Back to Journals » OncoTargets and Therapy » Volume 14
Partial Response to Pyrotinib Plus Capecitabine in an Advanced Breast Cancer Patient with HER2 Amplification and R157W Mutation After Anti-HER2 Treatment: A Case Report and Literature Review
Authors Qu Y, Liu Y ![]() , Ding K, Li Y, Hong X, Zhang H
, Ding K, Li Y, Hong X, Zhang H
Received 1 November 2020
Accepted for publication 7 January 2021
Published 2 March 2021 Volume 2021:14 Pages 1581—1588
DOI https://doi.org/10.2147/OTT.S289876
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Yanchun Qu,1– 3,* Yufeng Liu,3,* Kailin Ding,3 Yong Li,1,2 Xiaoyu Hong,4 Haibo Zhang1,2
1Department of Oncology, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China; 2Department of Oncology, Guangdong Provincial Hospital of Traditional Chinese Medicine, Guangzhou, People’s Republic of China; 3The Second Clinical Medical College of Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China; 4Nanjing Geneseeq Technology Inc, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Haibo Zhang
Department of Oncology, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, No. 111 Dade Road, Yuexiu District, Guangzhou, 510120, People’s Republic of China
Tel +86-020-81887233
Email [email protected]
Abstract: Human epidermal growth factor receptor2 (HER2) overexpression/amplification is associated with high malignancy, rapid disease progression and poor overall survival in breast cancer. The application of anti-HER2 drugs has greatly improved the survival of patients with HER2-positive breast cancer, but drug resistance issues affect the long-term efficacy. The HER2 mutation is considered to be one of the reasons for resistance to anti-HER2 therapy, and there is currently no standard treatment. We report for the first time the detection of HER2 amplification with R157W mutation by second-generation sequencing (NGS) in a 57-year-old hormone receptor-negative, HER2-positive woman with advanced breast cancer who was resistant to multi-line anti-HER2 therapies. She subsequently received pyrotinib combined with capecitabine treatment and achieved partial response. The small-molecule pan-HER family irreversible inhibitor pyrotinib combined with capecitabine has shown a promising effect in the treatment of HER2 mutation-induced resistance, but the molecular mechanism and efficacy need to be further verified.
Keywords: breast cancer, HER2 amplification, HER2 R157W mutation, pyrotinib plus capecitabine, anti-HER2 treatment
Introduction
Breast cancer is a malignant tumor with the highest morbidity and second highest mortality rate in women.1 HER2-positive (HER2 amplification) breast cancer accounts for about 15–20% of all breast cancers.2 Anti-HER2 drugs have greatly improved the survival of such patients. These drugs include humanized monoclonal antibodies like trastuzumab and pertuzumab, antibody-drug conjugates (ADC) like T-DM1, and novel oral small-molecule tyrosine kinase inhibitors like pyrotinib, neratinib, lapatinib and so on.
Acquired drug resistance is inevitable in patients with advanced HER2-positive breast cancer after first-line and second-line anti-HER2 treatments, and the mechanism is still under investigation. HER2 mutations are rare in breast cancer patients. A systematic review showed that the frequency of HER2 mutation in breast cancer patients was about 3%,3 while previous gene sequencing results suggested that patients with primary HER2-positive breast cancer had a lower frequency of HER2 mutation.4,5 Therefore, HER2 amplification and mutation are considered mutually exclusive in breast cancer patients. In recent years, studies have found that the HER2 gene mutation rate is higher in breast cancer patients treated with multi-line anti-HER2 therapies,6,7 and HER2 mutation is one of the causes of resistance to anti-HER2 treatment. There is no standard treatment for HER2 mutation patients yet. Of these, HER2 R157W mutation is a rare mutation and there are currently only two reports in the literature. One was somatic mutation found in micropapillary urothelial carcinoma (MPUC),8 which was not accompanied by HER2 overexpression and HER2 amplification. FATHMM prediction determined that it was a pathogenic mutation according to the COSMIC database. The other one was germline mutation found in breast cancer,9 of which the clinical significance is unknown. This is the first report of a patient with HER2-positive advanced breast cancer who developed HER2-amplification with R157W missense mutation after treatment of lapatinib plus trastuzumab and achieved partial response (PR) after using pyrotinib plus capecitabine.
