Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
PARP-1 and SIRT-1 are Interacted in Diabetic Nephropathy by Activating AMPK/PGC-1α Signaling Pathway
Authors Zhu H, Fang Z, Chen J, Yang Y, Gan J ![]() , Luo L, Zhan X
, Luo L, Zhan X ![]()
Received 18 November 2020
Accepted for publication 12 January 2021
Published 25 January 2021 Volume 2021:14 Pages 355—366
DOI https://doi.org/10.2147/DMSO.S291314
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Hengmei Zhu,1,2,* Zhi Fang,3,* Jiehui Chen,2 Yun Yang,2 Jiacheng Gan,4 Liang Luo,5 Xiaojiang Zhan1
1Department of Nephrology, The First Affiliated Hospital of Nanchang University, Nanchang 330006, People’s Republic of China; 2Department of Nephrology, Huazhong University of Science and Technology Union Shenzhen Hospital, Shenzhen 518000, People’s Republic of China; 3Department of Oncology, The First Affiliated Hospital of Nanchang University, Nanchang 330006, People’s Republic of China; 4Department of Nuclear Medicine, Huazhong University of Science and Technology Union Shenzhen Hospital, Shenzhen 518000, People’s Republic of China; 5Department of Cardiology, Ganzhou People’s Hospital, Ganzhou 341000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liang Luo
Department of Cardiology, Ganzhou People’s Hospital, Ganzhou 341000, People’s Republic of China
Tel/Fax +8613807979503
Email [email protected]
Xiaojiang Zhan
Department of Nephrology, The First Affiliated Hospital of Nanchang University, Nanchang 330006, People’s Republic of China
Tel/Fax +8613507919885
Email [email protected]
Introduction: Diabetic nephropathy (DN) is a metabolic disorder characterized by the accumulation of extracellular matrix (ECM). This study aims to investigate whether exists an interplay between poly (ADP-ribose) polymerase 1 (PARP-1) and sirtuin 1 (SIRT-1) in DN via AMP-activated protein kinase (AMPK)/peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α) signaling pathway.
Methods: Eight-week-old male obese leptin-resistant (db/db) mice and nondiabetic control male C57BLKs/J (db/m) mice were used in this study. Body weight and blood glucose were evaluated after 6 h of fasting, which continues for 4 weeks. The kidney tissues were dissected for Western blot, immunofluorescence (IF) assay. Besides, PARP activity assay, MTT assay, NAD+ qualification, Western blot and IF were also performed to detect the level and relation of PARP-1 and SIRT-1 in mouse mesangial cells (MCs) with or without high glucose followed by inhibiting or elevating PARP-1 and SIRT-1, respectively.
Results: Western blotting shows PARP-1 and ECM marker fibronectin (FN) are upregulated while SIRT-1 is downregulated in db/db mice (p< 0.05) or in mouse MCs with high glucose (p< 0.05), which are significantly restored by PARP-1 inhibitor (PJ34) (p< 0.05) and SIRT-1 lentiviral transfected treatment (p< 0.05), or worsened by SIRT-1 inhibitor EX527 (p< 0.05). PJ34 treatment (p < 0.05) or SIRT-1 overexpression (p < 0.05) could increase PGC-1α and p-AMPK levels, concomitant with down expression of FN, however, were reversed in the presence of EX527 (p< 0.05).
Discussion: Our results suggest an important relationship between PARP-1 and SIRT-1 through AMPK-PGC-1α pathway, indicating a potential therapeutic method for DN.
