Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Partial Thyroid Hormone-Binding Globulin Deficiency: A Case Report and Literature Review
Authors Liu X, Li S, Xiong J, Chen D, Jiang C, Zeng L, Qiu Y, Xia BW
Received 17 March 2023
Accepted for publication 21 June 2023
Published 26 July 2023 Volume 2023:16 Pages 2225—2232
DOI https://doi.org/10.2147/DMSO.S413048
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Xuefang Liu,* Suyan Li,* Jingni Xiong, Dandan Chen, Chan Jiang, Liankun Zeng, Youyan Qiu,* Bi-Wen Xia*
Department of Endocrine Medicine, The Fourth Affiliated Hospital of Guangzhou Medical University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Youyan Qiu; Bi-Wen Xia, Department of Endocrine Medicine, The Fourth Affiliated Hospital of Guangzhou Medical University, No. 1 Guangming East Road, Zengjiang Street, Zengcheng District, Guangzhou City, Guangdong, 511330, People’s Republic of China, Tel +86-020-62287186, Email [email protected]; [email protected]
Background: Thyroxine binding globulin (TBG) deficiency is a rare thyroid disease, mostly caused by genetic mutations and acquired by X-linked recessive inheritance. The clinical features of children with TBG deficiency and their family members were summarised and the Serpina7 gene mutation was analysed, providing a reference for the differentiation of TBG deficiency.
Methods: Thyroid function was detected in TBG deficient patients, and genetic analysis was performed using polymerase chain reaction (PCR) and direct DNA sequencing to detect the characteristics of TBG mutants. Using “thyroxine binding globulin, gene and mutation” as keywords, PubMed (biomedical literature database), Web of Science and other databases were searched for relevant studies to collect and summarise relevant information.
Results: The TBG (14.7 μg/mL), 70% triiodothyronine (T3) (< 0.3 nmol/L), total T3 (Tr3) (< 0.05 ng/mL) and thyroxine (T4) (14.72 nmol/L) values were lower than normal, while the thyrotropin (TSH) (2.33 uIU/mL), free T3 (FT3) (1.62 pmol/L), and free T4 (FT4) (11.39 pmol/L) values were normal. These values indicate a TBG partially deficient phenotype. Using PCR amplification and direct sequencing of the target gene, a missense mutation in exon 4 of the Serpina7 gene was found in the patient and the father, and the nucleic acid variant was C.909 (exon 4) g > T; the patient was heterozygous and the father was hemizygous. The literature search retrieved a total of 45 studies, most of which were related to mutations in the Serpina7 gene. The mutation locations included exons, introns, enhancers and promoters, with exons the predominant location. A total of 49 variants of the Serpina7 gene were identified.
Conclusion: Serpina7 C.909G (P.L303F) is a mutation acquired from the father by X-linked recessive inheritance. The main clinical features of TBG deficiency patients are low serum T4, T3 and TBG levels, normal TSH, FT3 and FT4 levels, and no clinical manifestations.
Keywords: thyroxine-binding globulin, deficiency, genes, mutation
Introduction
Thyroxine-binding globulin (TBG) is the main binding protein of the thyroid hormone in the human body. It combines around 75% of thyroxine (T4) and 70% of triiodofoxygen (T3).1 Thyroxine-binding globulin deficiency is a rare thyroid disease, mostly caused by a gene mutation, and is generally acquired through X-linked recessive inheritance.2 The gene encoding TBG is named the Serpina7 gene, while it is also known as the TBG gene. It is located on Xq22 2, spans 5.5 kb and contains five exons. This gene encodes a 54 kDa protein, a mature protein containing 395 amino acids, and is glycosylated by four glycosyl chains after translation.3 The Serpina7 gene mutation leads to the substitution of amino acid or the truncation of mature proteins, resulting in TBG deficiency or a decreased affinity with T4, or both.4 Genetic TBG defects, including changes in protein synthesis, secretion and stability, result in TBG variants.2 Reports from different countries have described how these mutations in the coding and non-coding regions of the Serpina7 gene lead to complete deficiency (TBG-CD) and partial deficiency (TBG-PD).5
Since the first report of the human Serpina7 gene in 1986,6 a series of mutations and polymorphisms have been identified. According to different serum TBG concentrations in patients, TBG deficiency has three phenotypes: TBG-CD, TBG-PD and excess (TBG-E).7 Thyroxine-binding globulin deficiency leads to a decrease in protein-bound thyroid hormones in the serum, resulting in a decrease in the total T4 (Tr4) and T3 (m) levels. However, since the concentrations of free T4 (FT4) and free T3 (FT3) are not affected by the binding protein,8 patients with TBG deficiency have normal FT4, FT3 and thyrotropin (TSH) levels, and there is no clinical manifestation of hypothyroidism. This paper summarises and analyses the clinical characteristics of a TBG-PD patient diagnosed through genetic analysis and investigates the Serpina7 gene mutation.