Case Description
A 57-year-old Chinese female was initially presented to an outside hospital and underwent radical resection of left breast cancer diagnosed with stage IIIC (pTxN3M0) in Jan 2013. Pathology indicated invasive ductal carcinoma of breast estrogen receptor (ER) (-), progesterone receptor (PR) (-), and HER2(3+) by immunohistochemistry. Then, she received one cycle of adjuvant chemotherapy with docetaxel and three cycles of oncolytic virus therapy in other hospital, but the patient could not provide detailed information. In March 2013, PET/CT showed bilateral lung metastases. However, the patient refused chemotherapy and received traditional Chinese medicine (TCM) decoction treatment. In March 2014, follow-up PET/CT revealed multiple metastases in liver, both lungs and bones. A liver biopsy conducted in our department suggested hepatic metastasis of the breast cancer (ER(-), PR(-), HER2(3+)). The patient received eight cycles of first-line-targeted treatment with trastuzumab (8mg/kg IV day 1 followed by 6mg/kg IV day 1 every 21 days) plus paclitaxel (135mg/m2 IV day 1 every 21 days), followed by maintenance treatment with trastuzumab (6mg/kg IV day 1 every 21 days), achieving partial response and progression-free survival (PFS) for approximately 1 year. Subsequently, she was treated with docetaxel (75mg/m2 IV day 1 every 21 days), xeloda (1000mg/m2 PO bid days 1–14 every 21 days), and trastuzumab (6mg/kg IV day 1 every 21 days) as the second-line treatment for six cycles, with the best efficacy of partial response and PFS for 10 months. Upon disease progression, the third-line treatment was gemcitabine (1000mg/m2 IV day 1 every 21 days) plus S-1 (40mg PO bid days 1–14 every 21 days) based chemotherapy for 4 cycles, with the best efficacy of stable disease (SD) and PFS for nearly 4 months. In the fourth-line treatment, she received epirubicin (80mg/m2 IV day 1 every 21 days) plus lapatinib (0.25g PO qd). However, after one cycle, due to impaired liver function (with her alanine aminotransferase [ALT] level reaching 1122U/L), use of epirubicin was discontinued. Liver function improved following liver protection therapy, and the patient then continued lapatinib monotherapy (0.5g PO qd), maintaining an SD with PFS of 2 months.
In October 2017, enlarged lesions revealed in the chest and abdomen CT indicated progressive disease (PD). Next-generation sequencing (NGS) of plasma detected HER2 and CDK12 amplification, NSD1 gene fusion, and TP53 L257R mutation. The patient began to receive trastuzumab (6mg/kg IV day 1 every 21 days) combined with lapatinib (1g PO qd) as the fifth-line treatment from November 2017. The best efficacy was SD, and PFS was about 9 months. In August 2018, plasma NGS showed R157W missense mutation in HER2 exon 4, along with HER2 amplification and TP53 L257R mutation. That September, a chest and abdomen CT confirmed PD. The patient began to receive pyrotinib(400mg PO qd) plus xeloda (1000mg/m2 PO bid days 1–14 every 21 days) as the sixth-line treatment, with the best efficacy of PR and PFS for 13 months. Plasma NGS monitoring during the period (December 2018) suggested a decrease in HER2 copy number and abundance of R157W mutation. In November 2019, when her disease progressed again, the re-examination of the plasma NGS showed CDK12 and HER2 amplification, DNMT3A inactivation mutation and TP53 L257R mutation. There was no HER2 mutation.
From November 2019 to January 2020, the patient received the seventh-line treatment with Abraxane (200mg IV day 1 every 14 days), with the optimal efficacy of SD. In February 2020, she began to accept the eighth-line treatment with pertuzumab (840mg IV day 1 followed by 420mg IV day 1 every 21 days) combined with trastuzumab (8mg/kg IV da y1 followed by 6mg/kg IV day 1 every 21 days) and xeloda (1000mg/m2 PO bid days 1–14 every 21 days). The efficacy has not been evaluated at the time of writing this paper.
Discussion
HER2-amplified breast cancer accounts for about 15–20% of all breast cancers2 and is often characterized by rapid progression and poor prognosis.10,11 The precise treatment of HER2 has completely changed the survival rate for breast cancer with HER2-amplification. However, as time goes by, anti-HER2 treatment will inevitably produce acquired drug resistance, which will affect the survival of patients. Therefore, it is imperative to clarify the mechanism of drug resistance and make targeted treatments. The resistance mechanism of anti-HER2 drugs is complex, and the resistance caused by HER2 mutation has attracted more and more attention and may become a new potential therapeutic target.12 We report a 57-year-old woman with advanced HER2-positive breast cancer who received multiple lines of anti-HER2 therapy and was confirmed to have the HER2 R157W missense mutation in exon 4 by plasma NGS, and obtained PR after treatment with pyrotinib and capecitabine. This case provides a new potential treatment option for HER2 mutations following resistance to HER2 therapy.
