Back to Journals » Cancer Management and Research » Volume 12
Pathogenic Heteroplasmic Somatic Mitochondrial DNA Mutation Confers Platinum-Resistance and Recurrence of High-Grade Serous Ovarian Cancer
Authors Ni J ![]() , Wang Y, Cheng X
, Wang Y, Cheng X ![]() , Teng F
, Teng F ![]() , Wang C, Han S
, Wang C, Han S ![]() , Chen X
, Chen X ![]() , Guo W
, Guo W ![]()
Received 21 August 2020
Accepted for publication 3 October 2020
Published 2 November 2020 Volume 2020:12 Pages 11085—11093
DOI https://doi.org/10.2147/CMAR.S277724
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Jing Ni,1,2 Yan Wang,3 Xianzhong Cheng,1 Fang Teng,4 Congyang Wang,3 Suping Han,2 Xiaoxiang Chen,1 Wenwen Guo3
1Department of Gynecologic Oncology, The Affiliated Cancer Hospital of Nanjing Medical University, Jiangsu Cancer Hospital, Jiangsu Institute of Cancer Research, Nanjing, 210009, People’s Republic of China; 2Department of Gynecology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, People’s Republic of China; 3Department of Pathology, The Second Affiliated Hospital of Nanjing Medical University, Nanjing 210011, People’s Republic of China; 4Department of Gynecology, Women’s Hospital of Nanjing Medical University (Nanjing Maternity and Child Health Care Hospital), Nanjing 210004, People’s Republic of China
Correspondence: Wenwen Guo
Department of Pathology, The Second Affiliated Hospital of Nanjing Medical University, 121# Jiangjiayuan Road, Nanjing, Jiangsu 210011, People’s Republic of China
Tel +86-25-58509909
Email [email protected]
Suping Han
Department of Gynecology, The First Affiliated Hospital of Nanjing Medical University, 300# Guangzhou Road, Nanjing, Jiangsu 210009, People’s Republic of China
Tel +86-25-68308083
Email [email protected]
Purpose: Platinum resistance is a primary barrier to improving the survival rate of ovarian cancer. The relationship between mtDNA somatic mutations and response to platinum-based chemotherapy in ovarian cancer has not been well clarified.
Patients and Methods: Here, we employed the next-generation sequencing (NGS) platform to identify mtDNA mutations of the unrelated high-grade serous ovarian cancer (HGSOC) patients.
Results: We identified 569 germline variants and 28 mtDNA somatic mutations, and found the platinum-sensitive relapsed HGSOC patients had more synonymous mutations while the platinum-resistant relapsed HGSOC patients had more missense mutations in the mtDNA somatic mutations. Meanwhile, we found that the HGSOC patients who harbored heteroplasmic pathogenic mtDNA somatic mutations had significantly higher prevalence of both platinum-resistance and relapse than those without (80.0% versus 16.7%, p=0.035). Additionally, we observed that the tumor tissues had significantly higher lactate-to-pyruvate (L/P) ratio than the paired nontumor tissues (p< 0.001), and L/P ratio of tumors with any heteroplasmic pathogenic mtDNA mutations was significantly higher than that of the tumors free of pathogenic mtDNA mutations (p=0.025).
Conclusion: Our findings indicate that these heteroplasmic pathogenic mtDNA somatic mutations may cause decreased respiratory chain activity and lead to the metabolism remodeling that seem to be beneficial for progression of both platinum-based chemotherapy resistance and relapse.
Keywords: high-grade serous ovarian cancer, chemotherapy, heteroplasmy, mitochondrial DNA mutation
Introduction
Ovarian cancer makes up about 4.5% of cancer-associated deaths in women worldwide and is the deadliest gynecologic malignant cancer.1 As a heterogeneous disease, ovarian cancer can originate form epithelial, germ cell or stromal components, at least 85% of all ovarian cancer is epithelial ovarian cancer (EOC). It is classified into five histological subtypes: serous, endometrioid, mucinous, clear cell and undifferentiated carcinomas.2 In fact, the majority (60–80%) of patients are diagnosed with serous ovarian cancer (SOC). Approximately 50% EOC patients are characterized as high-grade serous adenocarcinoma (HGSOC) who have advanced-stage disease (International Federation of Gynecology and Obstetrics/FIGO stage III or IV) at presentation.3
Currently the standard treatment for ovarian cancer remains debulking surgery and platinum-based chemotherapy. Although HGSOC is a chemo-sensitive disease, the major obstacle of platinum-based chemotherapy in HGSOC is the high incidence of intrinsic and acquired platinum resistance of tumor cells, which can result in treatment failure, disease relapse and even death.4 The platinum drugs are used extensively for treating many human cancers.5 The cytotoxic effect of the drugs is a multi-step process, and they work by entering cancer cells, targeting DNA molecules, producing various DNA adducts and double/single-strand breaks and interfering with DNA repair, subsequently blocking DNA replication and transcription and ultimately killing cancer cells by activating the apoptotic cascade.6 Therefore, factors, including the altered uptake, transport and metabolism of the drugs, enhanced repair capacity for platinum-related DNA damages, and the inhibition of apoptotic signaling, have been described as contributing to the platinum-resistance of cancer cells.6 In addition, research has shown that mitochondria can also play an important role in the oncogenesis as well as chemotherapy resistance,7–9 other studies have suggested that the heteroplasmic functional mutations in their own set of genetic material (mtDNA) are significant molecular events in behavior of cancers evolution including acquiring chemotherapy resistance.10−14 So far, the relationship between pathogenic heteroplasmic mtDNA mutations and their response to platinum-based chemotherapy remains largely unknown, and their is still a lack of systematic analysis regarding the spectrum of mtDNA mutation in ovarian cancer cells. The role of mtDNA somatic mutations in cancer development and progression, and drug resistance are not conclusive, and more work is needed to be done. In the present study, to further determine the spectrum of mtDNA variants and its association with platinum-resistance and recurrence, we sequenced the whole mtDNA genome and investigated the tumor cell metabolism in HGSOC. The results suggest that the pathogenic heteroplasmic mutations of mtDNA-encoded genes are frequently molecular events in HGSOC, and they could be related to the platinum chemoresistance and relapse of ovarian cancer.
