Back to Journals » Journal of Inflammation Research » Volume 16
Pathogenesis and Therapy of Coagulation Disorders in Severe Acute Pancreatitis
Authors Gui M ![]() , Zhao B, Huang J, Chen E, Qu H, Mao E
, Zhao B, Huang J, Chen E, Qu H, Mao E
Received 9 September 2022
Accepted for publication 12 November 2022
Published 6 January 2023 Volume 2023:16 Pages 57—67
DOI https://doi.org/10.2147/JIR.S388216
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Menglu Gui,1,* Bing Zhao,1,* Jun Huang,2,* Erzhen Chen,1 Hongping Qu,3 Enqiang Mao1
1Department of Emergency, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 2Department of Cardiovascular Medicine, State Key Laboratory of Medical Genomics, Shanghai Key Laboratory of Hypertension, Shanghai Institute of Hypertension, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 3Department of Critical Care Medicine, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Enqiang Mao, Department of Emergency, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, 197 Ruijin 2nd Road, Huangpu District, Shanghai, People’s Republic of China, Tel +86 13501747906, Email [email protected]
Abstract: Ischemia superimposed upon pancreatic edema leads to acute necrotizing pancreatitis. One possible mechanism contributing to ischemia is intravascular thrombogenesis since fibrin deposits have been detected in pancreatic capillaries by electron microscope. Current experimental and clinical data provided compelling evidence that the disorders in the blood coagulation system play a critical role in the pathogenesis of severe acute pancreatitis (SAP). This leads to microcirculatory failure of intra- and extrapancreatic organs and multiple organ failure and increases the case fatality rate. However, the mechanism of coagulopathy underlying SAP is not yet clear, although some anticoagulant drugs have entered clinical practice showing improvement in prognosis. Thus, enhanced understanding of the process might improve the treatment strategies with safety and high efficacy. Herein, the pathogenesis of the coagulation system of SAP was reviewed with a focus on the coagulation pathway, intercellular interactions, and complement system, thereby illustrating some anticoagulant therapies and potential therapeutic targets.
Keywords: coagulation, acute pancreatitis, endothelial cells, complement, anticoagulant therapy
Introduction
According to a survey of global epidemiology, the pooled incidence of AP is 34 cases per 100,000 individuals in the general population per year with 1.16 cases of mortality.1 The mortality among patients with persistent organ failure (OF) and pancreatic necrosis (PN) may be as high as 30–40%.2 AP has been viewed primarily as an autodigestion of the pancreas and its surroundings caused by the uncontrolled unleashing of pancreatic enzymes leading to the dysfunction of the gland, remote organs, and systems.3 Recently, it has been shown that ischemia superimposed upon pancreatic edema leads to the development of acute necrotizing pancreatitis (ANP).4 In addition, the microcirculation perfusion of extrapancreatic organs, such as the liver, kidney, is also affected.5 The possible mechanism is intravascular thrombogenesis since fibrin deposits have been detected in pancreatic capillaries by electron microscopy,4 ultimately leading to microcirculatory failure and deterioration of organ function and increasing the case fatality rate.6 Clinical studies revealed that the levels of plasma fibrinogen degradation products (FDP) are significantly higher in AP patients compared to healthy individuals, and higher levels of FDP are related to severity,6,7 and portosplenomesenteric venous thrombosis occurs in about 17.86% of patients with early-stage SAP.8 These phenomena indicated that the hypercoagulable state during AP and the therapeutic strategies aimed at enhancing microcirculation by anticoagulant therapy might improve the outcomes in AP even if the therapy is delayed and pancreatic necrosis cannot be influenced.5 Typically, with the progress of research on the coagulation mechanisms and drugs, safe and effective anticoagulant drugs, such as low molecular weight heparin (LMWH) and thrombomodulin (TM) have been administered in AP patients with satisfactory effects.9–11 However, the molecular mechanism of coagulation disorders underlying SAP and the choice of the anticoagulant drugs, the timing, and the amount is yet to be clarified.
With increasing incidence, AP has been associated with a significant socioeconomic burden since patients frequently suffer from diabetes mellitus, pancreatic exocrine insufficiency, and chronic pancreatitis after clinical resolution of AP; these events are associated with severity.1,12 This phenomenon highlights the urgent need for therapeutic agents with high safety and efficacy to improve the microcirculatory disturbances and prevent the progression of AP in the early phase. Herein, we summarized the disorders in the coagulation system during SAP with emphasis on the mechanism of anticoagulant therapies, potential therapeutic targets and their development that would provide a reference for clinical and basic science research.
Mechanism of Coagulation Disorders in Severe Acute Pancreatitis (SAP)
The microcirculation perfusion of intra- and extrapancreatic organs, such as the liver, kidney, are impaired in SAP.5 One reason may be the intravascular thrombosis, since fibrin deposits have been detected in pancreatic capillaries by electron microscopy,4 and the process of coagulation in blood samples collected from SAP patients starts in 3 min and culminates within 5 min.13 Thus, the local and systemic coagulopathy, which is in the range between scattered intravascular thrombosis in pancreatic microcirculation to disseminated intravascular coagulation (DIC),14,15 causes tissue damage or multiple organ failure (MOF).4 Previous studies have shown that inflammation shifts the hemostatic mechanisms in favor of thrombosis.15 In AP, one of the early events is the pancreas autodigestion due to premature trypsinogen activation.16 Injured acinar cells release chemokines, cytokines, and various adhesion molecules into the circulatory system, which in turn recruit and mediate the infiltration of immune cells to the site of injuries.17 This may encompass the key molecular and intercellular interactions related to triggers and pathways of the coagulation system that characterize the clotting disorders during SAP.