Proto-oncogene HER2, also known as ErbB2, belongs to the ERB family with ErbB1(EGFR), ErbB3(HER3), and ErbB4(HER4). No high-affinity ligand has been found in HER2 so far, so it must form homologously or as a heterodimer with other members of the family, binding with ATP to activate intracellular tyrosine kinases, thereby initiating the downstream signaling pathway and regulating the proliferation and differentiation of cells.13–15 HER2 is the most important driver gene of breast cancer, and the most common positive form is gene amplification. Amplification of HER2 would lead to increased protein synthesis (ie, overexpression of HER2 protein) or increased protein function, which would lead to overactivation of downstream signaling pathways and overgrowth of cells, playing an important role in the proliferation, invasion, metastasis, and evolution of breast cancer cells.16–18
H0648g,19 M77001,20 EGF100151,21 EGF10490022 et al studies have shown that patients with HER2-positive breast cancer can benefit from anti-HER2 humanized trastuzumab and double HER2-EGFR tyrosine kinase inhibitor (TKI) lapatinib. However, approximately 50% of HER2-positive patients developed resistance to trastuzumab 1 year after treatment.23 Lapatinib has achieved certain clinical efficacy in metastatic HER2-positive breast cancer treated with trastuzumab, but a significant proportion of patients develop disease progression due to innate or acquired resistance to lapatinib.24,25 Studies on the molecular mechanisms of trastuzumab and lapatinib resistance26 found that overexpression of other HER family receptors and their ligands, loss of PTEN leading to activation of the PI3K/Akt/mTOR pathway, PI3KCA mutation, and Akt mutation or amplification were common causes of drug resistance. Drug resistance has become an urgent problem.
The patient developed resistance to lapatinib combined with trastuzumab. The HER2 gene mutation was not detected before the patient received lapatinib plus trastuzumab treatment, while the R157W mutation was found in the disease progression after treatment. Moreover, by comparing the two gene test results (Table 1), only the HER2 mutation was acquired, so it was believed that the HER2 mutation was the main mechanism of drug resistance in this case. In recent years, with the gradual deepening of the understanding of HER2, it is believed that HER2 mutation plays an important role in the incidence, development and resistance of breast cancer.27,28 The primary HER2 mutation mostly occurred in HER2 negative conditions, while in HER2 positive breast cancer the HER2 mutation mostly occurred after anti-HER2 treatment. Fang et al6 performed HER2 full-length gene sequencing on the tissues of 198 patients with metastatic breast cancer (MBC) after multiple cycles of treatment and found that the rate of HER2 mutations in patients treated with trastuzumab was as high as 17.7%. Park et al7 also carried out NGS tests on the tissues of 36 refractory MBC patients after multi-cycle and multi-drug treatment, and found that 5 out of 6 patients with HER2 mutation were HER2 positive and developed drug resistance after receiving anti-HER2 drugs (trastuzumab, lapatinib).
