang R, et al. Methotrexate therapy of T-cell large granular lymphocytic leukemia impact of STAT3 mutation. Oncotarget. 2016;7:61419–61425. doi:10.18632/oncotarget.11360
11. Kwong YL, Wong KF, Chan LC, et al. Large granular lymphocyte leukemia. A study of nine cases in a Chinese population. Hematoparhology. 1995;103:76–81.
12. Fujishima N, Sawada K, Hirokawa M, et al. Long-term responses and outcomes following immunosuppressive therapy in large granular lymphocyte leukemia-associated pure red cell aplasia: a nationwide cohort study in Japan for the PRCA Collaborative Study Group. Haematologica. 2008;93:1555–1559. doi:10.3324/haematol.12871
13. Bareau B, Rey J, Hamidou M, et al. Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica. 2010;95:1534–1541. doi:10.3324/haematol.2009.018481
14. Hirokawa M, Sawada K, Fujishima N, et al. Long-term outcome of patients with acquired chronic pure red cell aplasia (PRCA) following immunosuppressive therapy: a final report of the nationwide cohort study in 2004/2006 by the Japan PRCA collaborative study group. Br J Haematol. 2015;169:879–886. doi:10.1111/bjh.13376
15. Zhao X, Zhou K, Jing L, et al. Treatment of T-cell large granular lymphocyte leukemia with cyclosporine A: expeience in a Chinese single institution. Leuk Res. 2013;37:547–551. doi:10.1016/j.leukres.2013.01.017
16. Kwong YL, Au WY, Leung AY, Tse EW. T-cell large granular lymphocyte leukemia: an Asian perspective. Ann Hematol. 2010;89:331–339. doi:10.1007/s00277-009-0895-3
17. Dhodapkar MV, Li CY, Lust JA, Tefferi A, Phyliky RL. Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood. 1994;84:1620–1627.
18. Balasubramanian SK, Sadaps M, Thota S, et al. Rational management approach to pure red cell aplasia. Haematologica. 2018;103:221–230. doi:10.3324/haematol.2017.175810
19. Wlodarski MW, O’Keefe C, Howe EC, et al. Pathologic clonal cytotoxic T-cell responses: non random nature of the T-cell-receptor restriction in granular lymphocyte leukemia. Blood. 2005;106:2769–2780. doi:10.1182/blood-2004-04-1622
20. Lacy MQ, Kurtin PJ, Tefferi A. Pure red cell aplasia: association with large granular lymphocyte leukemia and the prognostic value of cytogenetic abnormalities. Blood. 1996;87:3000–3006.
21. Ergas D, Resnitzky P, Berrebi A. Pure red blood cell aplasia associated with parvovirus B 19 infection in large granular lymphocyte leukemia. Blood. 1996;87:3523–3524.
22. Charles RJ, Sabo KM, Kidd PG, Abkowitz JL. The pathophysiology of pure red cell aplasia: implications for therapy. Blood. 1996;87:4831–4838.
23. Loughran TPJr. Clonal diseases of large granular lymphocytes. Blood. 1993;82:1–14.
24. Frickhofen N, Chen ZJ, Young NS, Cohen BJ, Heimpel H, Abkowitz JL. Parvovirus B19 as a cause of acquired chronic pure red cell aplasia. Br J Haematol. 1994;87:818–824. doi:10.1111/j.1365-2141.1994.tb06743.x
25. Kondo H, Mori A, Watanabe J, Takada J, Takahashi Y, Iwasaki H. Pure red cell aplasia associated with parvovirus B19 infection in T-large granular lymphocyte leukemia. Leuk Lymphoma. 2001;42:1439–1443. doi:10.3109/10428190109097777
26. Brown KE, Hibbs JR, Gallinella G, et al. Resistance to parvovirus B19 infection due to lack of virus receptor (erythrocyte P antigen). N Engl J Med. 1994;330:1192–1196. doi:10.1056/NEJM199404283301704
27. Brown KE, Young NS. Parvovirus B19 in human disease. Annu Rev Med. 1997;48:59–61. doi:10.1146/annurev.med.48.1.59
28. Sharma VR, Fleming DR, Slone SP. Pure red cell aplasia due to parvovirus B19 in a patient treated with rituximab. Blood. 2000;96:1184–1186.