Keywords: PARP-1, SIRT-1, diabetic nephropathy, AMPK/PGC-1α signaling pathway
Introduction
Diabetic nephropathy (DN), also referred to as diabetic kidney disease (DKD), is the leading cause of end-stage renal disease (ESRD).1,2 Characterized by high prevalence and poor prognosis, DN has become a research priority for scientists around the world.3 However, the precise pathogenesis of DN has not yet been fully elucidated. Recently, evidences show that the accumulation of extracellular matrix (ECM) has been involved in the pathogenesis of glomerulosclerosis, which is a characteristic pathological feature of DN.4 When subjected to metabolic, immunologic or hemodynamic injury, the kidney mesangial cells (MCs) are activated and then synthesize excessive ECM proteins.5 Hence, exploring the mechanism that regulates the accumulation of ECM could contribute to restoring kidney function. Previous study has shown that hyperglycemia is the major driving force of the progression from DN to ESRD.6,7 However, the link between higher levels of glucose and ECM accumulation is multifactorial and may include the overactivation of poly (ADP-ribose) polymerase 1 (PARP-1),7,8 and the reduction of sirtuin 1 (SIRT-1).9,10
PARPs constitute a family of several nicotinamide adenine dinucleotide (NAD+)-dependent nuclear enzymes which function as DNA damage sensor and signaling molecule.11 The activated PARP can recognize DNA breaks promptly along with the consumption of NAD+ and subsequently catalyze the synthesis of ADP-ribose polymers (PAR), which serves to initiate the DNA repair machinery and maintain genome integrity.12 However, overactivation of PARP-1 in the context of pathological extensive oxidative stress could reverse the protective role of PARP-1, resulting in cell necrosis or apoptosis.13 Increasing evidences show that the activation of PARP, especially of PARP-1 is involved in the development of multiple diseases, such as stroke,14 myocardial infarction,15 heart failure,16 vascular dysfunction,17 and mesenteric damage,18 muscle dysfunction,19,20 or renal ischemia-reperfusion injury,21 etc. Additionally, both pharmacological and genetic inhibition or deletion of PARP-1 produces striking reno-protection across several renal disease models.22–24 Thus, PARP-1 could be an attractive target for mechanistic and therapeutic investigation in DN.25
SIRT-1, a member of the silent information regulator 2 family, is the founding member of NAD+-dependent deacetylases, which is best known for its acknowledged relation to longevity regarding caloric restriction (CR).26,27 Recently, several investigators have found that SIRT-1 is associated with many other pathophysiological processes, particularly in age-related diseases, such as neurodegenerative disorders and osteoarthritis.28,29 In addition, evidences have shown that SIRT-1 can modulate the expression of target proteins and genes such as p53,29,30 peroxisome proliferator-activated receptor gamma coactivator 1 α (PGC-1α)31 and nuclear factor kappa B (NF-κB),32 which may participate in cellular pathways involving in metabolic diseases such as diabetes,33–35 although the contradictory consults which suggest the down-expression of SIRT-1 promotes the development of DN are also reported.36 Karbasforooshan et al37 and Wang et al32,38 elucidated again recently that the activity and expression of SIRT-1 were significantly decreased in diabetes and SIRT-1 may relate to the process of diabetes or diabetes-related diseases, indicating that SIRT-1 may also serve as a possible therapeutic target for DN. However, the way in which SIRT-1 contributes to kidney ECM accumulation and then leads to the development of DN remains to be determined.
The pathophysiological overactivation of PARP-1 consumes a large amount of NAD+, leading to NAD+ depletion sufficient to inhibit SIRT-1 and lead to cell death due to the same NAD+ they are competing for.39 Previously, Waldman et al40 illustrated that inhibiting PARP-1 by PARP-1 inhibitor can protect diabetic cardiomyocytes from extensive oxidative stress through the activation of SIRT-1-PGC-1α pathway. In addition, Lu et al14 also found that PARP-1-mediated NAD+ depletion inhibited the activity of SIRT-1, thereby leading to PGC-1α hyperacetylation that impacted mitochondrial function and subsequently caused neuron cell death. However, there are no current studies have confirmed whether exists a concrete cross talk between PARP-1 and SIRT-1 in DN. According to the researches, the AMPK/PGC-1α signaling pathway is thought to be the key mechanism to regulate the mitochondrial function, lipid metabolism and cell growth, which plays a crucial role in the pathophysiological mechanism of DN.41,42 Accumulating studies on AMPK/PCG-1α signaling pathway have shown that the treatment of many drugs significantly activated AMPK in type 2 diabetes (T2D) induced nephropathy, thereby activating PGC-1α, which was otherwise inhibited by T2D, such as 4-O-methylhonokiol,43 extracellular superoxide dismutase,44 okra,45 etc. However, evidences regarding the effects of PARP-1 and SIRT-1 through activation of AMPK/PGC-1α in DN are poorly understood.