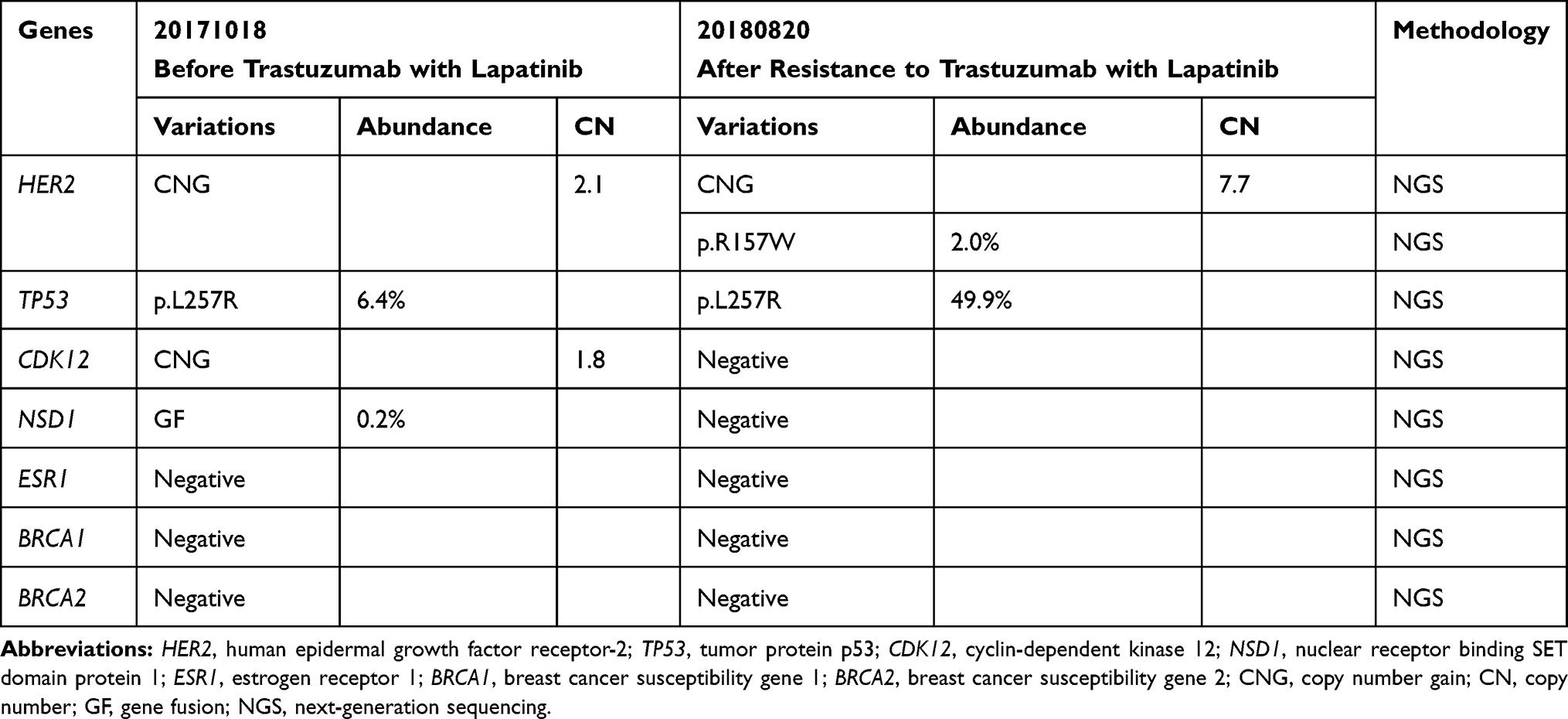 |
Table 1 NGS Results Detected Before and After Treatment with Trastuzumab and Lapatinib |
In the literature, the most common HER2 mutation sites in breast cancer were L755S, V777L, D769H, D769Y, G778_P780dup, Y772_A775dup in the kinase domain, and S310F and S310Y in the extracellular domain.3,29 There are no studies to prove the difference between HER2-negative combined mutation and HER2 positive-combined mutation, and most of the mutations can occur in both. The causes of resistance to HER2 therapy due to HER2 mutations are not fully understood. A cell-induced drug resistance test suggested that drug resistance of lapatinib might be related to the change of kinase structure caused by L755S and P780L mutations, which would activate the kinase activity and interfere with the inactive structure required for lapatinib binding.30,31 It may also be related to the production of larger amino acids after L726F, L785F, and other mutations, which interfered with the binding of lapatinib spatially.30 In addition, T789 mutation can stabilize the active conformation and directly compete with lapatinib for ATP binding sites, which may also be one of the causes of lapatinib resistance.30,31 Other studies have found that increased levels of EGFR-HER3 dimerization and expression of HER4 can also lead to lapatinib resistance. The extracellular resistance test of trastuzumab suggested that mutations in the HER2 kinase domain might alter the PI3K/AKT cascade signaling, thereby weakening the inhibition of trastuzumab to the PI3K/AKT pathway.32 It has also been observed in cell experiments that L755S mutation can lead to overactivation of PI3K/AKT/mTOR and MAPK pathways, leading to resistance to lapatinib and neratinib.33 Hanker B et al34 reported that a breast cancer patient with HER2 L869R mutation got new T798I mutation when neratinib resistance happened. Cell models confirmed that the presence of isoleucine at the 798 site leads to steric hindrance, reducing the binding affinity of neratinib. Smyth et al29 believed that HER2 mutation accompanied with other changes in HER family signaling pathways, such as HER3 mutation, might lead to drug resistance to neratinib.