Case Description
Case Data
This study was approved by the Medical Ethics Committee of the Fourth Affiliated Hospital of Guangzhou Medical University (No. 2022-H-002).
The patient, a 26-year-old female, was hospitalised for repeated palpitations, dizziness and shortness of breath for eight months and fatigue for ten days. The patient had no obvious inducement until eight months before admission, when she began to experience palpitations and shortness of breath, combined with paroxysmal swelling, and pain and discomfort in the anterior cervical area, which were subsequently relieved. The patient visited the local hospital for treatment. The serum examination findings are shown in Table 1.
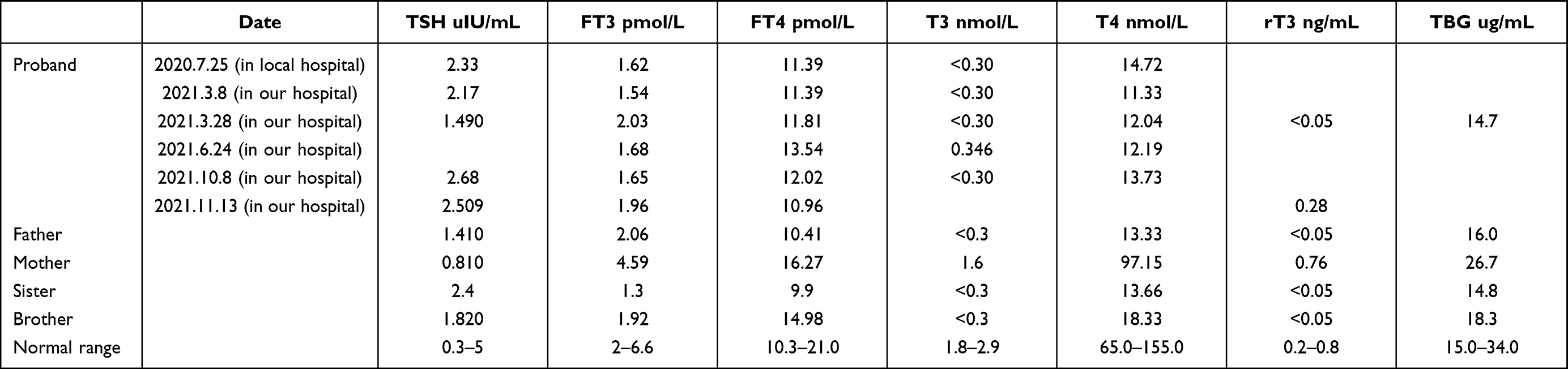 |
Table 1 Serum Examination Findings of Thyroid Function and Globulin Level in the Patient and the Families |
A cardiac colour Doppler ultrasound examination9 indicated a slight prolapse of the anterior mitral valve, while the left ventricular systolic and diastolic functions were normal. Following treatment with Youjiale (50 ug qd), the symptoms of palpitations and shortness of breath were relieved, as were the swelling and pain in the neck. The patient subsequently purchased and continued to take the Youjiale (50 mg qd), and while their symptoms of palpitations and shortness of breath recurred, they were tolerable. Ten days prior to admission, the patient had no obvious inducement or symptoms such as dizziness, nausea, vomiting or headache. The patient improved on her own but presented weakness in the limbs (mainly in both lower limbs) and pain and discomfort in both knees. There was no visual acuity decline and no visual field defect. The serum examination findings are shown in Table 1.
The details of the medical history of the patient were as follows:
- Previous history of fibrocystic breast disease.
- No history of hypertension.
- No history of cerebral infarction and coronary heart disease.
- No history of myocardial infarction.
- No history of atrial fibrillation.
Meanwhile, the mother had congenital heart disease and had undergone heart surgery (details unknown), while the father, brother and sister exhibited a decrease in FT3, TT3, TT4 and RT3, with the decrease in TT3 and TT4 more significant. The TBG detections across all three family members were normal at a low level and indicated a family genetic tendency.