Role of Tissue Factor (TF) in the Coagulation Disorder of SAP
TF is localized in all blood-tissue barriers, which starts the coagulation rapidly when the endothelial barrier is disrupted. During inflammation, the major source of TF is the circulatory system from monocytes.18 In AP patients, the procoagulant inflammatory cytokines, such as interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF),19 promote monocytes and endothelial cells (ECs) to secrete TF-containing microparticles (MPs).20 Proteinase 3 released by activated neutrophils induces TF from the ECs via protease-activated receptor-1.21 On the other hand, pancreatic disruption leads to direct exposure of TF to the blood.22 A high level of TF triggers the extrinsic pathway by binding factor VII/factor VIIa (FVII/FVIIa) to form TF-FVIIa complex, converting factor X (FX) to factor Xa (FXa). Then, FXa is incorporated into FXa-factor Va-Ca2+-phospholipids (FXa-FVa-Ca2+-PLs) complex known as the prothrombinase23 (Figure 1). Finally, thrombin is formed, leading to fibrin clots. Thus, the role TF plays inside blood vessels is consistent with the clinical studies, wherein high levels of FDP were associated with increased mortality and organ failure (OF) in SAP patients.6,24 Besides, TF-levels are raised early in SAP, and it is a favourable predictive marker of SAP.25 As the initiator of the coagulation cascades, TF might play a large part in the development of SAP. There needs to be a more basic experimental to explore their relationship.
 |
Figure 1 Hemostatic and inhibitor systems in plasma. The blood coagulation system is activated in two ways:23 TF-FVIIa activates FX; factor XII (FXII) via high molecular weight kininogen (HK), and plasma kallikrein (PK) activates factor XI (FXI) to factor XIa (FXIa), subsequently FXIa activate factor IX (FIX). Both pathways lead to FXa-FVa-Ca2+-PLs complex formation, catalyzing prothrombin (FII) to thrombin (FIIa) and ultimately, the synthesis of fibrin. Thrombin formation is regulated by three systems: (1) the tissue factor pathway inhibitor (TFPI) inactivates FX by inhibiting TF-FVIIa;29 (2) activated protein C (APC) along with its cofactor, thrombomodulin (TM) inactivates FVa and factor VIIIa (FVIIIa);34 (3) antithrombin (ATIII) and its cofactor heparin inhibits FXa, FIIa, TF-FVIIa, and factor IXa.44 The fibrinolytic system:50,53 tissue-type plasminogen activator (t-PA) and urokinase-type PA (u-PA) transform plasminogen into plasmin, which is inhibited by PA inhibitor-1 (PAI-1) and thrombin-activatable fibrinolysis inhibitor (TAFI), then the plasmin hydrolyzs fibrin. Warfarin inhibits FII, FVII, FIX, and FX by interfering with vitamin K epoxide reductase complex 1 (VKORC1).83 The coagulation is influenced by the increased release of TF and decreased TFPI, ATIII, and APC-TM mediate by SAP. |
Role of Endothelial Cells in the Coagulation Disorder of SAP
The endothelium lines the lumen of the entire circulatory system, separating blood and subendothelial, and maintaining vascular health by exerting antiplatelet, anticoagulant, and anti-inflammatory actions via TFPI, APC system, ATIII and fibrinolysis.26 During AP, the functions of the endothelium are impaired, leading to the evolution of pancreatic microcirculatory dysfunction.27
TFPI
TFPI is the inhibitor of TF-mediated coagulation, primarily synthesized by ECs.28 It binds to ECs via proteoglycans (PGs)/glycosaminoglycans (GAGs)28 and inactivates TF-FVIIa-FXa complex and prothrombinase in the early phase of the coagulation process29 (Figure 1). The deficiency of TFPI increases susceptibility to the development of DIC and thrombosis.30
However, in AP patients, the levels of TFPI are elevated, which is markedly associated with PN and OF. In one clinical study, the levels of free TFPI and free/total TFPI ratios were high in SAP patients, whereas the endogenous thrombin is not different between the patients in mild AP and severe AP,31 meaning that the TFPI loses the biological activity. Another study drew a similar conclusion since TF/TFPI ratio in non-survivors was lower than survivors in ANP.32 The major source of increased plasma TFPI is the injured ECs, which is an adequate marker of endothelial injury in AP.32 The raised level of TFPI cannot counteract hypercoagulability caused by vascular injury.
In conclusion, in the early stage of SAP, TF is highly upregulated.30,31 As the initiator of the coagulation, TF contributes to hypercoagulability,33 while the level of TFPI is also upregulated significantly. The high level of TFPI just represents the severity of ECs injuries that are the main and explicit reason for the progression of AP. As a result, TFPI is insufficient in contrast to the hypercoagulable state, and exogenous administration of recombinant TFPI may protect against thrombosis, impeding the progression from MAP to SNP.
Protein C (PC) System
The PC system harbors PC, its cofactor protein S, TM, and endothelial cell protein C receptor (EPCR), a transmembrane glycoprotein present on the surface of ECs. Activated PC (APC) exerts potent anticoagulation by inactivating FVa and FVIIIa34 (Figure 1). EPCR increases the efficiency of APC generation by presenting PC zymogen to thrombin/TM complex, of which TM acts as a critical cofactor in the activation of PC.35
However, the PC system is damaged in AP patients characterized by low levels of PC and APC, high APC/PC ratios,7,36 and significantly increased levels of plasma soluble TM (sTM) and EPCR.37 Two mechanisms might contribute to this phenomenon. First, elastase and proteinase 3 are released by neutrophils and cleave TM and EPCR from the cell surface, and TM is oxidized and consequently less active form.38,39 Second, the activation of the nuclear factor-kappa B pathway increases their shedding from the endothelial surface.40 As a result, the antithrombotic effectiveness of the PC system is impaired, simplifying the development of DIC.35 Surprisingly, sTM in plasma can still impair the propagation phase of coagulation via rapid activation of PC.41 This phenomenon is consistent with the clinical application of recombination TM (rTM) in AP patients complicated by DIC,10,11 which is highly beneficial. This phenomenon means that the function of PC is preserved partially. Coagulopathy in SAP patients is not alleviated by APC treatment but worsened compared to the placebo group.42 The reason may be that the APC leads to excessive anticoagulation during consumptive coagulopathy or disrupts the balance mediated by APC that is yet to be elucidated. Therefore, low PC level in plasma reflects the severity of AP,7,36,43 while recombinant APC is clinically infeasible.