In the current study, we demonstrated that there was an interesting relationship between PARP-1 and SIRT-1 that participated in the pathogenesis of DN via SIRT-1-AMPK-PGC-1α axis, which may open new avenues to help develop novel drugs for the prevention of DN.
Materials and Methods
Reagents
D-glucose was purchased from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum, DMEM/F12 medium and TRIzolTM reagent were purchased from Thermo Fisher scientific Invitrogen (Carlsbad, CA, USA). Rabbit anti-collagen-4 polyclonal antibody (ab6586) and rabbit anti-fibronectin polyclonal antibody (ab2413) were from Abcam (Cambridge, MA, USA). Rabbit anti-PARP-1 (46D11) polyclonal antibody (#9532), SIRT-1 antibody sampler Kit (#9787), Phospho-AMPKα (Thr172) (D4D6D) rabbit mAb (#50,081), AMPKα (D63G4) rabbit mAb (#5832), Acetylated-Lysine antibody (#9441) and β- Actin (8H10D10) mouse mAb (#3700) were from Cell Signaling Technology (Beverly, MA, USA). Rabbit anti- PGC-1α (AF5395) was from Affinity Biosciences (Cincinnati, OH, USA). PJ-34 hydrochloride hydrate/P4365 and EX-527/E7034 were from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). SIRT-1 lentiviral (sirtuin 1 Gene ID: 93,759, LV5 EF-1aF/GFP& Puro) was from gene pharma (Jiangsu, China). Cell lysis, mouse anti-rat β- actin monoclonal antibody and horseradish peroxidase-labeled secondary antibody against rabbit were purchased from Cell Signaling Technology (Beverly, MA, USA). HRP-labeled rabbit anti-mouse IgG were purchased from Beyotime (Shanghai, China). The Revert AidTM First Strand cDNA reverse transcription kit was purchased from Fermentas (Canada). The Universal PARP Colorimetric Assay Kit (4684–096-K) was purchased from Trevigen Inc. (Gaithersburg, MD, USA).
Establishment of Animal Model
Six 8-week-old male db/db (Lepr db/db) mice (36.5–38.9g) and six nondiabetic control male db/m mice C57BLKs/J (Jackson Laboratories, Bar Harbor, ME) (18.6–20.6g) were purchased from Nanjing Model Animal Center, Jiangsu, China. All animals were housed at proper room temperature (26°C) and humidity (70%) under a controlled light/dark cycle and had free access to water. All animals should be adaptively fed.46 The db/m mice and the db/db mice were fed with standard diet (12% fat, 28% protein and 60% carbohydrates). Blood glucose was randomly measured with Glucometer Elite (Bayer) after 6 h of fasting. Then, DN can be diagnosed when the urine albumin creatinine ratio (ACR) of the db/db mice was over 50 μg/mg and random blood glucose of the db/db mice was over 16.7 mM.46 After collecting the data of blood glucose and 24-hour urine for 4 weeks, animals were anesthetized and killed, then the renal tissues were dissected for the following experiments, respectively. All animal experiments were guided in accordance with the guidelines of the Care and Use of Laboratory Animals by the National Institutes of Health, and all efforts were made to minimize animal suffering.
Cell Culture and in vitro Experimental Design
Mouse MCs line SV40 MES13 that were purchased from the cell bank of Chinese Academy of Sciences were kindly provided by Associate Prof. Hu Wenxue, which was approved by the Department of Science and Technology of the First Affiliated Hospital of Nanchang University.