Thyroid Function Test
Serum T3, T4, FT3, FT4 and TSH levels were determined via electrochemiluminescence (Cobas 8000, Roche Diagnostics GmbH, Switzerland), with TBG determined via chemiluminescence (DPC2000, Siemens, Germany).10 The experimental procedure was as follows: 1) preparation of standards, reagents and samples prior to the experiment; 2) sample addition (standards and samples) (100 µL), incubation at 37°C for 1 h; 3) suctioning and discarding was performed, with detection solution A (100 µL) added, followed by incubation at 37°C for 1 h; 4) plate washing was performed three times; 5) detection solution B (100 µL) was added prior to incubation at 37°C for 30 min; 6) plate washing was performed five times; 7) the substrate was added (100 µL), followed by incubation at 37°C for 10 min; 8) the reading was performed. Thyroid function tests were performed on the proband and their family members.
Sequencing and Genotyping of the Serpina7 Gene and Its Mutation
The human genomic DNA was extracted from peripheral blood leukocytes using a standard phenol-chloroform method.11 The coding region of the Serpina7 gene (exons 1–4) and exon–intron junction of the proband’s family members were directly sequenced. In addition, the coding region of the Serpina7 gene (exons 1–4) and the exon–intron junction of 207 unrelated individuals were analysed through direct DNA sequencing and high-resolution melting (Tiangen Biotechnology Co., Ltd., Beijing, China).
Analysis Method
The patient was examined using thyroid-related antibodies and thyroid colour Doppler ultrasound, among other methods, and common thyroid diseases, such as Hashimoto’s thyroiditis, were excluded. A repeated examination of the thyroid function level showed the characteristics of a partial lack of TBG. Further gene sequencing revealed that the patient’s Serpina7 gene locus was abnormal. Relevant literature was retrieved to analyse whether the abnormal locus was related to the decrease in TBG expression.
Literature Search
The keywords “thyroxine-binding globulin, gene and mutation” were used to search the literature in PubMed (biomedical literature database), Web of Science and other databases from their date of establishment to February 2022. A total of 45 studies were retrieved.
Results
The clinical characteristics of children with TBG deficiency and Serpina7 gene mutation were studied. The thyroid function of the patients with TBG deficiency was tested, and genetic analysis, including in terms of family members, was conducted using polymerase chain reaction (PCR) analysis and DNA direct sequencing to detect the characteristics of TBG mutants.
Thyroid Function Test Results
The proband was a 26-year-old Chinese woman. The analysis of the venous blood indicated that the TBG (14.7 μg/mL), T3 (<0.3 nmol/L), RT3 (<0.05 ng/mL) and T4 (14.72 nmol/L) values were lower than normal. The T3 uptake was increased, indicating that the TBG-binding affinity for T3 is affected in vitro. However, the TSH (2.33 uIU/mL), FT3 (1.62 pmol/L) and FT4 (11.39 pmol/L) levels were normal, indicating a clinically euthyroid state. These values indicate the impaired binding of T3 and, thereby, also of T4 to TBG (TBG-PD phenotype). The T3 and T4 levels in the father and younger brother significantly decreased, indicating that this phenomenon is due to a familial inherited genetic mutation. Table 1 presents the test results.
Characteristics of Thyroxine Binding Globulin Mutants
The PCR amplification and direct sequencing of the target gene revealed that the patient and the father had a missense mutation in exon 4 of the Serpina7 gene, and the nucleic acid variation was C.909(exon4)G>T. The patient was heterozygous, and the father was hemizygous. Serpina7 C.909G(P.L303F) is a mutation and was acquired from the father through X-linked recessive inheritance (Figure 1).