ATIII
ATIII is a serine protease inhibitor produced by the liver. It inactivates FXa, thrombin, TF-FVIIa, and FIXa (Figure 1). The cofactor heparin alters its conformation by direct binding to inhibit thrombin by 1000-fold. It also exerts an anticoagulatory effect by binding to specific heparan sulfates of PGs/GAGs that cover the endothelial surface, protecting the PGs/GAGs and maintaining the endothelial barrier function.44
However, in AP patients, the level of ATIII decreases as severity increases, which is rather pronounced in cases of biliary AP.7 This phenomenon could be ascribed to a combination of impaired synthesis because of the negative acute-phase response, degradation by elastase, and consumption because of thrombin generation.30,45 ATIII deficiency can result in severe venous thromboembolism46 and is positively correlated with the severity of DIC.47 On the other hand, ATIII (1500 units/day for 3 days when baseline plasma ATIII activity ≤69%) alone does not improve the DIC score in patients with acute cholangitis.48 Conversely, in septic patients complicated by DIC, high-dose ATIII (30,000 IU in total over 4 days) without concomitant heparin significantly reduces septic coagulative response or mortality compared with the placebo group.49 These clinical studies may guide the use of ATIII in AP patients. The application of ATIII individually cannot effectuate its anticoagulation function. Thus, increasing the dose improves excessive coagulative response; however, the risk of hemorrhage cannot be ignored. Thus, clinical studies using ATIII combined with an appropriate dose of heparin are recommended.
Fibrinolysis
Tissue-type PA (t-PA) and u-PA released by ECs are the main activators in the fibrinolytic system, which transform plasminogen into plasmin. PAI-1 and TAFI are the regulators of the fibrinolytic systems, of which PAI-1 is the principal inhibitor50,51 (Figure 1).
In AP patients, the level of TAFI rises at the onset of the disease.45 Patients with OF have significantly higher plasma levels of PAI-1, and non-survivors demonstrate more potent suppression of fibrinolysis than survivors.24,52 The high levels of fibrinolytic inhibitors may be the stimulation of proinflammatory factors and hypoxia.53 The marked increase in PAI-1 level causes fibrinolytic shutdown, subsequently failing to counteract the systemic deposition of fibrin clots during system inflammatory reaction syndrome, leading to thrombosis and DIC.50 However, DIC further developed into hyperfibrinolytic states.54 Hence, the fibrinolysis system with orchestrated regulation is the same complex as the coagulation system and is poorly understood. Strikingly, global assays of fibrinolysis are promising in predicting the risk of thrombosis but not bleeding.54 A better understanding of the fibrinolytic system alterations during SAP allows us to set up an effective plan for thromboprophylaxis and decrease the incidence of malignant coagulopathy.
In conclusion, during SAP, the endothelial function is disrupted due to the disorders in TFPI, APC system, ATIII, and fibrinolysis. These indexes predict the severity of the SAP, and they can also be potential targets that prevent the progress of SAP.
Roles of Platelets, Leukocyte, and Their Interactions in Coagulation Disorder of SAP
Platelets are essential cellular components of the coagulation system. Activated by thrombin and inflammatory mediators,55 platelets mediate vascular permeability, leukocyte chemotaxis, and synthesis of inflammatory factors, ultimately leading to platelet-leukocyte-endothelial interactions27,56 caused by P-selectin, and neutrophil extracellular traps (NETs), which partially account for coagulopathy in SAP patients.
P-Selectin
P-selectin stored in granular structures of ECs and platelets can be quickly mobilized towards the cell surface upon stimulation.57 Its ligand P-selectin glycoprotein ligand-1 (PSGL-1) expressed on platelets, monocytes, and neutrophils mediate leukocyte and platelet rolling on the vascular wall58,59 as well as platelet-neutrophil and platelet–platelet aggregations to link inflammatory infiltration and thrombus formation58 (Figure 2). Notably, monocyte-derived, TF-containing MPs fail to incorporate in thrombi when infused into P-selectin null mice, indicating that the accumulation of leukocyte-derived TF in growing thrombi is mediated by PSGL-1 on the MPs.60
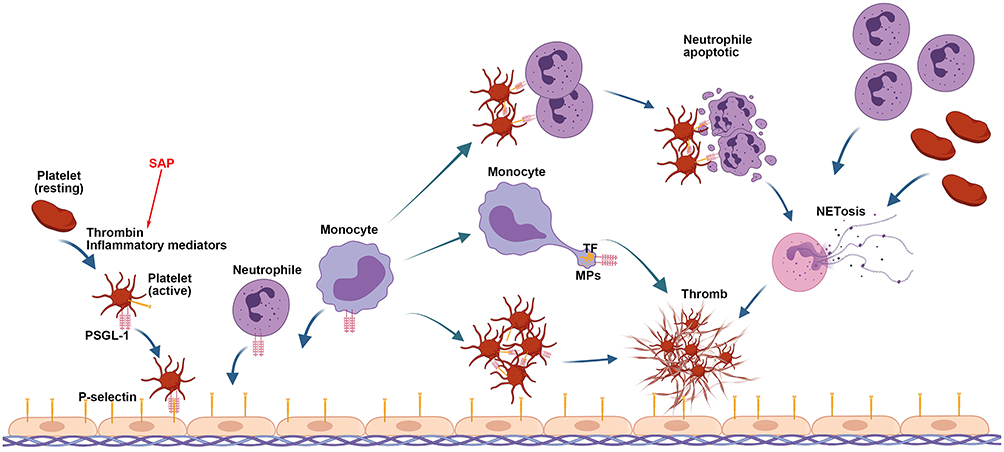 |
Figure 2 Role of P-selectin and NETs in thrombosis. The interaction of P-selectin, expressed on stimulated endothelial cells and activated platelets, with PSGL-1 mediates the rolling of these cells on vascular endothelial layers, platelet–neutrophil, platelet-platelet aggregations, and incorporation in thrombi of monocyte-derived TF in capillary venules. The activated platelets mediate neutrophil necroptosis, NET formation, and NET-MP aggregates to induce thrombin generation. In turn, NETs recruit more platelets and neutrophils to reinforce each other, damaging the endothelium. The initiator is inflammatory mediator released in the early stage of SAP. |
In AP patients, enhanced P-selectin expression is related to severity.61 The elevated level of P-selectin markedly strengthens the leukocyte–endothelium interaction and the thrombosis.62 Suppressing P-selectin inhibits leukocyte and platelet rolling in postcapillary venules of the inflamed pancreas,63 protecting against thrombosis64 and improving pancreatic microcirculation and histopathology of acinar necrosis without causing any bleeding complications.65 Thus, P-selectin plays a critical role in thrombosis by mediating the aggregation of platelets and leukocytes on the stimulated ECs, thereby contributing to the development of AP. Animal studies have shown that PSGL-1 or P-selectin is a safe and effective target against thrombosis that is promising in improving the prognosis of AP patients.