29. Young N, Harrison M, Moore J, Mortimer P, Humphries RK. Direct demonstration of the human parvovirus in erythroid progenitor cells infected in vitro. J Clin Invest. 1984;74:2024–2032. doi:10.1172/JCI111625
30. Young NS, Mortimer PP, Moore JG, Humphries RK. Characterization of a virus that causes transient aplastic crisis. J Clin Invest. 1984;73:224–230. doi:10.1172/JCI111195
31. Ishida F, Matsuda K, Sekiguchi N, et al. STAT3 gene mutations and their association with pure red cell aplasia in large granular lymphocyte leukemia. Cancer Sci. 2014;105:342–346. doi:10.1111/cas.12341
32. Dhodapkar MV, Lust JA, Phyliky RL. T-cell large granular lymphocytic leukemia and pure red cell aplasia in a patient with type I autoimmune polyendocrinopathy: response to immunosuppressive therapy. Mayo Clin Proc. 1994;69:1085–1088. doi:10.1016/s0025-6196(12)61377-9
33. Hara T, Mizuno Y, Nagata M, et al. Human gamma delta T-cell receptor-positive cell-mediated inhibition of erythropoiesis in vitro in a patient with type I autoimmune polyglandular syndrome and pure red blood cell aplasia. Blood. 1990;75:941–950.
34. Abkowitz JL, Kadin ME, Powell JS, Adamson JW. Pure red cell aplasia: lymphocyte inhibition of erythropoiesis. Br J Haematol. 1986;63:59–67. doi:10.1111/j.1365-2141.1986.tb07495.x
35. Hirayama Y, Nagai T, Ohta H, et al. A case of pure red cell aplasia accompanied with granular lymphocytic leukemia the tumor cells of which suppressed colony formation of BFU-E, and which was successfully treated by cyclophosphamide and cyclosporin A. Jpn J Clin Hematol. 1997;38:1206–1211.
36. Nagasawa T, Abe T, Nakagawa T. Pure red cell aplasia and hypogam-maglobulinemia associated with T-cell chronic lymphocytic leukemia. Blood. 1981;57:1025–1031.
37. Mangan KF, Chikkappa G, Farley PC. T gamma (Tγ) cells suppress growth of erythroid colony forming units in vitro in the pure red cell aplasia of B-cell chronic lymphocytic leukemia. J Clin Invest. 1982;70:1148–1156. doi:10.1172/jci110713
38. Partanen S, Ruutu T, Vuopio P, Andersson LC. Acquired pure red cell aplasia: a consequence of increased natural killer cell activity? Leuk Res. 1984;8:117–122. doi:10.1016/0145-2126(84)90039-0
39. Tefferi A, Windebank KP, Veeder MH, Kiely JM. Steroid-responsive pure red cell aplasia associated with natural killer cell lymphocytosis. Am J Hematol. 1989;31:211–212. doi:10.1002/ajh.2830310313
40. Lipton JM, Nadler LM, Canellos GP, Kudisch M, Reiss CS, Nathan DG. Evidence for genetic restriction in the suppression of erythropoiesis by a unique subset of T lymphocytes in man. J Clin Invest. 1983;72:694–706. doi:10.1172/JCI111019
41. Hansen RM, Lerner H, Abrams RA, Patrick CW, Malik MI, Keller R. T-cell chronic lymphocytic leukemia with pure red-cell aplasia: laboratory demonstration of persistent leukemia in spite of apparent complete clinical remission. Am J Hematol. 1986;22:79–86. doi:10.1002/ajh.2830220112
42. Zoumbos NC, Gascon P, Djeu FY, Trost SR, Young NS. Circulating activated suppressor T lymphocytes in aplastic anemia. N Engl J Med. 1985;312:257–265. doi:10.1056/NEJM198501313120501
43. Mangan KF. T-cell-mediated suppression of hematopoiesis. N Engl J Med. 1985;312:306–307. doi:10.1056/NEJM198501313120509
44. Kannourakis G, Begley CG, Johnson GR, Werkmeister JA, Burns GF. Evidence for interactions between monocytes and natural killer cells in the regulation of in vitro hemopoiesis. J Immunol. 1988;140:2489–2494.