Patients and Methods
Subjects and Tissue Samples
All patients were clinically diagnosed sporadic HGSOC and underwent primary treatment in the Department of Gynecologic Oncology, Jiangsu Cancer Hospital and signed their informed consent for the study. The experimental procedure was approved by the Ethics Committee of Jiangsu Cancer Hospital. Tissue samples were obtained upon informed consent from the patients (age 44–68 years) at the time of primary debulking surgery. Tumor and corresponding surrounding non-tumor tissue samples were confirmed by senior oncologists and pathologists, and immediately removed, dissected, snap-frozen in liquid nitrogen and stored at −80°C. In addition, whole blood samples were collected in citrate-anticoagulated tube from each patient and used to confirm the identified mtDNA variants.
The clinical evaluation criterion for platinum response of patients was as follows: the patients who initially respond to first-line platinum-based chemotherapy and subsequently relapse more than 6 months after the initial chemotherapy have been categorized as “platinum-sensitive”, and the patients who relapse/progress within 6 months after first-line platinum-based chemotherapy have been identified as “platinum-resistant, relapsed”.
Whole Mitochondrial Genome Sequence Analysis and Pathogenic Assessment
We adopted a commercialized next-generation sequencing (NGS) technique, VariantPro Mitochondrial Panel (LC Sciences, Hangzhou, China) to rapidly determined the possible mtDNA sequence variants. Firstly, the NGS mtDNA-targeted library preparation was accomplished by multiplex PCR-based technology with mitochondrial specific primer sets provided by LC Sciences (Hangzhou, China). The PCR amplified products were purified by Agen-court AMPure XP beads (Beckman Coulter Genomics, UK), and diluted to 20 pmol/L, then directly sequenced on the Illumina Hiseq X Ten platform based on the Paired-End 150 (PE150) strategy.
Prior to aligning sequence reads, the cleaned, paired-end sequence reads were produced by removing the low-quality reads with a cutoff score of Q20. Then we used this workflow to process the raw reads that included mapping the sequence reads to mitochondrial genome reference sequence (rCRS reference) with Burrows–Wheeler Alignment (BWA) program, combining mapped and unmapped reads to do a local realignment around the edges of base indels often results in misaligned bases creating false positive variant calls, recording the aligned reads that were uniquely mapped to the mitochondrial genome in order to avoid possible effects of nuclear mitochondrial sequences (NumtS), applying Genome Analysis ToolKit (GATK) caller to identify mtDNA variants with recalibration of gaussian mixture model, and utilizing ANNOVAR to annotate bio-functional information of the identified variants.
The sequence variants identified in both tumor and matched non-tumor tissues were scored as germline variants, and any mtDNA sequence differences between tumor and matched non-tumor tissues were scored as somatic mutations. All mtDNA somatic mutations were crosschecked and confirmed by comparing with the mtDNA sequences of blood cells form the same patients. The sequence variants have taken an approach to determine homoplasmic and heteroplasmic levels based on allele counts of sequence reads via GATK recalibration. Each mtDNA variant was checked against the MITOMAP database.15,16 The variants that were not recorded in the MITOMAP were labeled as novel mtDNA variants. Instead, the variants that appeared in the MITOMAP were registered as polymorphisms or mutations.
The pathogenic potential of each possible non-synonymous change was inferred based on the rCRS and predicted using SIFT and PolyPhen-2 platforms. The pathogenic effect of newly observed tRNA mutations were evaluated by MitoTIP system.
Lactic Acid and Pyruvate Measurements
The levels of lactic acid and pyruvate were measured using the commercial kit (Nanjing Jiancheng Bioengineering Institute, China) as directed by the manufacturer’s instructions.
Statistical Analysis
The data was analyzed using SPSS statistical software (SPSS Inc.). Continuous variable comparison was conducted by Student’s t-test or Mann–Whitney test. The categorical variable was analyzed using Chi-squared or Fisher’s exact test. In all cases, p-value less than 0.05 was considered statistically significant.