This case reported that R157W missense mutation happened in the extracellular domain belonging to exon4 of HER2 which could be found in the cBioPortal database35,36 within the TCGA data set (Figure 1). Prior to this, only two cases of HER2 R157W mutation have been reported in the literature, and that which was found in breast cancer harbored germline mutations. As this residue is presented in the extracellular domain, we think the drug-resistant mechanism may be related to the change of the extracellular domain receptor structure, which interfered with the binding of trastuzumab. It may also strengthen dimerization with other family receptors, such as HER2/HER3, and HER2/HER4 polymerization, thereby weakening the inhibition of lapatinib, which activated downstream pathways and caused a cascade reaction, eventually leading to tumor progression.
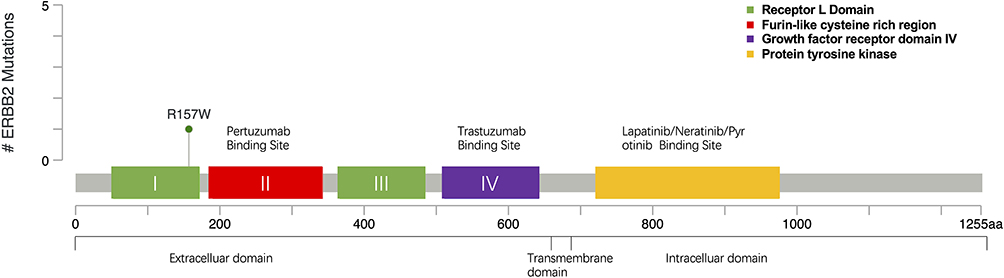 |
Figure 1 Mutation site map of R157W gene in TCGA dataset from cBioPortal database.35,36 |
There is no standard treatment for HER2 mutation, but many studies have begun to explore treatment strategies. A number of studies have demonstrated that the pan-HER2 irreversible inhibitor neratinib has a good inhibitory effect on certain mutations. Zuo et al28 found that neratinib had a strong inhibitory effect on drug-resistant mutant cell lines bearing K753E or L755S mutations. Cocco et al37 demonstrated that neratinib could not only effectively overcome the acquired resistance to HER2 therapy in breast cancer cells carrying both HER2 amplification and L755S somatic mutation but also significantly inhibit tumor growth in mice which are resistant to trastuzumab and lapatinib as well as carrying both D769Y mutation and HER2 amplification. At the same time, it was observed that neratinib had clinical activity in breast cancer patients with both HER2 gene amplification and mutation. SUMMIT is a Phase II basket clinical trial involving patients with cancers featuring HER2 mutations, and the result shows that neratinib has a good clinical effect on patients with breast cancer accompanied by HER2 mutations but without amplification, with an ORR of 32% at 8 weeks,4 but resistance to T798-related mutations.33 Overall, neratinib has a certain clinical efficacy against common HER2 mutations, but has not yet been marketed in China.
Pyrotinib is a new generation of small-molecule irreversible pan-ErbB receptor TKI, covalently binding with the ATP binding site in the intracellular kinase regions of HER1, HER2, and HER4, which prevents the formation of homodimers/heterodimers in the HER family, inhibits phosphorylation itself, blocks the activation of downstream signaling pathways, and inhibits tumor cell growth.38 Its structure and mechanism of action are similar to neratinib, but it has stronger inhibitory activity in vitro. In 2019, Jiang et al, reported a phenix study orally at the ASCO conference, confirming that to HER2 positive MBC females who previously received paclitaxel and trastuzumab, pyrotinib plus capecitabine, compared to placebo plus capecitabine, can effectively improve the median PFS. In this case, the patient developed disease progression in September 2018 after receiving trastuzumab combined with lapatinib, and received pyrotinib plus capecitabine. Three months later, she got partial response, (Figure 2) and symptoms were relieved. Plasma NGS indicated that both the copy number of HER2 amplification and the abundance of R157W mutations were decreased compared with previous results (Table 2). The relationship between gene dynamics and efficacy is shown in Figure 3.