45. Hansson M, Kiessling R, Andersson B. Human fetal thymus and bone marrow contain target cells for natural killer cells. Eur J Immunol. 1981;11:8–12. doi:10.1002/eji.1830110103
46. Hansson M, Beran M, Andersson B, Kiessling R. Inhibition of in vitro granulopoiesis by autologous and allogeneic human NK cells. J Immunol. 1982;129:126–132.
47. Fisch P, Handgretinger R, Schaefer HE. Pure red cell aplasia. Br J Haematol. 2000;111:1010–1022. doi:10.1046/j.1365-2141.2000.02429.x
48. Sieff C, Bicknell D, Caine G, Robinson J, Lam G, Greaves MF. Changes in cell surface antigen expression during hemopoietic differentiation. Blood. 1982;60:703–713.
49. Robinson J, Sieff C, Delia D, Edwards PA, Greaves M. Expression of cell-surface HLA-DR, HLA-ABC and glycophorin during erythroid differentiation. Nature. 1981;289:68–71. doi:10.1038/289068a0
50. Handgretinger R, Geiselhart A, Moris A, et al. Pure red-cell aplasia associated with clonal expansion of granular lymphocytes expressing killer-cell inhibitory receptor. N Engl J Med. 1999;340:278–284. doi:10.1056/NEJM199905133401909
51. Messner HA, Fauser AA, Curtis JE, Dotten D. Control of antibody-mediated pure red-cell aplasia by plasmapheresis. N Engl J Med. 1981;304:1334–1338. doi:10.1056/NEJM198105283042205
52. Krantz SB, Moore WH, Zaentz SD. Studies on red cell aplasia. V. Presence of erythroblast cytotoxicity in G-globulin fraction of plasma. J Clin Invest. 1973;52:324–336. doi:10.1172/JCI107188
53. Casadevall N, Dupuy E, Molho-Sabatier P, Tobelem G, Varet B, Mayeux P. Autoantibodies against erythropoietin in a patient with pure red-cell aplasia. N Engl J Med. 1996;334:630–633. doi:10.1056/NEJM199603073341004
54. Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905–1913. doi:10.1056/NEJMoa1114885
55. Jerez A, Clemente MJ, Makishima H, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120:3048–3057. doi:10.1182/blood-2012-06-435297
56. Rajala HL, Olson T, Clemente MJ, et al. The analysis of clonal diversity and therapy responses using STAT3 mutations as a molecular marker in large granular lymphocytic leukemia. Haematologica. 2015;100:91–99. doi:10.3324/haematol.2014.113142
57. Jerez A, Clemente MJ, Makishima H, et al. STAT3-mutations indicate the presence of subclinical T cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood. 2013;122:2453–2459. doi:10.1182/blood-2013-04-494930
58. Qiu ZY, Fan L, Wang L, et al. STAT3 mutations are frequent in T-cell large granular lymphocytic leukemia with pure red cell aplasia. J Hematol Oncol. 2013;6:82–84. doi:10.1186/1756-8722-6-82
59. Kawakami T, Sekiguchi N, Kobayashi J, et al. Frequent STAT3 mutations in CD8+ T cells from patients with pure red cell aplasia. Blood Adv. 2018;2:2704–2712. doi:10.1182/bloodadvances.2018022723
60. Shi M, He R, Feldman AL, et al. STAT3 mutation and its clinical and histopathologic correlation in T-cell large granular lymphocytic leukemia. Hum Pathol. 2018;73:74–81. doi:10.1016/j.humpath.2017.12.014
61. Savola P, Kelkka T, Rajala HL, et al. Somatic mutations in clonally expanded cytotoxic T lymphocytes in patients with newly diagnosed rheumatoid arthritis. Nat Commun. 2017;8:15869.