 |
Figure 1 Characteristics of TBG mutants. (A): gene sequence of this patient; (B): gene sequence of the patient’s father; (C): gene sequence of the patient’s mother. |
Bibliography Retrieval
A total of 45 studies that met the requirements were retrieved from databases at home and abroad, with 45 articles retrieved from foreign databases and two from a Chinese database. An individual case of the Serpina7 gene mutation with various types of mutations, including point mutation, deletion mutation, joint mutation, intron splice-site mutation and TBG extra gene mutation was found. Except for one report of autosomal inheritance, the remainder were X-linked inheritances. Fifty-three TBG variants were identified, of which 46 were found to be mutant genes.2–5,11–34
Discussion
Thyroxine binding globulin is the major thyroid hormone transport protein in the serum. Using a complementary DNA clone for the analysis of somatic cell hybrids and in-situ hybridisation, Trent et al assigned the TBG locus to Xq21–q22.35 Elsewhere, Murata et al stated that the five types of inherited TBG variants have been described, all of which are X-linked. Three of these are thought to be quantitative variants and are widely distributed. It would appear that TBG is completely absent in the first form, while it appears to be physically, antigenically and functionally normal in the second and third forms. An alteration in the rate of TBG synthesis is believed to be responsible since the rates of degradation are normal.36 Takamatsu and Refetoff (1986) described a sixth type of inherited TBG variant, TBG-Chicago (314200.0010), which differs from all five of the previously reported variants in being markedly resistant to heat denaturation. The other five variants have a variable degree of increased sensitivity to denaturation by heat and acid.37 Takamatsu et al measured denatured TBG in sera from 32 unrelated families with inherited TBG deficiency. High levels were found in the serum samples from two out of 16 families with TBG-PD. Further studies revealed that these two families had TBG variants with alteration in both stability and isoelectric focusing patterns, thus indicating the presence of structural gene mutations.38 Elsewhere, undetermined electrophoretic slow variants were found in 4% to 12% of black and Pacific Island populations, respectively.39 The characteristic cathode shift of all subtypes was detected on isoelectric focusing, indicating that the difference exists in the core protein. In addition, the heat resistance of TBG-s is slightly higher, and the average undetermined human serum is low, thus reducing the T4 concentration.
The Serpina7 gene is located in Xq22.2. The TBG deficiency caused by the Serpina7 gene mutation is a rare thyroid disease, which is generally acquired through X-linked recessive inheritance.2 The proband in this study was diagnosed with TBG-PD and was found to have a missense mutation of the Serpina7 gene: C.909(exon4)G>T(P.L303F). The thyroid functions of her father, brother and sister indicated that the FT3, T3, T4 and RT3 levels decreased, with the decrease in T3 and T4 more substantial, which was consistent with the clinical phenotype of the patient. The TBG detection was at a normal level among all three family members. All these data indicated a family genetic tendency. Based on the nucleotide variation at this site in the human genome database, it can be concluded that the genetic variation does not belong to single nucleotide polymorphism (SNP). The amino acid sequence of the missense mutation site is relatively conservative in various species. Analyses performed using pathogenicity prediction software, such as PolyPhen-2, SIFT and Mutation Taster, indicate that the mutation will affect the protein function and has pathogenic potential. Results obtained via DynaMut server prediction indicated that the p.E91K, p.I92T and p.L303F variants reduced the protein stability, and that the p.L303F variants had a significant effect on the protein stability.13 The more mutation sites, the greater the impact of TBG. Fang Yanlan et al13 concluded that polymorphisms in the Serpina7 gene can cause the disease, especially when multiple SNP sites are present at the same time, which can lead to decreased expression. The more SNPs carried, the lower the TBG and the higher the likelihood of complete TBG.
Therefore, it is believed that the Serpina7 gene mutation found in the proband is the cause of the disease. In addition, the father’s Serpina7 gene’s exon 4 also appeared to be a missense. As noted, the patient was heterozygous, and the father was hemizygous, meaning the gene mutation was acquired through X-linked recessive inheritance. The function of its variant protein needs to be further confirmed. The clinical manifestations of the proband in this study are completely consistent with those of TBG-PD reported in the literature.34 The serum TBG, T3, RT3 and T4 values of the proband were lower than the normal values, but the TSH, FT3 and FT4 values were normal. These values are indicative of the TBG-PD phenotype. Following treatment with levothyroxine tablets, the symptoms were relieved.
Among the 16 reported TBG missense mutations, most are benign, while two are of little importance.38,39 The TBG-Poly mutation has no considerable effect on TBG molecules,40 but it has a high allele frequency in different populations. The TBG mutation exhibited an X-linked genetic pattern. Most patients are hemizygous men or homozygous women. Due to the inactivation of the X chromosome, heterozygous women typically have more moderate TBG levels than both affected and unaffected men.