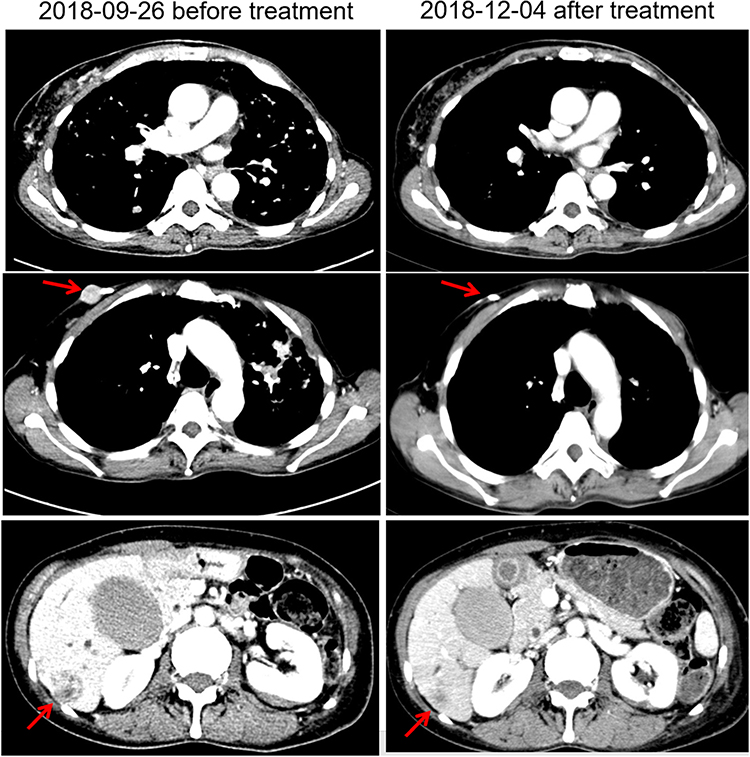 |
Figure 2 Changes of images before and after treatment. The red arrows indicate the metastatic tumor. |
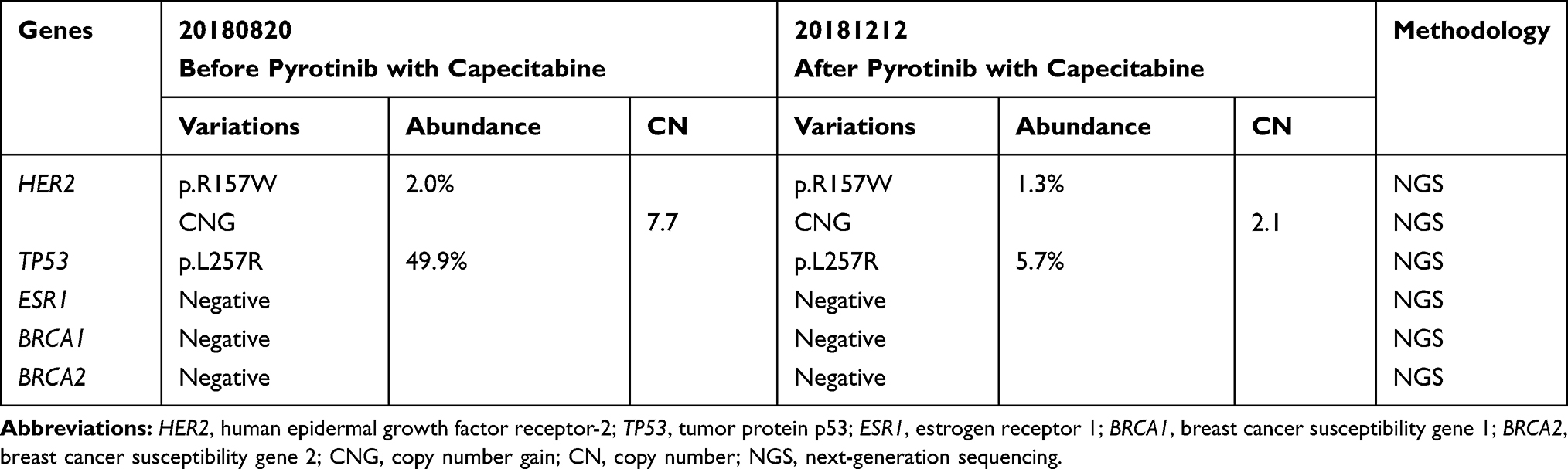 |
Table 2 NGS Results Detected Before and After Treatment with Pyrotinib and Capecitabine |
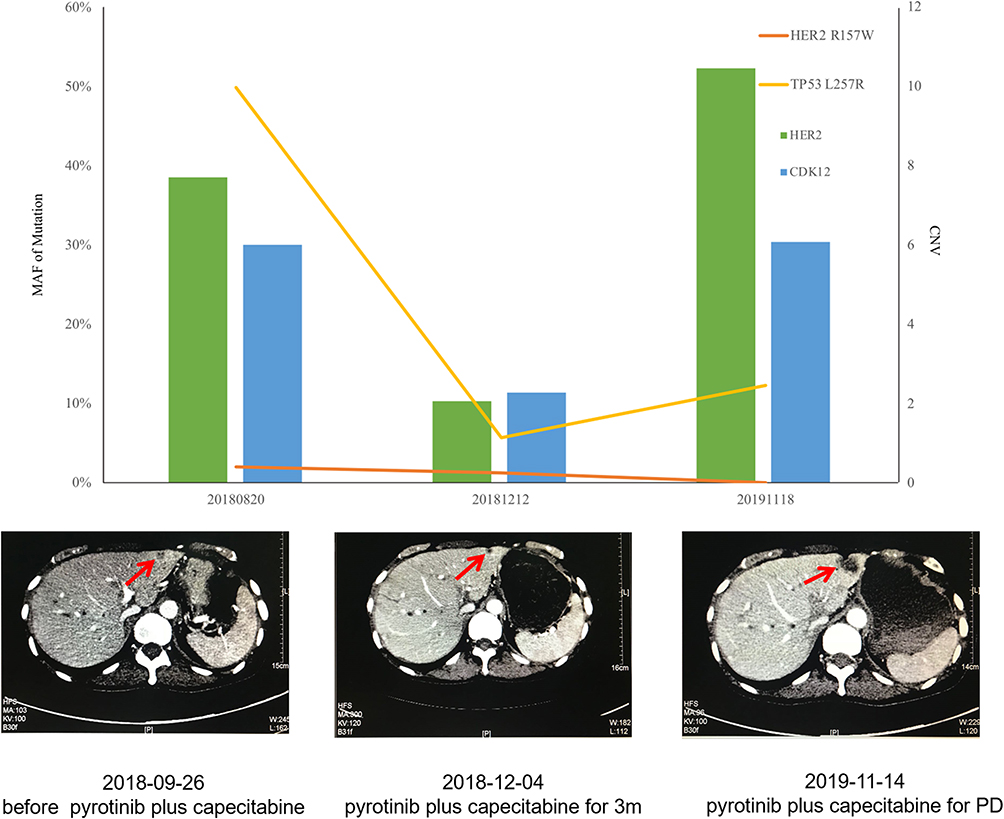 |
Figure 3 The relationship between gene dynamics and efficacy. The red arrows indicate the metastatic tumor. |
Conclusion
Drug resistance has affected the efficacy of anti-HER2 therapy in breast cancer, and HER2 mutation is one of the causes. However, there is no standard treatment yet. We report for the first time the occurrence of HER2 amplification accompanied by R157W mutation after anti-HER2 treatment. This case is the first clinical report of pyrotinib plus capecitabine effective for HER2 mutation, which shows a good clinical efficacy against HER2 resistance accompanied with mutation in MBC patients, and provides a possible new management strategy of anti-HER2 treatment for patients with HER2-positive breast cancer.
Ethical Approval
Institutional approval was not required to publish the case details.
Patient Informed Consent
Written informed consent was obtained from the patient for the publication of her case details and images.
Acknowledgments
The authors thank the patient and her family. This study did not receive any funding support from external sources. Yanchun Qu and Yufeng Liu are co-first authors for this study.
Disclosure
Xiaoyu Hong is the employee of Nanjing Geneseeq Technology Inc. The other authors have no conflicts of interest that are directly relevant to the content of this article.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi:10.3322/caac.21590
2. Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321:288–300.
3. Petrelli F, Tomasello G, Barni S, Lonati V, Passalacqua R, Ghidini M. Clinical and pathological characterization of HER2 mutations in human breast cancer: a systematic review of the literature. Breast Cancer Res Treat. 2017;166:339–349.
4. Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352.
5. Hyman DM, Piha-Paul SA, Won H, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. 2018;554:189–194.
6. Fang Y, Jiang Y, Wang X, Yang X, Gao Y, Wang J. Somatic mutations of the HER2 in metastatic breast cancer. Tumour Biol. 2014;35:11851–11854.