Results
Spectrum of Mitochondrial Genome Variants in HGSOC
We sequenced the entire mitochondrial genome of the 16 unrelated HGSOC tumors and their corresponding matched non-tumor cells. By comparing the rCRS, 597 sequence variants at 256 different nucleotide loci were identified (Table 1, S1-2). Of which, the 569 sequence variants at 237 different nucleotide loci were supposed to be the germline variants (Table S1). All observed germline variants were reported as the polymorphisms in the MITOMAP with recording of GeneBank frequency. Note that the nucleotide position (np) 514–523 region of rCRS is a typical microsatellite sequence containing a stretch of five CA dinucleotide repeats, termed np514-523 CA repeats. However, a stable variant containing four CA-repeats at this sequence region was found in five cases of soc5, soc11, soc14, soc17 and soc19 (Table S1).
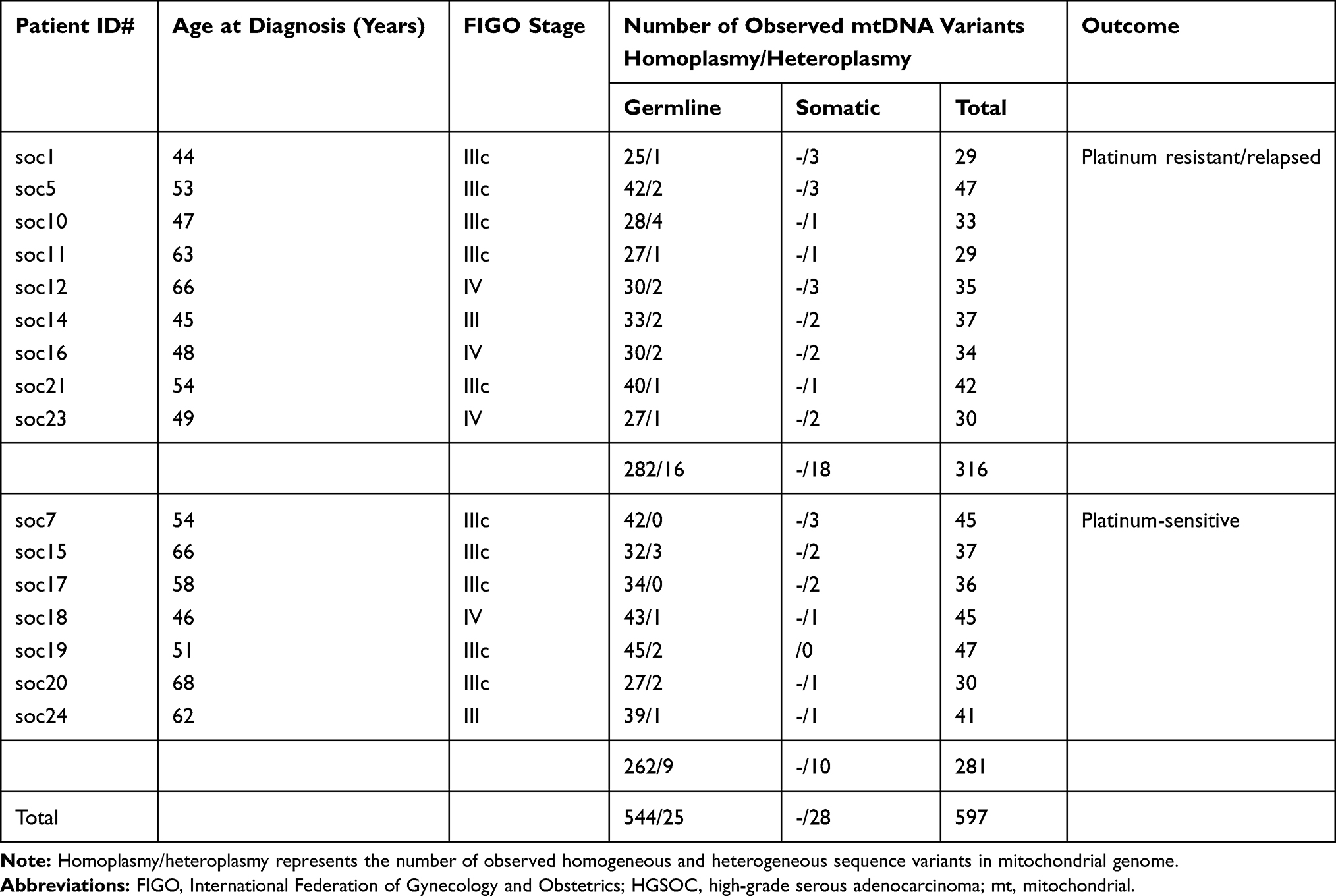 |
Table 1 Baseline Data of the Study Patients with HGSOC |
Additionally, 28 sequence variants at 24 different nucleotide loci of mtDNA only presented in the tumor tissues (Table S2), not in any matched non-tumor cells. These variants could be categorized as mtDNA somatic mutations, and 15 variants were recorded in the MITOMAP, and 13 variants were new-found mutations (Table S2).
Characteristics of Germline mtDNA Variants
Most of the germline variants (95.6%, 544/569) were homoplasmic, and the remaining 4.4% variants (25/569) were heteroplasmic (Table 1). Especially at the np 303–309 region, which was a polycytidine stretch (C-tract, C7) from np303 to np309 in rRCS and termed D310 C-tract, the 12 of 16 cases carrying heteroplasmic variants were in this sequence region (Table S1). The mtDNA of the 12 cases represented a mixture of variant repeats with various length of consecutive stretch of C residues (C8, C9, C11, or C12) (Table S1).