The cell was cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Grand Island, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, CA, USA), 100 U/mL penicillin and 100 mg/mL streptomycin under standard culture conditions (37°C in 5% CO2). For all following experiments, cells were cultured in serum-free conditions for 24 hours when they reached 80% confluence. Growth-arrested MCs were incubated with normal glucose (Control, 5 mM) or high glucose (HG, 25 mM) medium for 48 hours, respectively (see Supplementary data 1 and Figure 1). And then the HG-treated-cells were pretreated with the PARP-1 inhibitor PJ34 (3 µL) and/or the SIRT-1 inhibitor EX527 (2 µL) for 2 h (See Supplementary data 2 and Figure 2). Moreover, the HG group should be transfected with SIRT-1 lentiviral (See Supplementary data 3 and Figure 3) with or without PJ34 (3 µL). All doses and times of these treatments were determined in our pre-experimental results and previous study.47 As described above, MCs were then divided into the following groups for subsequent experiments: G5, G25, G25+PJ34, G25+EX527, G25+PJ34+EX527, G25+SIRT-1 lentivirus transfected (G25+Lv-SIRT-1), G25+SIRT-1 lentivirus transfected+PJ34 (G25+Lv-SIRT-1+PJ34).
Lentiviral Treatment of MC Cultures
To achieve SIRT-1 overexpression in MCs, we transfected HG-treated-MCs with SIRT-1 overexpression-lentiviral (purchased from gene pharma) according to the manufacturer’s instructions.48 Control cells were transfected with negative controls. MCs were infected at 1 d in vitro with a multiplicity of infection (MOI) of 1 in the presence of 1 μg/mL polybrene for 6 h. Then, the transfection medium was replaced with fresh DMEM. Subsequent assays were conducted 48 h after transfection. The overexpression of SIRT-1 was confirmed by fluorescence-inverted microscope.
Western Blot Analysis
Protein samples were extracted from cells or kidney tissues as described previously.24 Briefly, treated cells or kidney tissues were lysed in RIPA lysis buffer containing protease inhibitor and the protein concentrations were finally determined by BCA kit. Then, the protein samples were fractionated by SDS-PAGE (10–15% polyacrylamide gels) and then transferred to PVDF membranes, which were blocked and then incubated with the appropriate primary antibodies and secondary antibody. Primary antibodies were used to detect PARP-1 (1:1000; Cell Signaling Technology, Beverly, MA), SIRT-1 (1:1000; Cell Signaling Technology, Beverly, MA), p-AMPK (Thr172) (1:1000; Cell Signaling Technology, Beverly, MA), FN (1:500; Abcam, Cambridge, MA), PGC-1α (1:500; Affinity Biosciences, Cincinnati, OH).35 β-Actin (1:1000; Cell Signaling Technology, Beverly, MA) was used as an internal control. The protein expression levels of the target genes were quantified by relative densitometry performed by ImageJ (National Institutes of Health) and normalized to the protein expression levels of β-actin. The values were shown as changes relative to those of the control sample.49
Immunofluorescence (IF) Assay
MCs on 35 mm glass dishes were treated as described previously.50 The plates were washed with cold PBS and fixed with 4% paraformaldehyde for 20 min at room temperature. After permeabilizing with 0.3% of triton, the unspecific sites of the cells were blocked by 1% BSA + 0.3% triton. Then, the cells were incubated with primary antibody experimenters used before,35 rabbit anti-SIRT-1 diluted 1:10 (Santa Cruz Biotechnology) and mouse anti-PARP-1 diluted 1:10 (Enzo Life Sciences, Farmingdale NY) overnight at 4°C and incubated with fluorescence conjugated secondary antibodies (Beyotime, China) counterstained with DAPI next day. Images were analyzed under a fluorescence-inverted microscope.