62. Valori M, Jansson L, Kiviharju A, et al. A novel class of somatic mutations in blood detected preferentially in CD8+ cells. Clin Immunol. 2017;175:75–81. doi:10.1016/j.clim.2016.11.018
63. Shvidel L, Duksin C, Tzimanis A, et al. Cytokine release by activated T-cells in large granular lymphocytic leukemia associated with autoimmune disorders. Hematol J. 2002;3:32–37. doi:10.1038/sj/thj/6200149
64. Kwong YL, Wong KF. Association of pure red cell aplasia with T large granular lymphocyte leukaemia. J Clin Pathol. 1998;51:672–675. doi:10.1136/jcp.51.9.672
65. DeZern AE, Pu J, McDevitt MA, Jones RJ, Brodsky RA. Burst-forming unit-erythroid assays to distinguish cellular bone marrow failure disorders. Exp Hematol. 2013;41:808–816. doi:10.1016/j.exphem.2013.04.013
66. Coutinho J, Lima M, dos Anjos Teixeira M, et al. Pure red cell aplasia associated to clonal CD8+ T-cell large granular lymphocytosis: dependence on cyclosporin A therapy. Acta Haematol. 1998;100:207–210. doi:10.1159/000040906
67. Masuda M, Arai Y, Okamura T, Wada M, Mizoguchi H. Pure red cell aplasia (PRCA) with thymoma: a possible distinct clinical entity distinct from large granular lymphocyte (LGL) leukemia. Am J Hematol. 2000;63:102–106. doi:10.1002/(sici)1096-8652(200002)63:2<102::aid-ajh9>3.0.co;2-p
68. Ergas D, Tsimanis A, Shtalrid M, Duskin C, Berrebi A. T-gamma large granular lymphocyte leukemia associated with amegakaryocytic thrombocytopenic purpura, sjogren’s syndrome, and polyglandular autoimmune syndrome type II, with subsequent development of pure red cell aplasia. Am J Hematol. 2002;69:132–134. doi:10.1002/ajh.10024
69. Kouides PA, Rowe JM. Large granular lymphocyte leukemia presenting with both amegakaryocytic thrombocytopenic purpura and pure red cell aplasia: clinical course and response to immunosuppressive therapy. Am J Hematol. 1995;49:232–236. doi:10.1002/ajh.2830490309
70. Handa SI, Schofield KP, Sivakumaran M, Short M, Pumphrey RS. Pure red cell aplasia associated with malignant thymoma, myasthenia gravis, polyclonal large granular lymphocytosis and clonal thymic T cell expansion. J Clin Pathol. 1994;47:676–679. doi:10.1136/jcp.47.7.676
71. Zhou K, Wang HJ, Zhang L, et al. The clinical characteristics of T cell large granular lymphocyte leukemia associated with pure red cell aplasia. Zhonghua Xue Ye Xue Za Zhi. 2008;29:312–315.
72. Saitoh T, Karasawa M, Sakuraya M, et al. Improvement of extrathymic T cell type of large granular lymphocyte (LGL) leukemia by cyclosporin A: the serum level of fas ligand is a marker of LGL leukemia activity. Eur J Haematol. 2000;65:272–275. doi:10.1034/j.1600-0609.2000.065004272.x
73. Mori S, Suzushima H, Nishikawa K, et al. Smoldering gamma delta T-cell granular lymphocytic leukemia associated with pure red cell aplasia. Acta Haematol. 1995;94:32–35. doi:10.1159/000203967
74. Oshimi K, Hoshino S, Takahashi M, et al. Ti (WT31)-negative, CD3-positive, large granular lymphocyte leukemia with nonspecific cytotoxicity. Blood. 1988;71:923–931.
75. Tanaka Y, Matsui K, Yamashita K, et al. T-large granular lymphocyte leukemia preceded by pure red cell aplasia and complicated with hemophagocytic syndrome caused by Epstein-Barr virus infection. Intern Med. 2006;45:631–635. doi:10.2169/internalmedicine.45.1594
76. Oshimi K. Granular lymphocyte proliferative disorders: report of 12 cases and review of the literature. Leukemia. 1988;2:617–627.
77. Sandberg Y, Almeida J, Gonzalez M, et al. TCRγδ+ large granular lymphocyte leukemias reflect the spectrum of normal antigen-selected TCRγδ+ T-cells. Leukemia. 2006;20:505–513. doi:10.1038/sj.leu.2404112
78. Bourgault-Rouxel AS, Loughran TP
79. Spour EF, Leemhuis T, Jenski L, Redmond R, Fillak D, Jansen J. Characterization of normal human CD3+CD5- and gamma delta T cell receptor positive T lymphocytes. Clin Exp Immunol. 1990;80:114–121. doi:10.1111/j.1365-2249.1990.tb06450.x
80. Tefferi A, Li CY, Witzig TE, et al. Chronic natural killer cell lymphocytosis: a descriptive clinical study. Blood. 1994;84:2721–2725.