NETs
NETs are a meshwork of DNA fibers comprising histones, antimicrobial proteins, and high-mobility group box 1 (HMGB1), exhibiting antimicrobial functions by trapping and killing extracellular pathogens in infectious diseases.66 During inflammation, neutrophils are recruited first after endothelial injury and activation;67,68 subsequently, activated platelets induce neutrophil necroptosis and NET formation69 (Figure 2). NETs enhance thrombin generation,68 promote granulocyte-platelet and platelet aggregation via a collaborative interaction between histone, platelets, and inorganic polyphosphate,66,69 and further activate the platelets.70
In AP patients, the plasma levels of NET components increase significantly compared to the controls.71 Platelets regulate the formation of NETs and NET-MP aggregations, inducing thrombin generation,71,72 and in turn, NETs recruit platelets and neutrophils, reinforcing each other and injuring the endothelium within pancreatic microvasculature71,73 (Figure 2). These phenomena reveal that except for strengthening the antimicrobial potential in infectious diseases and contributing to normal hemostasis, NETs also impair organ perfusion during AP by amplifying the systemic or local thrombosis. Neutrophil recruitment and activation at the site of endothelial damage is considered as the initial and indispensable event in thrombus formation.67 Therefore, the inhibition of the recruitment of neutrophils or the formation of NETs can serve as a therapeutic target to improve the outcomes in AP.
Therefore, platelets are cellular components of the coagulation system and act as a bridge between coagulation and inflammation by interacting with the leukocyte, strengthening each other, ultimately aggravating the severity of SAP. Suppressing these key intersections may improve the outcomes.
Influence of the Complement System in SAP Coagulation
The complement system shares a common origin with the coagulation system and influences each other. It is activated through proteolytic cascades,74 leading to the formation of membrane attack complexes, ultimately polymerizing and inducing lysis of the cellular target.75 In the early stage of AP, C3 and C4 decrease significantly, suggesting the participation of the complement system.76 Recent studies have shown specific crosstalk between complement and coagulation in AP patients. First, in addition to activation by serine proteases, granzyme B and trypsin also cleave the central complement components, generating C3a and C5a.77 Second, C3a and C5a release TF from monocytes and ECs74,75 and promote platelet activation, leading to thrombogenesis.75,78 Moreover, C3 is essential for the recruitment of neutrophils into the pancreas and NET formation.79 The products of the complement system create a procoagulant environment, enhancing the activation of platelets, granulocytes, and ECs, increasing the microcirculation thrombosis and pancreatic injury. Thus, inhibiting some intersections that magnify the clotting response might improve the microcirculatory perfusion without increasing the risk of bleeding.
Interactions Between Platelets, Leukocyte and Complement System
The complement and coagulation systems do not function independently. Platelets and complement systems activate and enhance mutually via P-selectin, a receptor for C3b, reinforcing platelet adhesion, spreading, and aggregation.78 After C3a and C5a complement activation, the direct products also stimulate the platelets and promote coagulation by stimulating phosphatidylserine exposure.75,78 Platelets and C3 are indispensable for NET formation and granulocyte–platelet aggregation66,69 by mediating the recruitment and necrosis of neutrophils.79 Subsequently, NETs recruit more neutrophils, activate trypsin, and injury normal tissues.73 P-selectin and NETs emerge as intersection points between the complement cascade and coagulation system, forming a vicious cycle in the development of SAP. Animal studies have suggested that inhibition of P-selectin and NETs ameliorates tissue damage and pancreatic microcirculatory perfusion by reducing leukocyte-platelet-endothelium interaction63,80 and improves survival.81 Currently, these therapies are useful as they do not disrupt the normal hemostasis in experimental models but need an in-depth exploration before clinical trials.
Anticoagulation Therapy in AP Patients
Overactivation of clotting cascades and deterioration of anticoagulation and fibrinolysis functions aggravate the microcirculatory disturbance, leading to pancreatic necrosis and venous thrombosis.82 Pharmaceutical management of the coagulation system may ameliorate the severity and mortality of AP. In clinical practice, some anticoagulation therapies have made significant progress.
Vitamin K Antagonists
Warfarin inhibits the generation of vitamin K-dependent coagulation factors (Figure 1).83 The pretreatment with low doses of warfarin (90 or 180 µg/kg/day once a day for 7 days before induction of AP) inhibits the development of ischemia/reperfusion-induced AP in rats, reduces the activity of serum lipase and amylase and levels of D-Dimer, and pancreatic damage by reversing the decline in pancreatic blood flow without hemorrhage;84 however, the delayed effects of warfarin treatment should also be considered. The use of vitamin K antagonists in animals harvests favorable results, thereby necessitating the translation of these findings into clinical trials.
APC
In animal studies, APC improves the severity of pancreatic histology and serum markers of inflammation in ANP rats.43 Although the PC pathway defects are associated with the progress of MOF,36 the coagulopathy in SAP with APC treatment is not improved in patients, and the normal homeostasis of coagulation is slower than the placebo group.42 Clinical studies did not find any specific benefits of the APC therapy, and it is infeasible to supply exogenous APC to AP patients to ameliorate prognosis.
ATIII
In AP patients, the level of ATIII decreases. The clinical studies of ATIII treatment are yet lacking. In an animal study, ATIII (500 μg/kg) was injected intravenously 30 min before or after the induction of SAP in rats, which in turn attenuated oxidative stress by decreasing TNF-α mRNA, and intercellular adhesion molecule-1 expression and protected against SAP-induced renal dysfunction by inhibiting renal cell apoptosis but did not have beneficial effects on pancreatic or liver injury.85 Animal study has shown the potential benefits of ATIII therapy, but more data are warranted to evaluate the benefits and risks in the future.
Thrombomodulin
TM functions are impaired in SAP patients.37 In a clinical study consisting of 38 patients with SAP complicated by DIC, rTM was administered to 7 patients at 130 IU/kg/day and to 6 patients at 380 IU/kg/day, for 1–6 days. rTM administration was started, on average, 58 hours after the onset of severe acute pancreatitis. No differences were detected between the groups except a significantly low platelet count at the beginning in the rTM treatment group. On day 6, DIC scores were significantly lower, and on day 60, the mortality rate was also significantly lower in the rTM group compared to conventional treatment group.10 In a retrospective analysis consisting of 54 SAP patients, 24 were treated with rTM at a dosage of 380 U/kg/day or 130 U/kg/day for patients on hemodialysis once these patients were diagnosed with DIC. The rTM treatment was maintained until DIC scores improved to a Japanese Association for Acute Medicine score of ≤3. Thus, patients who received rTM had severe disease, with high Acute Physiology and Chronic Health Evaluation (APACHE) II and sepsis-related organ failure assessment scores, FDP, and D-dimer, and low platelet count on admission. After a week, the differences disappeared, and no serious adverse events, such as bleeding, occurred in the treatment group even if the platelet count was markedly reduced.11 These findings suggested that rTM might improve the prognosis of survival in SAP patients accompanied by DIC, and no adverse reactions occurred.