Studies have shown that TBG deficiency can inhibit the growth and development of bones and reduce weight.29 The deficiency also leads to a decrease in thyroid hormone bound to the serum protein, but there is no clinical manifestation of hypothyroidism since the functional FT4 and FT3 concentrations are not affected by the binding protein. Therefore, normal levels of FT4 and FT3 are also an important distinguishing factor between TBG deficiency and central hypothyroidism.
This study involves certain limitations. First, the number of cases is relatively small, and while this is largely due to the rarity of the disease, the results may not be fully representative. Second, the content of the study may not be deep enough, with the lack of relevant analysis of the mechanism and function of gene mutations resulting in less abundant content. In further studies, we will adjust and deepen the study content accordingly and strive to expand the number of cases and improve the representativeness of the study.
Conclusion
The proband of this study has Serpina7 C.909G(P. L303F), which is a mutation that was acquired from the father through X-linked recessive inheritance. The main clinical characteristics of patients with TBG deficiency are as follows: the levels of serum T4, T3 and TBG are low; the levels of TSH, FT3 and FT4 are normal; and there is no clinical manifestation of hypothyroidism. Therefore, for patients with low T4, T3 and TBG but normal TSH, the serum levels of FT3 and FT4 need to be checked in addition to the conventional consideration of central hypothyroidism. If necessary, the serum TBG level and related genes should be tested to identify TBG deficiency. This will help avoid misdiagnosis and inappropriate medical treatment and is of great significance to improving the quality of life of TBG patients.
Data Sharing Statement
All data generated or analyzed during this study are included in this article.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the declaration of Helsinki.This study was conducted with approval from the Ethics Committee of The Fourth Affiliated hospital of Guangzhou Medical University (No. 2022-H-002). Written informed consent has been provided by the patient to have the case details published.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors had no personal, financial, commercial, or academic conflicts of interest in this work.
References
1. Alshehri B, Pagnin M, Lee JY, Petratos S, Richardson SJ. The role of transthyretin in oligodendrocyte development. Sci Rep. 2020;10(1):
2. Gawandi S, Jothivel K, Kulkarni S. Identification of a novel mutation in thyroxine-binding globulin (TBG) gene associated with TBG-deficiency and its effect on the thyroid function. J Endocrinol Invest. 2022;45(4):731–739. doi:10.1007/s40618-021-01697-z
3. Pappa T, Moeller LC, Edidin DV, Pannain S, Refetoff S. A novel mutation in the TBG gene producing partial thyroxine-binding globulin deficiency (glencoe) identified in 2 families. Eur Thyroid J. 2017;6(3):138–142. doi:10.1159/000455097
4. Gomes-Lima CJ, Maciel AAFL, Andrade MO, et al. Thyroxine-binding globulin deficiency due to a novel SERPINA7 mutation: clinical characterization, analysis of X-chromosome inactivation pattern and protein structural modeling. Gene. 2018;666:58–63. doi:10.1016/j.gene.2018.05.018
5. Dang PP, Xiao WW, Shan ZY, et al. Novel frameshift mutation causes early termination of the thyroxine-binding globulin protein and complete thyroxine-binding globulin deficiency in a Chinese family: a case report. World J Clin Cases. 2019;7(22):3887–3894. doi:10.12998/wjcc.v7.i22.3887
6. Flink IL, Bailey TJ, Gustafson TA, Markham BE, Morkin E. Complete amino acid sequence of human thyroxine-binding globulin deduced from cloned DNA: close homology to the serine antiproteases. Proc Natl Acad Sci USA. 1986;83(20):7708–7712. doi:10.1073/pnas.83.20.7708