PARP Activity Assay
PARP activity was tested by detecting the quantity of biotinylated-NAD+ converting into nicotinamide and biotinylated-ADP ribose polymers via the universal colorimetric PARP assay kit (Trevigen Inc., Gaithersburg, MD). The activity of PARP-1 was detected in accordance with the manufacturer’s instructions. Cell lysates from MCs which contains about 50 µg of protein were added into a 96-well plate coated with histones and biotinylated poly-ADP-ribose, after 1h-incubation, treated the cells with streptavidin-horse-radish peroxidase, and read at 450 nm in a spectrophotometer.51,52
MTT Assay
Presently, the MTT (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay is widely used to access the viability and the cytotoxicity of cells.53 To detect MCs viability, MTT assay was performed according to the manufacturer’s instructions. MCs were seeded in 96-well culture plates at a density of 1×104 cells per well. After an-overnight adsorption, the cells were incubated with medium either containing 5 mM glucose or 25 mM glucose, with PJ34 and/or EX527, respectively. And 25 mM glucose medium containing SIRT-1 overexpression lentiviral with or without PJ34 was also considered as experimental groups. Followed by incubation with 100 µL MTT solution (10 mg/mL in PBS) for 4h, the MCs were further lysed, and the purple formazan crystals were solubilized with 100 µL DMSO (Sigma-Aldrich). The wells containing only medium without MCs were set as a blank. Finally, we used an enzyme-linked immunosorbent assay reader (Thermo LabSystems, OY, Helsinki, Finland) to analyze the absorbance at 570 nm.50
NAD+ Qualification
NAD+ in db/db kidney tissues and HG-induced MCs was estimated colorimetrically using an NAD+/NADH assay kit (Abcam, Cambridge, MA) as described previously.50 The standard was prepared according to the manufacturer’s protocol.
Statistical Analysis
All data are presented as mean ± SD deviation from at least three independent experiments. Differences between two groups were evaluated using Student’s t-test, and multiple comparisons were performed using one-way analysis of variance (ANOVA) followed by a Student–Newman–Keuls (SNK) test. A p value <0.05 was considered statistically significant.
Results
PARP-1 Activation and SIRT-1 Inhibition in db/db Mice Induced by Hyperglycemia
To study the interplay between SIRT-1 and PARP-1 in vivo, we induced hyperglycemia by using a mouse model of insulin resistance, which has been shown to generate ROS and DNA damage in kidney tissues.23 As shown in Figure 1A and B, the activity of PARP-1 increased markedly in the renal tissues from db/db mice (DN group) compared to control db/m mouse (NC group) (p < 0.05), which followed by increasing expression of ECM protein fibronectin (FN) (p < 0.001) and decreasing expression of SIRT-1 (p < 0.05). Considering the results of many researchers in explaining the association between overexpression of PARP-1 and down-expression of SIRT-1 in diabetic cells, it is reasonable to consider the decrease expression of SIRT-1 in the kidney of db/db mice significant.14,40 In addition, the fluorescence of PARP-1 was stronger in the glomeruli of DN mice; however, the fluorescence of SIRT-1 was stronger in the NC mice, which demonstrated that the expression of PARP-1 was significantly higher in the glomeruli of the untreated DN mice compared to that of NC mice, on the contrary, the level of SIRT-1 in DN group was significantly lower compared to the NC group (Figure 1C). These results strongly suggested that hyperglycemia may lead to the accumulation of FN and increase of PARP-1 along with the decline of SIRT-1, which indicates a potential link between PARP-1 and SIRT-1 in diabetic mice.