The details of all subjects’ germline mtDNA variant distribution are illustrated in Figure 1. 46.7% variants (266/569) were detected in non-protein coding region, and 53.3% variants (303/569) were found in protein-coding region. Of the 266 variants in non-coding region, 67.3% (179), 27.8% (74), 3.8% (10) and 1.1% (3) variants located in D-loop, rRNA gene, tRNA gene and intergenic regions, respectively. In the 303 germline variants of protein-coding region, most of them (64.4%, 195/303) were synonymous mutations and the remaining variants (35.6%, 108/303) were non-synonymous mutations that included non-synonymous variants, and missense and truncating mutations.
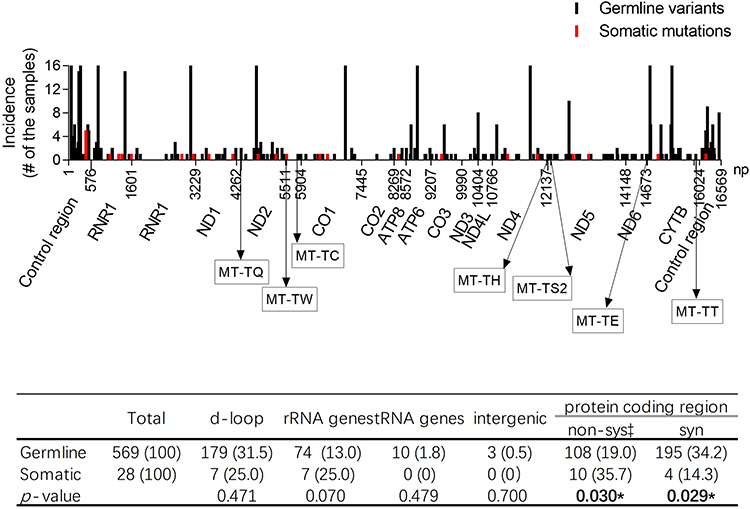 |
Figure 1 The distribution of the identified sequence variants in mitochondrial gnome. *p-values less than 0.05. |
Seen from the perspective of DNA base changes, the subjects had base transitions amount to 87.3% (497/569) of all germline variants, while base transversions and small indels only made up 5.1% (29/569) and 7.6% (43/569), respectively (Table S3). Of these, base transitions had considerably more base changes of A:T to G:C than G:C to A:T, 62.6% (311/497) against 37.4% (186/497) (Table S3). Compared with the A:T to G:C mutations (transitions), the G:C to A:T transitions were mostly located in protein coding regions (49.8% vs 71.5%, p < 0.001), in those cases of transitions were mainly synonymous mutations (Table S3). There were four types for transversions, A:T to C:G, A:T to T:A, G:C to T:A and G:C to C:G, account for 2.1%, 1.2%, 1.2% and 0.5%, respectively (Table S3). Of those transversions, 14/29 and 15/29 of variants resided in d-loop and protein-coding regions, respectively (Table S3). However, all 43 small indels were resided in non-protein coding regions (Table S3).
Characteristics of Somatic mtDNA Mutations
With the exception of soc19, each tumor sample harbored one or more somatic mutations, and all identified mtDNA somatic mutations were heteroplasmic, and their heteroplasmic level varied from 0.08 to 0.82 (Table S2). The observed somatic mutations that were resided in d-loop (7 mutations), rRNA gene (7 mutations), and protein-coding regions (14 mutations), respectively (Figure 1). In the d-loop region, 5 cases harbored the A to C mutation positioned at np439 (m.439 A>C) (Table S2). Among those five samples, a tumor sample from soc12 had both m.439 A>C and m.16182 A>C mutations (Table S2). The remaining somatic mutation of the d-loop region was m.16183A>C (Table S2).
Seven somatic mutations were found in non-protein coding regions (rRNA and tRNA genes) (Table S2). Of which, 4 mutations were situated in the MT-RNR1 gene, 2 mutations were in MT-RNR2 gene, while the other one was in tRNA MT-TW gene. The somatic mutation in MT-TW gene, m.5540 G>A, was heteroplasmic, scored 16.11 by MitoTIP system and graded it as possibly pathogenic mutation (Table S2). Fourteen heteroplasmic somatic mutations of protein-coding genes were detected from 12 cases, including 4 synonymous mutations (MT-ATP8 p.T6T, MT-ND4 p.A131A, MT-ND4 p.V381V and MT-CYB p.N74N), 6 missense mutations (MT-ND2 p.V115A, MT-CYB p.I78T, MT-ND2 p.I267T, MT-CO1 p.G160E, MT-ND5 p.A160T, and MT-ND5 p.A289T; the latter three mutations were new-found variants in MITOMAP), 2 nonsense mutations (MT-ND1 p.W290X and MT-CO1 p.G226X), and 2 frameshift mutations (MT-ND1 p.T87fs and MT-CO3 p.V91fs). The observed 6 missense mutations were scored by PolyPhen2 and SIFT, and assessed by evolutionary conservation of amino acids sequences. Five observed missense mutations were able to lead to the non-conservative replacement of an evolutionarily conserved amino acid (Figure S1). Those missense mutations had highly pathogenic potential and were considered the probably damaging mutations. Interestingly, the tumor cells with those heteroplasmic possible pathogenic mtDNA mutations had a significantly higher lactate/pyruvate ratio than which without those pathogenic mtDNA mutations and the non-tumor cells (p = 0.025 and p < 0.001, respectively) (Figure 2). The data suggested that the high lactate/pyruvate ratio in the tumor cells was significantly associated with the heteroplasmic pathogenic mtDNA somatic mutations.