LMWH
There are several small-scale clinical investigations about the administration of LMWH in SAP patients. In a multicenter prospective clinical study, 265 SAP patients were randomized to LMWH treatment group or conventional treatment group, all the parameters in the two groups were not significantly different. LMWH was administered at 100 µg/kg/day from the time of admission for one week. After treatment, in LMWH treatment group, the clinical presentation improvement rate (MOF 68.4% vs 32.8%, Symptoms 53.3% vs 26.7%) and laboratory parameter improvement (blood amylase 913±281 vs 1738±346) were significantly higher than those in conventional treatment group, and the acute physiology and chronic health evaluation (APACHE) II score (8.5 ± 1.8 vs 9.6± 2.4), complication rate (acute respiratory distress syndrome 12.6% vs 35.4%, pancreatic encephalopathy 2.2% vs 10.0%, GI bleeding 3.7% vs 4.6%), CT score (3.8 ± 2.2 vs 4.9 ± 2.4), mortality (10.4%, 14/135 vs 30.6%, 40/130) and mean hospital stay (30±8 days vs.43±11 days) in LMWH treatment group were obviously lower than those in conventional treatment group.9
In another single-center, prospective, randomized controlled study, a total of 100 patients whose symptoms started within 24 h with moderately severe and severe AP (MSAP) diagnosed by the revised Atlanta criteria were randomized to receive either conventional treatment or conventional treatment plus LMWH. There were 50 patients in each group. LMWH was administered at 1 mg/kg twice a day between days 1 and 7. After treatment, local and systemic complications (14% vs 34%) and PN (6.1% vs 22.9%) developed significantly less frequently in the LMWH group. There was a trend for shorter hospital stay and lower mortality in the LMWH group while they were not significantly different between the groups. No hemorrhagic complication occurred.86 Several other clinical studies and drew close conclusions.87,88
Thus, LMWH might be used as a simple, safe, economical, and effective method for the treatment of SAP with improved prognosis.
Other Therapies
Targeting NETs or P-Selectin
NETs and P-selectin play a critical role in microcirculatory pancreatic hypoperfusion. In animal studies, inhibition of P-selectin or NET formation attenuates organ injury and neutrophil recruitment in the inflamed pancreas80 and improves survival by improving pancreatic microcirculation.63,65,81 Presently, NET targeting is only carried out in animals as the risks are yet to be elucidated. Thus, targeting NETs or P-selectin in AP might be a promising approach in the management of thrombotic disorders.
Discussion
AP is a common and potentially life-threatening disease as the morbidity has been increasing dramatically due to ascending incidence of obesity that promotes gallstone formation, alcohol consumption, hypertriglyceridemia, and smoking.2 AP is associated with a large socioeconomic, psychological, and physiological burden.1,12 Reportedly, pancreatic microcirculatory disturbances aggravate histopathologic tissue damage and worsen the outcomes of SAP; deterioration of coagulation is a key to this pathological process.4 During AP, overactivation of clotting cascades and upregulation of procoagulant substances, insufficient anticoagulation and fibrinolysis increases fibrin formation and deposition leading to thrombosis of the microvasculature, which can impair organ perfusion and result in the production of organ system dysfunction. So, improvement of pancreatic and systemic microvascular disturbance by interrupting the activation of the clotting system is very important in blocking the pathological and clinical process of the development of pancreatitis.
The present review illustrates that LMWH treatment in SAP patients or rTM treatment in SAP patients complicated by DIC improves the severity and mortality rate in both short- and long-term, and peripheral perfusion and systemic oxygenation without serious adverse events.9–11 While these clinical trials were too small to demonstrate a statistical survival benefit, the results were encouraging enough to carry out larger, multicenter clinical trials designed to evaluate the potential treatment benefit of LMWH and rTM replacement in SAP. In preclinical studies, ATIII and low doses of warfarin reverse the decreased blood flow.84,85 Animal studies have shown favorable effects, while studies of standard therapy regimens are lacking in AP patients, necessitating the translation of these findings into clinical trials under intensive monitoring. Taken together, the activation of the hemostatic system has been ascribed a critical pathophysiological role in AP, which causes pancreatic perfusion failure, ischemia, and tissue necrosis. Thus, a single-target therapy may be insufficient, requiring novel drugs. Large-scale clinical trials are needed to identify the appropriate drug and the adequate dose under various clinical situations.
Data Sharing Statement
All data generated or analysed during this study are included in this published article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Menglu Gui, Bing Zhao, and Jun Huang contributed equally to this article as co–first authors.
Funding
The authors report no funding was received for this work.
Disclosure
The authors declare that they have no conflicts of interest in relation to this work.