7. Refetoff S. Inherited thyroxine-binding globulin abnormalities in man. Endocr Rev. 1989;10(3):275–293. doi:10.1210/edrv-10-3-275
8. Mayerl S, Alcaide Martin A, Bauer R, Schwaninger M, Heuer H, Ffrench-Constant C. Distinct actions of the thyroid hormone transporters Mct8 and Oatp1c1 in murine adult hippocampal neurogenesis. Cells. 2022;11(3):524. doi:10.3390/cells11030524
9. Donovan LE, Metcalfe A, Chin A, et al. A practical approach for the verification and determination of site- and trimester-specific reference intervals for thyroid function tests in pregnancy. Thyroid. 2019;29(3):412–420. doi:10.1089/thy.2018.0439
10. Leong KW, Yu F, Makrigiorgos GM. Mutation enrichment in human DNA samples via UV-mediated cross-linking. Nucleic Acids Res. 2022;50(6):e32. doi:10.1093/nar/gkab1222
11. Carvalho GA, Weiss RE, Vladutiu AO, Refetoff S. Complete deficiency of thyroxine-binding globulin (TBG-CD Buffalo) caused by a new nonsense mutation in the thyroxine-binding globulin gene. Thyroid. 1998;8(2):161–165. doi:10.1093/nar/gkab1222
12. Chen LD, Lu HJ, Gan YL, et al. Partial thyroxine-binding globulin deficiency in a family with coding region mutations in the TBG gene. J Endocrinol Invest. 2020;43(12):1703–1710. doi:10.1007/s40618-020-01245-1
13. Fang Y, Chen H, Chen Q, Wang C, Liang L. Compound hemizygous variants in SERPINA7 gene cause thyroxine-binding globulin deficiency. Mol Genet Genomic Med. 2021;9(2):e1571. doi:10.1002/mgg3.1571
14. Hengeveld RCC, Albersen M, Hadders MAH, et al. A newborn falsely suspected of congenital hypothyroidism due to mutated thyroxine-binding globulin with low binding affinity. Horm Res Paediatr. 2021;94(1–2):76–80. doi:10.1159/000516691
15. Moeller LC, Appiagyei-Dankah Y, Köhler B, Biebermann H, Janssen OE, Führer D. Two novel mutations in the Serpina7 gene are associated with complete deficiency of thyroxine-binding globulin. Eur Thyroid J. 2015;4(Suppl 1):108–112. doi:10.1159/000381093
16. Miura Y, Hershkovitz E, Inagaki A, Parvari R, Oiso Y, Phillip M. A novel mutation causing complete thyroxine-binding globulin deficiency (TBG-CD-Negev) among the bedouins in southern Israel. J Clin Endocrinol Metab. 2000;85(10):3687–3689. doi:10.1210/jcem.85.10.6899
17. Reutrakul S, Dumitrescu A, Macchia PE, Moll GW Jr, Vierhapper H, Refetoff S. Complete thyroxine-binding globulin (TBG) deficiency in two families without mutations in coding or promoter regions of the TBG genes: in vitro demonstration of exon skipping. J Clin Endocrinol Metab. 2002;87(3):1045–1051. doi:10.1210/jcem.87.3.8275
18. Moeller LC, Fingerhut A, Lahner H, et al. C-terminal amino acid alteration rather than late termination causes complete deficiency of thyroxine-binding globulin CD-NeuIsenburg. J Clin Endocrinol Metab. 2006;91(8):3215–3218. doi:10.1210/jc.2005-2261
19. Mannavola D, Vannucchi G, Fugazzola L, et al. TBG deficiency: description of two novel mutations associated with complete TBG deficiency and review of the literature. J Mol Med. 2006;84(10):864–871. doi:10.1007/s00109-006-0078-9
20. Berger HR, Creech MK, Hannoush Z, Watanabe Y, Kargi A, Weiss RE. A novel mutation causing complete thyroid binding globulin deficiency (TBG-CD MIA) in a male with coexisting graves disease. AACE Clin Case Rep. 2017;3(2):e134–e139. doi:10.4158/EP161421.CR
21. Su CC, Wu YC, Chiu CY, Won JG, Jap TS. Two novel mutations in the gene encoding thyroxine-binding globulin (TBG) as a cause of complete TBG deficiency in Taiwan. Clin Endocrinol. 2003;58(4):409–414. doi:10.1046/j.1365-2265.2003.01730.x
22. Domingues R, Font P, Sobrinho L, Bugalho MJ. A novel variant in Serpina7 gene in a family with thyroxine-binding globulin deficiency. Endocrine. 2009;36(1):83–86. doi:10.1007/s12020-009-9202-2
23. Hickey RD, Mao SA, Glorioso J, et al. Curative ex vivo liver-directed gene therapy in a pig model of hereditary tyrosinemia type 1. Sci Transl Med. 2016;8(349):349ra99. doi:10.1126/scitranslmed.aaf3838