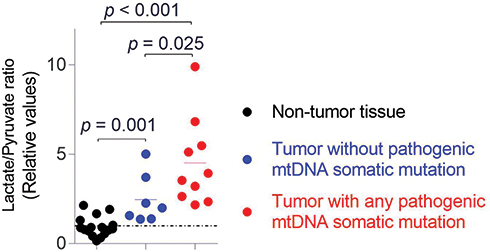 |
Figure 2 High lactate/pyruvate ratio was significantly associated with the pathogenic heteroplasmic mtDNA somatic mutations in the HGSOC tumors. The p-value was calculated by Mann–Whitney test. |
Characteristic Differences Between the Germline and Somatic Mutations in HGSOC Patients
We observed that the germline variants of protein-coding regions had higher proportion of synonymous mutations and lower ratio of functional sequence variants (included non-synonymous variants, and missense and truncating mutations) when compared with somatic mutations of the same regions (34.2% vs 14.3%, p = 0.029; 19.0% vs 35.7%, p = 0.030) (Figure 1). The G>T and A>C mutations were especially prone in the mtDNA somatic mutations of tumor cells, and they had significant higher mutation rate than those in the germline variants (25.0% vs 3.3%, p < 0.001) (Table S3).
Heteroplasmic Pathogenic mtDNA Somatic Mutation Contributes to Platinum Resistance and Relapse of HGSOC
In the platinum-resistant relapsed HGSOC patients, we observed that 298 germline variants ranged from 26 to 44 variants per patient with a median of 32, and 18 somatic mutations ranged from 1 to 3 mutations per sample (Table 1). A total of 271 germline variants were identified in the platinum-sensitive HGSOC patients ranged from 29 to 47 variants per patient with a median of 40, and 10 somatic mutations were found in tumor samples of those patients ranged from 1 to 3 mutations per sample (Table 1).
We found that the tumor cells from platinum-sensitive patients had more somatic synonymous mutations when compared with those from platinum-resistant relapsed patients (p = 0.004) (Table 2). We further compared the mutation frequency of various types of base-substitutions between the two groups, and assessed the relationship between mtDNA base-substitutions and chemotherapy outcome of HGSOC. In both mtDNA germline variants and somatic mutations, the occurrence frequency of molecular events for transversions, transitions, or