References
1. Petrov MS, Yadav D. Global epidemiology and holistic prevention of pancreatitis. Nat Rev Gastroenterol Hepatol. 2019;16(3):175–184. doi:10.1038/s41575-018-0087-5
2. Iannuzzi JP, King JA, Leong JH, et al. Global incidence of acute pancreatitis is increasing over time: a systematic review and meta-analysis. Gastroenterology. 2022;162(1):122–134. doi:10.1053/j.gastro.2021.09.043
3. Padureanu V, Florescu D, Pădureanu R, et al. Role of antioxidants and oxidative stress in the evolution of acute pancreatitis (review). Exp Ther Med. 2022;23(3):197. doi:10.3892/etm.2022.11120
4. Cuthbertson CM, Christophi C. Disturbances of the microcirculation in acute pancreatitis. Br J Surg. 2006;93(5):518–530. doi:10.1002/bjs.5316
5. Foitzik T, Eibl G, Hotz B, et al. Persistent multiple organ microcirculatory disorders in severe acute pancreatitis: experimental findings and clinical implications. Dig Dis Sci. 2002;47(1):130–138. doi:10.1023/A:1013284008219
6. Liu C, Zhou X, Ling L, et al. Prediction of mortality and organ failure based on coagulation and fibrinolysis markers in patients with acute pancreatitis: a retrospective study. Medicine. 2019;98(21):e15648. doi:10.1097/MD.0000000000015648
7. Yang N, Hao J, Zhang D, Antithrombin III. D-dimer levels as indicators of disease severity in patients with hyperlipidaemic or biliary acute pancreatitis. J Int Med Res. 2017;45(1):147–158. doi:10.1177/0300060516677929
8. Ding L, Deng F, Yu C, et al. Portosplenomesenteric vein thrombosis in patients with early-stage severe acute pancreatitis. World J Gastroenterol. 2018;24(35):4054–4060. doi:10.3748/wjg.v24.i35.4054
9. Lu XS, Fu Q, Jie-Qin L, et al. Low molecular weight heparin in the treatment of severe acute pancreatitis: a multiple centre prospective clinical study. Asian J Surg. 2009;32(2):89–94. doi:10.1016/S1015-9584(09)60017-8
10. Yano T, Taniguchi M, Shirasaka T, et al. Effectiveness of soluble recombinant human thrombomodulin in patients with severe acute pancreatitis complicated by disseminated intravascular coagulation. Turk J Anaesthesiol Reanim. 2019;47(4):320–326. doi:10.5152/TJAR.2019.42709
11. Eguchi T, Tsuji Y, Yamashita H, et al. Efficacy of recombinant human soluble thrombomodulin in preventing walled-off necrosis in severe acute pancreatitis patients. Pancreatology. 2015;15(5):485–490. doi:10.1016/j.pan.2015.08.002
12. Hollemans RA, Hallensleben NDL, Mager DJ, et al. Pancreatic exocrine insufficiency following acute pancreatitis: systematic review and study level meta-analysis. Pancreatology. 2018;18(3):253–262. doi:10.1016/j.pan.2018.02.009
13. Zhu R, Wei S, Wu C, et al. Utility of clot formation and lysis assay to monitor global coagulation state of patients with severe acute pancreatitis. Dig Dis Sci. 2012;57(5):1399–1403. doi:10.1007/s10620-012-2034-6
14. Maduzia D, Ceranowicz P, Cieszkowski J, et al. Administration of warfarin accelerates the recovery in ischemia/reperfusion-induced acute pancreatitis. J Physiol Pharmacol. 2020;71(3). doi:10.26402/jpp.2020.3.13.
15. Dumnicka P, Kuśnierz-Cabala B, Sporek M, et al. Serum concentrations of angiopoietin-2 and soluble fms-like tyrosine kinase 1 (sFlt-1) Are Associated with coagulopathy among patients with acute pancreatitis. Int J Mol Sci. 2017;18(4):753. doi:10.3390/ijms18040753
16. Saluja A, Dudeja V, Dawra R, et al. Early intra-acinar events in pathogenesis of pancreatitis. Gastroenterology. 2019;156(7):1979–1993. doi:10.1053/j.gastro.2019.01.268
17. Lee PJ, Papachristou GI. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol. 2019;16(8):479–496. doi:10.1038/s41575-019-0158-2
18. Macey MG, Wolf SI, Wheeler-Jones CPD, et al. Expression of blood coagulation factors on monocytes after exposure to TNF-treated endothelium in a novel whole blood model of arterial flow. J Immunol Methods. 2009;350(1–2):133–141. doi:10.1016/j.jim.2009.08.007
19. Pereda J, Sabater L, Aparisi L, et al. Interaction between cytokines and oxidative stress in acute pancreatitis. Curr Med Chem. 2006;13(23):2775–2787. doi:10.2174/092986706778522011
20. Grignani G, Maiolo A. Cytokines and hemostasis. Haematologica. 2000;85(9):967–972.
21. Dugina TN, Kiseleva EV, Chistov IV, et al. Receptors of the PAR family as a link between blood coagulation and inflammation. Biochemistry. 2002;67(1):65–74. doi:10.1023/A:1013952114485
22. Lisman T, Porte RJ. Activation and regulation of hemostasis in acute liver failure and acute pancreatitis. Semin Thromb Hemost. 2010;36(4):437–443. doi:10.1055/s-0030-1254052
23. Schmaier AH. Transferrin: a blood coagulation modifier. Cell Res. 2020;30(2):101–102. doi:10.1038/s41422-020-0275-z
24. Radenkovic D, Bajec D, Ivancevic N, et al. D-dimer in acute pancreatitis a new approach for an early assessment of organ failure. Pancreas. 2009;38(6):655–660. doi:10.1097/MPA.0b013e3181a66860
25. Andersson E, Axelsson J, Eckerwall G, Ansari D, Andersson R. Tissue factor in predicted severe acute pancreatitis. World J Gastroenterol. 2010;16(48):6128–6134. doi:10.3748/wjg.v16.i48.6128
26. Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. 2019;133(9):906–918. doi:10.1182/blood-2018-11-882993