81. Kurt H, Jorgensen JL, Amin HM, et al. Chronic lymphoproliferative disorder of NK-cells: a single-institution review with emphasis on relative utility of multimodality diagnostic tools. Eur J Haematol. 2018;100:444–454. doi:10.1111/ejh.13038
82. Sivakumaran M, Bhavnani M, Stewart A, Roberts BE, Geary GC. Is pure red cell aplasia (PRCA) a clonal disorder? Clin Lab Haematol. 1993;15:1–5.
83. Motoji T, Yamada O, Takahashi M, Oshimi K, Mizoguchi H. Granular lymphocyte leukemia with pure red cell aplasia: usefulness of gene analysis in assessing therapeutic effect. Am J Hematol. 1992;39:212–219. doi:10.1002/ajh.2830390311
84. Yamada O, Mizoguchi H, Oshimi K. Cyclophosphamide therapy for pure red cell aplasia associated with granular lymphocyte-proliferative disorders. Br J Haematol. 1997;97:392–399. doi:10.1046/j.1365-2141.1997.282672.x
85. Go RS, Lust JA, Phyliky RL. Aplastic anemia and pure red cell aplasia associated with large granular lymphocyte leukemia. Semin Hemato. 2003;40:196–200.
86. Tabata R, Tabata C. Distinct effect of cyclophosphamide and cyclosporine on pure red cell aplasia associated with T-cell large granular lymphocyte leukemia. Int Immunopharmacol. 2014;23:391–394. doi:10.1016/j.intimp.2014.10.013
87. Sawada K, Fujishima N, Hirokawa M. Acquired pure red cell aplasia: updated review of treatment. Br J Haematol. 2008;142:505–514. doi:10.1111/j.1365-2141.2008.07216.x
88. Go RS, Tefferi A, Li CY, Lust JA, Phyliky RL. Lymphoproliferative disease of granular T lymphocytes presenting as aplastic anemia. Blood. 2000;96:3644–3646.
89. Ru X, Liebman HA. Successful treatment of refractory pure red cell aplasia associated with lymphoproliferative disorders with the anti-CD52 monoclonal antibody alemtuzumab (campath-1H). Br J Haematol. 2003;123:278–281.
90. Mavromatis B, Cheson BD. Monoclonal antibody therapy of chronic lymphocytic leukemia. J Clin Oncol. 2003;21:1874–1881. doi:10.1200/JCO.2003.09.113
91. Lamy T, Loughran JTP. How I treat LGL leukemia. Blood. 2011;117:2764–2774. doi:10.1182/blood-2010-07-296962
92. Schützinger C, Gaiger A, Thalhammer R, et al. Remission of pure red cell aplasia in T-cell receptor gamma delta-large granular lymphocyte leukemia after therapy with low-dose alemtuzumab. Leukemia. 2005;19:2005–2008. doi:10.1038/sj.leu.2403956
93. Au WY, Lam CC, Chim CS, et al. Alemtuzumab induced complete remission of therapy-resistant pure red cell aplasia. Leuk Res. 2005;29:1213–1215. doi:10.1016/j.leukres.2005.02.018
94. Chow JK, Chan TK. Low-dose subcutaneous alemtuzumab is a safe and effective treatment for chronic acquired pure red cell aplasia. Hong Kong Med J. 2013;19:549–552. doi:10.12809/hkmj133798
95. Rosenblum MD, LaBelle JL, Chang CC, et al. Efficacy of alemtuzumab treatment for refractory T-cell large granular lymphocytic leukemia. Blood. 2004;103:1969–1971. doi:10.1182/blood-2003-11-3951
96. Bible KC, Tefferi A. Cyclosporine A alleviates severe anaemia associated with refractory large granular lymphocytic leukaemia and chronic natural killer cell lymphocytosis. Br J Haematol. 1996;93:406–468. doi:10.1046/j.1365-2141.1996.5061047.x
97. Martin S, Marty F, Fiumara K, et al. Infectious complications associated with alemtuzumab use for lymphoproliferative disorders. Clin Infect Dis. 2006;43:16–24. doi:10.1086/504811
98. Epling-Burnette PK, Liu JH, Catlett-Falcone R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest. 2001;107:351–362. doi:10.1172/JCI9940
99. Teramo A, Gattazzo C, Passeri F, et al. Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T-type large granular lymphocyte leukemia. Blood. 2013;121:3843–3854. doi:10.1182/blood-2012-07-441378
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.