small indels in platinum-resistant relapsed patients had no significant difference to those in platinum-sensitive patients (Figure 3A-B). More importantly, we found that, the HGSOC patients harbored heteroplasmic pathogenic mtDNA somatic mutations experienced a higher incidence of platinum-resistance and relapse than patients who did not carry those pathogenic mtDNA somatic mutations (80% vs 16.7%, p = 0.035) (Table 3).
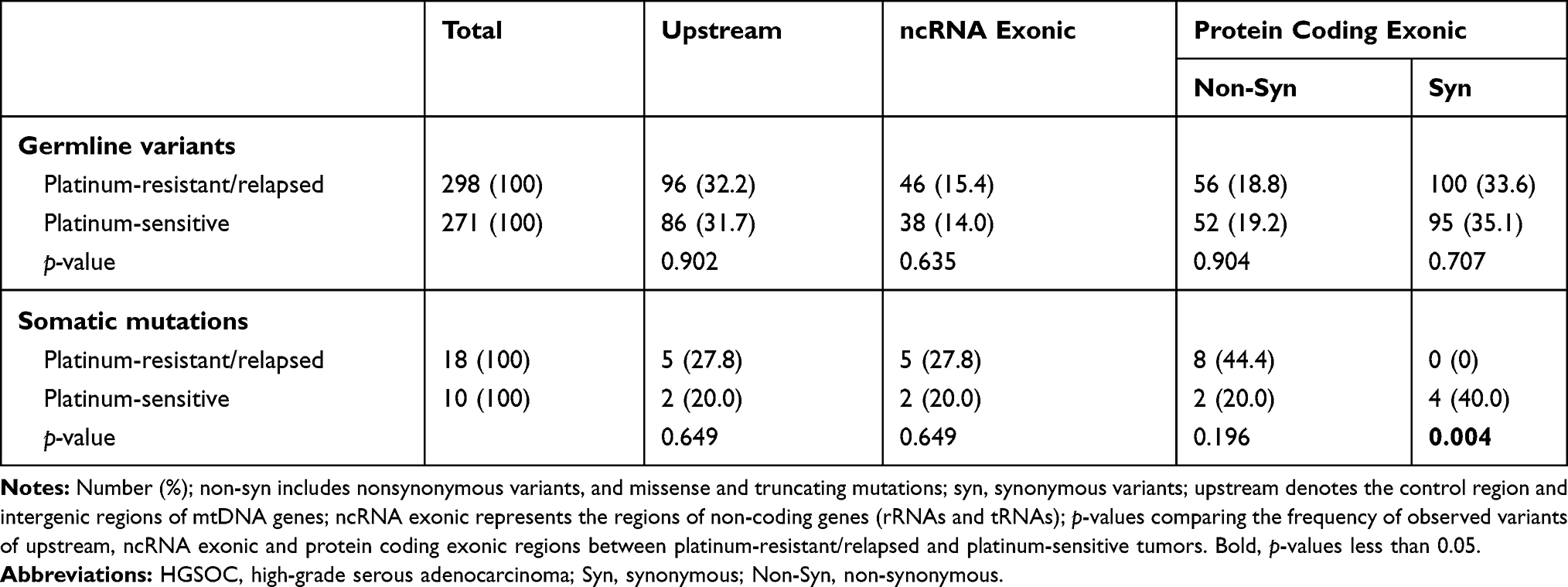 |
Table 2 Distribution of mtDNA Variants (Germline and Somatic) in Mitochondrial Genome, Stratified According to the Prognosis of HGSOC Patients Undergoing Platinum-Based Chemotherapy |
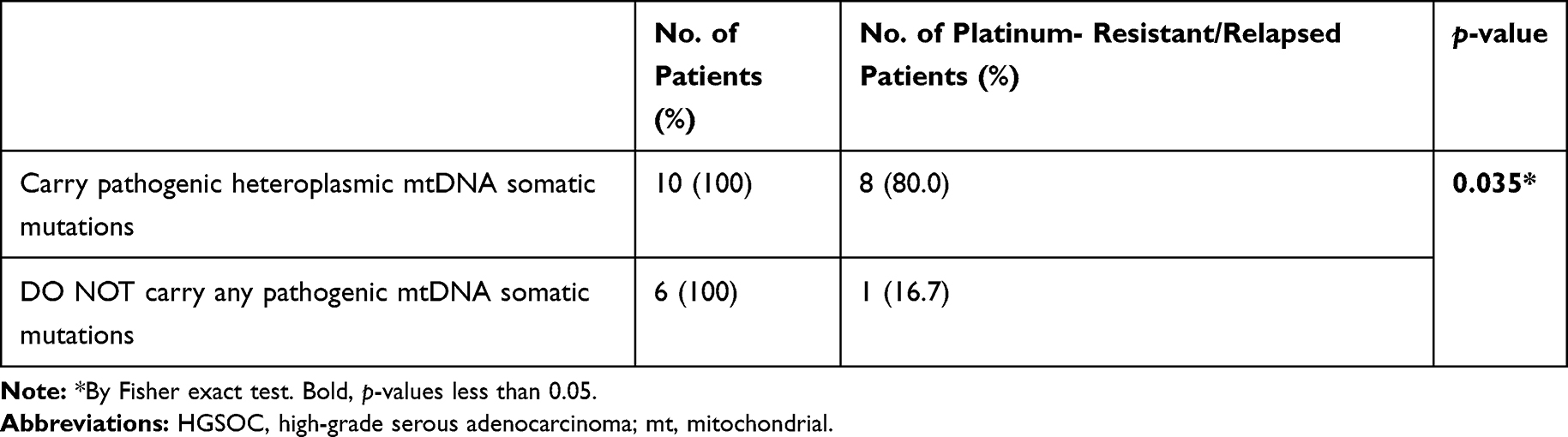 |
Table 3 The Relationship of Pathogenic mtDNA Somatic Mutations with Platinum Resistance and Relapse of HGSOC |
 |
Figure 3 Base change characteristic differences between the mtDNA germline variants (A) and the mtDNA somatic mutations (B), stratified according to the prognosis of HGSOC patients undergoing platinum-based chemotherapy.Abbreviations: HGSOC, high-grade serous adenocarcinoma; mt, mitochondrial. |
Discussion
Mitochondria are described as the powerhouse in all nucleated cells, and they provide much of energy through oxidative phosphorylation (OXPHOS). They are regarded as the key signaling hub that participate in multiple cellular processes such as intermediate metabolism, integrated cellular stress responses and the regulation of cell death.17 Studies have documented mitochondria as the master regulator of danger signaling fundamentally implicated in cancer biology.7,17,18 Mitochondrial dysfunction is a significant feature in kinds of cancer cells, and it has been suspected to play a crucial role in cancer development and progression, including metabolism reprogramming, aggressive behavior, and response to chemotherapy drugs for a long period.7,17,18 As the semiautonomous organelles, mitochondria possesses their own genome, mtDNA, along with their own RNA transcription and protein translation and assembly machinery.