7. Park YH, Shin HT, Jung HH, et al. Role of HER2 mutations in refractory metastatic breast cancers: targeted sequencing results in patients with refractory breast cancer. Oncotarget. 2015;6:32027–32038.
8. Ross JS, Wang K, Gay LM, et al. A high frequency of activating extracellular domain ERBB2 (HER2) mutation in micropapillary urothelial carcinoma. Clin Cancer Res. 2014;20:68–75.
9. Wang T, Xu Y, Sheng S, et al. HER2 somatic mutations are associated with poor survival in HER2-negative breast cancers. Cancer Sci. 2017;108:671–677.
10. Pegram M, Slamon D. Biological rationale for HER2/neu (c-erbB2) as a target for monoclonal antibody therapy. Semin Oncol. 2000;27:13–19.
11. Harbeck N, Gnant M. Breast cancer. Lancet. 2017;389:1134–1150. doi:10.1016/S0140-6736(16)31891-8
12. Connell CM, Doherty GJ. Activating HER2 mutations as emerging targets in multiple solid cancers. ESMO Open. 2017;2:e279. doi:10.1136/esmoopen-2017-000279
13. Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284:99–110. doi:10.1016/S0014-4827(02)00099-X
14. Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–475. doi:10.1038/nrc2656
15. Helikar T, Kochi N, Kowal B, et al. A comprehensive, multi-scale dynamical model of ErbB receptor signal transduction in human mammary epithelial cells. PLoS One. 2013;8:e61757. doi:10.1371/journal.pone.0061757
16. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi:10.1126/science.3798106
17. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi:10.1038/35052073
18. Hsu JL, Hung MC. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016;35:575–588.
19. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792.
20. Marty M, Cognetti F, Maraninchi D, et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J Clin Oncol. 2005;23:4265–4274.
21. Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743.
22. Blackwell KL, Burstein HJ, Storniolo AM, et al. Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 Study. J Clin Oncol. 2012;30:2585–2592.
23. Mohd SM, Crown J, Hennessy BT. Overcoming resistance and restoring sensitivity to HER2-targeted therapies in breast cancer. Ann Oncol. 2012;23:3007–3016.
24. Takeda T, Yamamoto H, Kanzaki H, et al. Yes1 signaling mediates the resistance to Trastuzumab/Lap atinib in breast cancer. PLoS One. 2017;12:e171356.
25. Eustace AJ, Conlon NT, McDermott M, et al. Development of acquired resistance to lapatinib may sensitise HER2-positive breast cancer cells to apoptosis induction by obatoclax and TRAIL. BMC Cancer. 2018;18:965.
26. Esteva FJ, Yu D, Hung MC, Hortobagyi GN. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol. 2010;7:98–107.
27. Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–237.
28. Zuo WJ, Jiang YZ, Wang YJ, et al. Dual characteristics of novel HER2 kinase domain mutations in response to HER2-targeted therapies in human breast cancer. Clin Cancer Res. 2016;22:4859–4869.
29. Smyth LM, Piha-Paul SA, Won HH, et al. Efficacy and determinants of response to HER kinase inhibition in HER2-mutant metastatic breast cancer. Cancer Discov. 2020;10:198–213.
30. Trowe T, Boukouvala S, Calkins K, et al. EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res. 2008;14:2465–2475.
31. Kancha RK, von Bubnoff N, Bartosch N, Peschel C, Engh RA, Duyster J. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS One. 2011;6:e26760.
32. Kong X, Zhang K, Wang X, et al. Mechanism of trastuzumab resistance caused by HER-2 mutation in breast carcinomas. Cancer Manag Res. 2019;11:5971–5982.
33. Li J, Xiao Q, Bao Y, et al. HER2-L755S mutation induces hyperactive MAPK and PI3K-mTOR signaling, leading to resistance to HER2 tyrosine kinase inhibitor treatment. Cell Cycle. 2019;18:1513–1522.
34. Hanker AB, Brewer MR, Sheehan JH, et al. An acquired HER2(T798I) gatekeeper mutation induces resistance to neratinib in a patient with HER2 mutant-driven breast cancer. Cancer Discov. 2017;7:575–585.
35. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404.
36. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:l1.
37. Cocco E, Javier CF, Razavi P, et al. Neratinib is effective in breast tumors bearing both amplification and mutation of ERBB2 (HER2). Sci Signal. 2018;11.
38. Zhu Y, Li L, Zhang G, et al. Metabolic characterization of pyrotinib in humans by ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1033–1034:117–127.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.