27. Eibl G, Buhr HJ, Foitzik T. Therapy of microcirculatory disorders in severe acute pancreatitis: what mediators should we block? Intensive Care Med. 2002;28(2):139–146. doi:10.1007/s00134-001-1194-1
28. Yuan HQ, Hao Y-M, Ren Z, et al. Tissue factor pathway inhibitor in atherosclerosis. Clin Chim Acta. 2019;491:97–102. doi:10.1016/j.cca.2019.01.024
29. Mast AE. Tissue factor pathway inhibitor: multiple anticoagulant activities for a single protein. Arterioscler Thromb Vasc Biol. 2016;36(1):9–14. doi:10.1161/ATVBAHA.115.305996
30. Sungurlu S, Kuppy J, Balk RA. Role of antithrombin III and tissue factor pathway in the pathogenesis of sepsis. Crit Care Clin. 2020;36(2):255–265. doi:10.1016/j.ccc.2019.12.002
31. Lindstrom OK, Tukiainen EM, Kylänpää M-L, et al. Thrombin generation in vitro and in vivo, and disturbed tissue factor regulation in patients with acute pancreatitis. Pancreatology. 2011;11(6):557–566. doi:10.1159/000333481
32. Yasuda T, Ueda T, Kamei K, et al. Plasma tissue factor pathway inhibitor levels in patients with acute pancreatitis. J Gastroenterol. 2009;44(10):1071–1079. doi:10.1007/s00535-009-0096-9
33. Zelaya H, Rothmeier AS, Ruf W. Tissue factor at the crossroad of coagulation and cell signaling. J Thromb Haemost. 2018;16(10):1941–1952. doi:10.1111/jth.14246
34. Pendurthi UR, Rao LVM. Endothelial cell protein C receptor-dependent signaling. Curr Opin Hematol. 2018;25(3):219–226. doi:10.1097/MOH.0000000000000416
35. Dinarvand P, Moser KA. Protein C deficiency. Arch Pathol Lab Med. 2019;143(10):1281–1285. doi:10.5858/arpa.2017-0403-RS
36. Lindstrom O, Kylanpaa L, Mentula P, et al. Upregulated but insufficient generation of activated protein C is associated with development of multiorgan failure in severe acute pancreatitis. Crit Care. 2006;10(1):R16. doi:10.1186/cc3966
37. Chen Y, Ke L, Meng L, et al. Endothelial markers are associated with pancreatic necrosis and overall prognosis in acute pancreatitis: a preliminary cohort study. Pancreatology. 2017;17(1):45–50. doi:10.1016/j.pan.2016.12.005
38. Esmon CT. Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost. 2006;32(Suppl 1):49–60. doi:10.1055/s-2006-939554
39. Villegas-Mendez A, Montes R, Ambrose LR, et al. Proteolysis of the endothelial cell protein C receptor by neutrophil proteinase 3. J Thromb Haemost. 2007;5(5):980–988. doi:10.1111/j.1538-7836.2007.02480.x
40. Song D, Ye X, Xu H, et al. Activation of endothelial intrinsic NF-{kappa}B pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood. 2009;114(12):2521–2529. doi:10.1182/blood-2009-02-205914
41. Dargaud Y, Scoazec JY, Wielders SJH, et al. Characterization of an autosomal dominant bleeding disorder caused by a thrombomodulin mutation. Blood. 2015;125(9):1497–1501. doi:10.1182/blood-2014-10-604553
42. Kyhala L, Lindström O, Kylänpää L, et al. Activated protein C retards recovery from coagulopathy in severe acute pancreatitis. Scand J Clin Lab Invest. 2016;76(1):10–16. doi:10.3109/00365513.2015.1084041
43. Yamanel L, Mas M, Comert B, et al. The effect of activated protein C on experimental acute necrotizing pancreatitis. Crit Care. 2005;9(3):R184–90. doi:10.1186/cc3485
44. Schlommer C, Brandtner A, Bachler M. Antithrombin and its role in host defense and inflammation. Int J Mol Sci. 2021;22:8. doi:10.3390/ijms22084283
45. Fidan S, Erkut M, Cosar A, et al. Higher thrombin-antithrombin III complex levels may indicate severe acute pancreatitis. Dig Dis. 2018;36(3):244–251. doi:10.1159/000485613
46. Wang D, Tian M, Cui G, et al. Antithrombin deficiency and decreased protein C activity in a young man with venous thromboembolism: a case report. Front Med. 2018;12(3):319–323. doi:10.1007/s11684-017-0553-4
47. Iba T, Saitoh D. Efficacy of antithrombin in preclinical and clinical applications for sepsis-associated disseminated intravascular coagulation. J Intensive Care. 2014;2(1):66. doi:10.1186/s40560-014-0051-6
48. Nakahara K, Okuse C, Adachi S, et al. Use of antithrombin and thrombomodulin in the management of disseminated intravascular coagulation in patients with acute cholangitis. Gut Liver. 2013;7(3):363–370. doi:10.5009/gnl.2013.7.3.363
49. Kienast J, Juers M, Wiedermann CJ, et al. Treatment effects of high-dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. J Thromb Haemost. 2006;4(1):90–97. doi:10.1111/j.1538-7836.2005.01697.x
50. Madoiwa S. Recent advances in disseminated intravascular coagulation: endothelial cells and fibrinolysis in sepsis-induced DIC. J Intensive Care. 2015;3:8. doi:10.1186/s40560-015-0075-6
51. Urano T, Suzuki Y, Iwaki T, Sano H, Honkura N, Castellino FJ. Recognition of plasminogen activator inhibitor type 1 as the primary regulator of fibrinolysis. Curr Drug Targets. 2019;20(16):1695–1701. doi:10.2174/1389450120666190715102510
52. Nakajima T, Ueda T, Takeyama Y, et al. Protective effects of vascular endothelial growth factor on intestinal epithelial apoptosis and bacterial translocation in experimental severe acute pancreatitis. Pancreas. 2007;34(4):410–416. doi:10.1097/mpa.0b013e3180335c64
53. Kaji H. Adipose tissue-derived plasminogen activator inhibitor-1 function and regulation. Compr Physiol. 2016;6(4):1873–1896.
54. Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015;29(1):17–24. doi:10.1016/j.blre.2014.09.003
55. Fender AC, Rauch B, Geisler T, et al. Protease-activated receptor PAR-4: an inducible switch between thrombosis and vascular inflammation? Thromb Haemost. 2017;117(11):2013–2025. doi:10.1160/TH17-03-0219
56. Lordan R, Tsoupras A, Zabetakis I, et al. Forty years since the structural elucidation of platelet-activating factor (paf): historical, current, and future research perspectives. Molecules. 2019;24:23. doi:10.3390/molecules24234414
57. Zinellu A, Mangoni AA, Review S. and meta-analysis of the effect of statins on circulating E-selectin, L-selectin, and P-Selectin. Biomedicines. 2021;9:11. doi:10.3390/biomedicines9111707
58. Zhang X, Zhu M, Jiang XL, et al. P-selectin glycoprotein ligand 1 deficiency prevents development of acute pancreatitis by attenuating leukocyte infiltration. World J Gastroenterol. 2020;26(41):6361–6377. doi:10.3748/wjg.v26.i41.6361
59. Rossaint J, Zarbock A. Platelets in leucocyte recruitment and function. Cardiovasc Res. 2015;107(3):386–395. doi:10.1093/cvr/cvv048
60. Zwicker JI, Trenor CC, Furie BC, et al. Tissue factor-bearing microparticles and thrombus formation. Arterioscler Thromb Vasc Biol. 2011;31(4):728–733. doi:10.1161/ATVBAHA.109.200964
61. Tsaroucha AK, Schizas D, Vailas MG, et al. E and P selectins as potential markers in the assessment of the severity of acute pancreatitis. Pancreas. 2018;47(4):406–411. doi:10.1097/MPA.0000000000001009
62. Setiadi H, Yago T, Liu Z, et al. Endothelial signaling by neutrophil-released oncostatin M enhances P-selectin-dependent inflammation and thrombosis. Blood Adv. 2019;3(2):168–183. doi:10.1182/bloodadvances.2018026294
63. Hartman H, Abdulla A, Awla D, et al. P-selectin mediates neutrophil rolling and recruitment in acute pancreatitis. Br J Surg. 2012;99(2):246–255. doi:10.1002/bjs.7775
64. Jin H, Gebska MA, Blokhin IO, et al. Endothelial PPAR-gamma protects against vascular thrombosis by downregulating P-selectin expression. Arterioscler Thromb Vasc Biol. 2015;35(4):838–844. doi:10.1161/ATVBAHA.115.305378
65. Hackert T, Sperber R, Hartwig W, et al. P-selectin inhibition reduces severity of acute experimental pancreatitis. Pancreatology. 2009;9(4):369–374. doi:10.1159/000212098
66. Carestia A, Kaufman T, Rivadeneyra L, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol. 2016;99(1):153–162. doi:10.1189/jlb.3A0415-161R
67. Darbousset R, Thomas GM, Mezouar S, et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012;120(10):2133–2143. doi:10.1182/blood-2012-06-437772
68. Carminita E, Crescence L, Panicot-Dubois L, Dubois C. Role of neutrophils and NETs in animal models of thrombosis. Int J Mol Sci. 2022;23:3.
69. Nakazawa D, Desai J, Steiger S, et al. Activated platelets induce MLKL-driven neutrophil necroptosis and release of neutrophil extracellular traps in venous thrombosis. Cell Death Discov. 2018;4:6. doi:10.1038/s41420-018-0073-2
70. Zucoloto AZ, Jenne CN. Platelet-neutrophil interplay: insights into neutrophil extracellular trap (NET)-driven coagulation in infection. Front Cardiovasc Med. 2019;6:85. doi:10.3389/fcvm.2019.00085
71. Merza M, Hartman H, Rahman M, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. 2015;149(7):1920–1931 e8. doi:10.1053/j.gastro.2015.08.026
72. Madhi R, Rahman M, Taha D, et al. Platelet IP6K1 Regulates Neutrophil Extracellular Trap-Microparticle Complex Formation in Acute Pancreatitis. JCI Insight. 2019. doi:10.1172/jci.insight.129270
73. Hu J, Kang H, Chen H, et al. Targeting neutrophil extracellular traps in severe acute pancreatitis treatment. Therap Adv Gastroenterol. 2020;13:1756284820974913. doi:10.1177/1756284820974913
74. Dzik S. Complement and coagulation: cross talk through time. Transfus Med Rev. 2019;33(4):199–206. doi:10.1016/j.tmrv.2019.08.004
75. Conway EM. Complement-coagulation connections. Blood Coagul Fibrinolysis. 2018;29(3):243–251. doi:10.1097/MBC.0000000000000720
76. Zhang L, Qiao Z, Feng H, et al. The early predictive role of complement C3 and C4 in patients with acute pancreatitis. J Clin Lab Anal. 2020;34(5):e23205. doi:10.1002/jcla.23205
77. Bettac L, Denk S, Seufferlein T, et al. Complement in pancreatic disease-perpetrator or savior? Front Immunol. 2017;8:15. doi:10.3389/fimmu.2017.00015
78. Del Conde I, Cruz MA, Zhang H, et al. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201(6):871–879. doi:10.1084/jem.20041497
79. Linders J, Madhi R, Mörgelin M, King BC, Blom AM, Rahman M. Complement component 3 is required for tissue damage, neutrophil infiltration, and ensuring NET formation in acute pancreatitis. Eur Surg Res. 2021;2021:1–14.
80. Madhi R, Rahman M, Taha D, et al. Targeting peptidylarginine deiminase reduces neutrophil extracellular trap formation and tissue injury in severe acute pancreatitis. J Cell Physiol. 2019;234(7):11850–11860. doi:10.1002/jcp.27874
81. Murthy P, Singhi AD, Ross MA, et al. Enhanced neutrophil extracellular trap formation in acute pancreatitis contributes to disease severity and is reduced by chloroquine. Front Immunol. 2019;10:28. doi:10.3389/fimmu.2019.00028
82. Easler J, Muddana V, Furlan A, et al. Portosplenomesenteric venous thrombosis in patients with acute pancreatitis is associated with pancreatic necrosis and usually has a benign course. Clin Gastroenterol Hepatol. 2014;12(5):854–862. doi:10.1016/j.cgh.2013.09.068
83. Lin L, Zhao L, Gao N, et al. From multi-target anticoagulants to DOACs, and intrinsic coagulation factor inhibitors. Blood Rev. 2020;39:100615. doi:10.1016/j.blre.2019.100615
84. Maduzia D, Ceranowicz P, Cieszkowski J, et al. Pretreatment with warfarin attenuates the development of ischemia/reperfusion-induced acute pancreatitis in rats. Molecules. 2020;25:11. doi:10.3390/molecules25112493
85. Kong Y, Yin J, Cheng D, et al. Antithrombin III attenuates AKI following acute severe pancreatitis. Shock. 2018;49(5):572–579. doi:10.1097/SHK.0000000000000946
86. Tozlu M, Kayar Y, Ince AT, et al. Low molecular weight heparin treatment of acute moderate and severe pancreatitis: a randomized, controlled, open-label study. Turk J Gastroenterol. 2019;30(1):81–87. doi:10.5152/tjg.2018.18583
87. Lu XS, Qiu F, Li Y-X, et al. Effect of lower-molecular weight heparin in the prevention of pancreatic encephalopathy in the patient with severe acute pancreatitis. Pancreas. 2010;39(4):516–519. doi:10.1097/MPA.0b013e3181c3c954
88. Patil B, Meena LN, Sharma DC, et al. Impact of low-molecular-weight heparin in the treatment of moderately severe and severe acute pancreatitis; a randomized, single blind, Phase 3 control trial. Int J Surg. 2022;101:106621. doi:10.1016/j.ijsu.2022.106621
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.