Human mtDNA is a 16,569-base pair (bp) long closed circular genome with 37 genes coding 13 oxidative respiratory chain subunits, 22 transfer RNAs (tRNAs) and 2 ribosomal RNAs (rRNAs). Mutations of mtDNA, germline and somatic, have been reported as frequent events in various types of human cancer.11,19,20 Multiple studies have indicated that some germline polymorphisms in mtDNA d-loop region may be related to an increased risk of the malignancies.21,22 Cancer cells are very different from normal cells, and they use aerobic glycolysis, called warburg effect, to generate energy and metabolic intermediates for the rapid proliferation and aggressiveness.23 A series of studies suggested that some pathogenic somatic mtDNA mutations of cancer cells can confer cancerogenesis, and promote aggressiveness in vitro and in vivo by changing mitochondrial metabolism, and activating anti-apoptotic and oncogenic signaling pathways.7,10,13,24–28 Theoretically, the pathogenic mtDNA mutations can cause damage to mitochondria, and the degree for damaged mitochondrial depends on burden of the mtDNA mutations that is known as mutation load. The different levels of mtDNA mutation loads have been linked to diametrically opposite bioenergetic and biological consequences.29 Studies demonstrated that cancer cells with homoplasmic disruptive mutations or high mutational load of mtDNA-encoded genes presented with a consequent heavy mitochondrial respiration impairment and displayed the features of sluggish growth and indolent biological behavior.30,31 In contrast to that a mild heteroplasmic burden of pathogenic mutations in mtDNA cannot completely injure mitochondrial metabolism and their biological function, and they promote the reprograming of bioenergetic/biosynthetic profile of cancer cells and push tumor progression through mitochondrial stress response and oncogenic signaling pathway, respectively, activated by pathogenic mtDNA mutations-involved mitochondrial dysfunctions.10−14 There was evidence that the mild heteroplasmic mutational burden was able to confer the aggressive and chemoresistant phenotype of cancer cells. All these explained that mtDNA genes whose disruptive mutations have the janiform janus-faced impact on the tumor development and progression, including metabolic reprogramming, metastatic capability acquisition, and the chemotherapeutic response.18 Consistent with these, we observed that platinum-resistance and relapse are more prevalent in patients who had a mild heteroplasmic pathogenic somatic mtDNA mutation than in those without (p = 0.035). (Table 3). It indicats that mitochondrial dysfunction caused by heteroplasmic pathogenic mtDNA somatic mutation might contribute to platinum chemotherapy resistance and recurrence of HGSOC.
In order to understand whether mtDNA mutations and dysfunctional mitochondria underlay the platinum-resistance and recurrence of ovarian cancer, whole mtDNA sequencing was performed. We observed those unrelated HGSOC patients carrying as many as 36 germline variants each, and most of the germline variants (95.6%) were homoplasmic (Table 1). It is accommodated with the viewpoint that mtDNA presents a highly recognizable identity to molecular evolution.32,33 Compared with the germline variants, each tumor sample from the HGSOC patients had fewer somatic mtDNA mutations (Table 1), and all of them were heteroplasmic. It is interesting that the G>T and A>C mutations were especially prone in the tumor cells (Table S3). Considering that the A:T→C:G and G:C→T:A base-substitution mutations mainly caused by oxidative conversion of guanine to 8-oxo-guanine (GO) during DNA replication process, the phenomenon of accumulation of oxidative damage-derived G>T and A>C somatic mutations in the tumor samples suggested that the tumor cells are facing even severe challenge of oxidative stress.
Among the somatic mutations in tumor cells, 64.3% (9/14) of somatic mutations in protein encoding region (77.8% of them are new-found mutations) were predominantly detected in the conserved regions of the targeted protein that had a high potential of causing damage to OXPHOS activities (Table S2). Predictors of amino acid changed pathogenic potential PolyPhen2 and SIFT revealed all 5 tumor-specific missense mutations to be probably damaging. Proof of mitochondrial functional impairment was obtained through assessment of lactate/pyruvate ratio, which was used to reflect a metabolic shift of the aerobic oxidation to glycolysis of tumor cells. We found that the tumor cells with any possibly pathogenic mtDNA mutations, including missense, nonsense, frame-shift mutations and tRNA mutation with a high MitoTIP score, had a significantly higher lactate/pyruvate ratio than those without them and the non-tumor cells (p = 0.025 and p < 0.001, respectively) (Figure 2). It indicated that metabolic shift of the aerobic oxidation to glycolysis in tumor cells was associated with heteroplasmic pathogenic somatic mtDNA mutations. It is worth mentioning that 62.5% (10/16) of tumor samples harbored such pathogenic heteroplasmic mtDNA somatic mutations. Obviously, the result is consistent with the previously reported that heteroplasmic possibly damaging mtDNA mutations are high frequent molecular events in human cancers.11,19,20 By comparing and analyzing the characteristics of sequence variants between platinum-resistant relapsed and platinum-sensitive patients, the result has established a high frequency of pathogenic heteroplasmic mtDNA somatic mutations in HGSOC.
Conclusion
Consistent with the mentioned theoretical analysis, these results suggested that mild pathogenic heteroplasmic mtDNA mutations frequently occur in the HGSOC and likely confer a selective advantage for tumor cells in development and progression such as acquisition of platinum-based chemotherapy resistance. Further molecular evolution and genetic research through single-cell analysis in the area would be needed. Moreover, our results also implied that the mild load of pathogenic mtDNA mutations in the tumor cells may be a potential biomarker for predicting development of chemoresistance and recurrence.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
The committee’s reference number was Jiangsu Cancer Hospital’s Ethical Committee 2020-05-01.
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 81501205 and 81472441), the Six Major Talent Summit of Jiangsu (grant number 2018-WSW-063), Youth Medical Talent of Jiangsu Province (grant number QNRC2016665), and the Institute level project of Jiangsu Cancer Hospital (grant number ZM201804).