24. Soheilipour F, Fazilaty H, Jesmi F, Gahl WA, Behnam B. First report of inherited thyroxine-binding globulin deficiency in Iran caused by a known de novo mutation in SERPINA7. Mol Genet Metab Rep. 2016;8:13–16. doi:10.1016/j.ymgmr.2016.06.001
25. Wang F, Quan HB, Ji Q, Mo ZW, Liu HW, Chen KN. Three cases of complete thyroxine-binding globulin deficiency in a family and literature review. Chin J Endocrinol Metab. 2021;37(07):653–656. doi:10.3760/cma.j.cn311282-20200906-00618
26. Fang YL, Wang CL, Liang L. Cases of thyroglobulin deficiency were reviewed. Chin J Pediatr. 2016;54(06):428–432. doi:10.3760/cma.j.issn.0578-1310.2016.06.008
27. Mok SF, Loh TP, Venkatesh B, Deepak DS. Elevated free thyroxine and non-suppressed thyrotropin. BMJ Case Rep. 2013;2013:bcr2013201527. doi:10.1136/bcr-2013-201527
28. Domingues R, Bugalho MJ, Garrão A, Boavida JM, Sobrinho L. Two novel variants in the thyroxine-binding globulin (TBG) gene behind the diagnosis of TBG deficiency. Eur J Endocrinol. 2002;146(4):485–490. doi:10.1530/eje.0.1460485
29. Ferrara AM, Pappa T, Fu J, et al. A novel mechanism of inherited TBG deficiency: mutation in a liver-specific enhancer. J Clin Endocrinol Metab. 2015;100(1):E173–81. doi:10.1210/jc.2014-3490
30. Lacka K, Nizankowska T, Ogrodowicz A, Lacki JK. A novel mutation (del 1711 G) in the TBG gene as a cause of complete TBG deficiency. Thyroid. 2007;17(11):1143–1146. doi:10.1089/thy.2007.0023
31. Mannavola D, Vannucchi G, Fugazzola L, Cerutti N, Persani L, Beck-Peccoz P. Genetic analyses and evaluation of peripheral parameters of thyroid hormone action for the differential diagnosis of RTH. A novel heterozygous missense mutation (M334T) discovered. J Endocrinol Invest. 2002;25(2):RC4–6. doi:10.1007/BF03343969
32. Cao L, Lou X, Zhou L, Wu Y. The decrease of T3 / T4 is not hypothyroidism - A new mutation of Serpina7 gene results in partial thyroglobulin deficiency. Pharmazie. 2021;76(9):428–430. doi:10.1691/ph.2021.1559
33. Trent JM, Flink IL, Morkin E, van Tuinen P, Ledbetter DH. Localization of the human thyroxine-binding globulin gene to the long arm of the X chromosome (Xq21-22). Am J Hum Genet. 1987;41(3):428–435.
34. Murata Y, Takamatsu J, Refetoff S. Inherited abnormality of thyroxine-binding globulin with no demonstrable thyroxine-binding activity and high serum levels of denatured thyroxine-binding globulin. N Engl J Med. 1986;314(11):694–699. doi:10.1056/NEJM198603133141107
35. Takamatsu J, Refetoff S. Inherited heat-stable variant thyroxine-binding globulin (TBG-Chicago). J Clin Endocrinol Metab. 1986;63(5):1140–1144. doi:10.1210/jcem-63-5-1140
36. Geno KA, Nerenz RD. Evaluating thyroid function in pregnant women. Crit Rev Clin Lab Sci. 2022;1–20. doi:10.1080/10408363.2022.2050182
37. Waltz MR, Pullman TN, Takeda K, Sobieszczyk P, Refetoff S. Molecular basis for the properties of the thyroxine-binding globulin-slow variant in American blacks. J Endocrinol Invest. 1990;13(4):343–349. doi:10.1007/BF03349576
38. Bhatkar SV, Rajan MG, Velumani A, Samuel AM. Thyroid hormone binding protein abnormalities in patients referred for thyroid disorders. Indian J Med Res. 2004;120(3):160–165.
39. Ascoli P, Cavagnini F. Hypopituitarism. Pituitary. 2006;9(4):335–342. doi:10.1007/s11102-006-0416-5
40. Ferrara AM, Cakir M, Henry PH, Refetoff S. Coexistence of THRB and TBG gene mutations in a Turkish family. J Clin Endocrinol Metab. 2013;98(6):E1148–51. doi:10.1210/jc.2013-1413
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Alpha-1 Antitrypsin Gene Variants in Patients without Severe Deficiency Diagnosed with Pulmonary Emphysema on Chest CT
Laviña E, Lumbreras S, Bravo L, Soriano JB, Izquierdo JL, Rodríguez JM
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:353-361
Published Date: 3 February 2024