Disclosure
The authors declare no conflicts of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492
2. Prat J, D’Angelo E, Espinosa I. Ovarian carcinomas: at least five different diseases with distinct histological features and molecular genetics. Hum Pathol. 2018;80:11–27. doi:10.1016/j.humpath.2018.06.018
3. Kim J, Park EY, Kim O, et al. Cell origins of high-grade serous ovarian cancer. Cancers (Basel). 2018;10:433.
4. Davis A, Tinker AV, Friedlander M. “Platinum resistant” ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol Oncol. 2014;133:624–631.
5. Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257.
6. Li H, Li J, Gao W, et al. Systematic analysis of ovarian cancer platinum-resistance mechanisms via text mining. J Ovarian Res. 2020;13:27. doi:10.1186/s13048-020-00627-6
7. Feeley KP, Bray AW, Westbrook DG, et al. Mitochondrial genetics regulate breast cancer tumorigenicity and metastatic potential. Cancer Res. 2015;75:4429–4436. doi:10.1158/0008-5472.CAN-15-0074
8. Badrinath N, Yoo SY. Mitochondria in cancer: in the aspects of tumorigenesis and targeted therapy. Carcinogenesis. 2018;39:1419–1430. doi:10.1093/carcin/bgy148
9. Chang JC, Chang HS, Wu YC, et al. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J Exp Clin Cancer Res. 2019;38:30. doi:10.1186/s13046-019-1028-z
10. Park JS, Sharma LK, Li H, et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum Mol Genet. 2009;18:1578–1589. doi:10.1093/hmg/ddp069
11. Larman TC, DePalma SR, Hadjipanayis AG, et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci U S A. 2012;109(35):14087–14091. doi:10.1073/pnas.1211502109
12. Maybury BD. Mitochondrial DNA damage is uncommon in cancer but can promote aggressive behaviour. Anticancer Res. 2013;33:3543–3552.
13. Kenny TC, Hart P, Ragazzi M, et al. Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPR(mt) to promote metastasis. Oncogene. 2017;36:4393–4404. doi:10.1038/onc.2017.52
14. Nissanka N, Moraes CT. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020;21:e49612. doi:10.15252/embr.201949612
15. Lott MT, Leipzig JN, Derbeneva O, et al. mtDNA variation and analysis using mitomap and mitomaster. Curr Protoc Bioinformatics. 2013;44::
16. Brandon MC, Lott MT, Nguyen KC, et al. MITOMAP: a human mitochondrial genome database–2004 update. Nucleic Acids Res. 2005;33:D611–613. doi:10.1093/nar/gki079
17. Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. 2012;13:780–788. doi:10.1038/nrm3479
18. Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. 2017;1858:686–699. doi:10.1016/j.bbabio.2017.01.012
19. Liu VW, Shi HH, Cheung AN, et al. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res. 2001;61:5998–6001.
20. VanTrappen PO, Cullup T, Troke R, et al. Somatic mitochondrial DNA mutations in primary and metastatic ovarian cancer. Gynecol Oncol. 2007;104(1):129–133. doi:10.1016/j.ygyno.2006.07.010
21. Earp MA, Brooks-Wilson A, Cook L, et al. Inherited common variants in mitochondrial DNA and invasive serous epithelial ovarian cancer risk. BMC Res Notes. 2013;6:425. doi:10.1186/1756-0500-6-425
22. Han SW, Yang X, Pan YF. Can mitochondria DNA provide a novel biomarker for evaluating the risk and prognosis of colorectal cancer? Dis Markers. 2017;2017:5189803.
23. Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3:1. doi:10.1186/s40170-015-0128-2
24. Sharma LK, Fang H, Liu J, et al. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet. 2011;20:4605–4616. doi:10.1093/hmg/ddr395
25. Petros JA, Baumann AK, Ruiz-Pesini E, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719–724. doi:10.1073/pnas.0408894102
26. Ishikawa K, Takenaga K, Akimoto M, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi:10.1126/science.1156906
27. Imanishi H, Hattori K, Wada R, et al. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PloS One. 2011;6:e23401. doi:10.1371/journal.pone.0023401
28. Singh RK, Saini S, Verma D, et al. Mitochondrial ND5 mutation mediated elevated ROS regulates apoptotic pathway epigenetically in a P53 dependent manner for generating pro-cancerous phenotypes. Mitochondrion. 2017;35:35–43. doi:10.1016/j.mito.2017.05.001
29. Iommarini L, Kurelac I, Capristo M, et al. Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum Mol Genet. 2014;23:1453–1466. doi:10.1093/hmg/ddt533
30. Gasparre G, Porcelli AM, Bonora E, et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci U S A. 2007;104:9001–9006. doi:10.1073/pnas.0703056104
31. Guerra F, Perrone AM, Kurelac I, et al. Mitochondrial DNA mutation in serous ovarian cancer: implications for mitochondria-coded genes in chemoresistance. J Clin Oncol. 2012;30:e373–378. doi:10.1200/JCO.2012.43.5933
32. Girolimetti G, De Iaco P, Procaccini M, et al. Mitochondrial DNA sequencing demonstrates clonality of peritoneal implants of borderline ovarian tumors. Mol Cancer. 2017;16:47. doi:10.1186/s12943-017-0614-y
33. Perrone AM, Girolimetti G, Procaccini M, et al. Potential for mitochondrial DNA sequencing in the differential diagnosis of gynaecological malignancies. Int J Mol Sci. 2018;19:2048. doi:10.3390/ijms19072048
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.