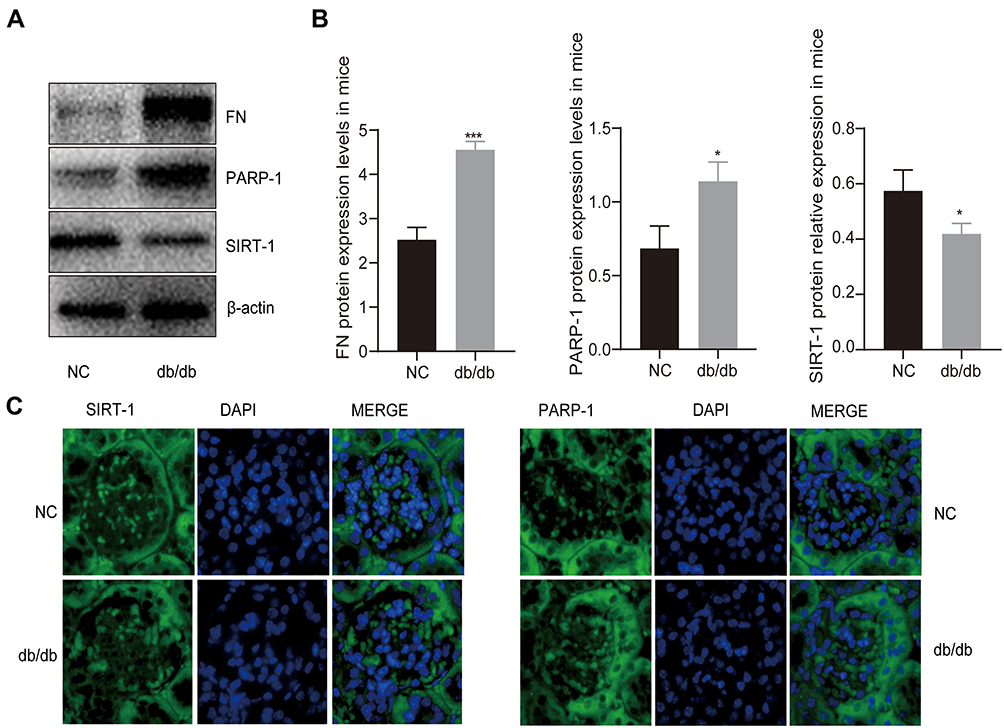 |
Figure 1 The expression of PARP-1 and SIRT-1 in db/db mice and db/m mice. (A and B) Western blotting results showed hyperglycemia decreased the levels of SIRT-1 but increased the activation of PARP-1 and the expression of FN in db/db mice compared to db/m group. (C) Immunofluorescence images showed that the expression of PARP-1 was increased in db/db group mice glomerulus compared with db/m group mice glomerulus, which was contrary to the expression of SIRT-1. Blue, nuclear staining (DAPI); green, target protein staining. Data are presented as mean ± SD. We conduct 3 independent experiments. *Compared with NC, P < 0.05; ***compared with NC, P < 0.001. |
The Effect of PARP-1 and SIRT-1 in High Glucose-Induced Activating MCs
To further investigate the role of SIRT-1 and PARP-1 in high glucose-stimulated MCs in vitro, we applied PJ34, EX527 or Lv-SIRT-1 to co-incubate MCs with high glucose according to the above research. We postulated that PARP-1 inhibition will lead to the activation of SIRT-1 since both PARP-1 and SIRT1 are NAD+ dependent (See Supplementary data 4 and Figure 4).39 As is shown in Figure 2A and B, high glucose (25 mM) over-activated PARP-1 (p < 0.05) and declined SIRT-1 protein expression (p < 0.001), thus leading to increase FN protein expression (p < 0.05), compared to that of normal glucose (5 mM). PARP-1 inhibitor PJ34 alleviated the protein level of PARP-1 (p < 0.05), promoted the expression SIRT-1 (p < 0.05) and FN (p < 0.01) while SIRT-1 inhibitor EX527 worsened the above change (p < 0.05) (Figure 2A and B). Moreover, when PJ34 was incubated with EX527, the changes in PARP-1, SIRT-1 and FN protein levels were between the PJ34 and EX527 groups (Figure 2A and B). In addition, Lv-SIRT-1 totally reversed the protein level of PARP-1, SIRT-1 and FN, while incubation PJ34 with Lv-SIRT-1 did even better (p < 0.05) (Figure 2A and B). The above results are consistent with the results in immunofluorescence (Figure 2C), which indicates that PARP-1 and SIRT-1 may exert a completely different role in DN.
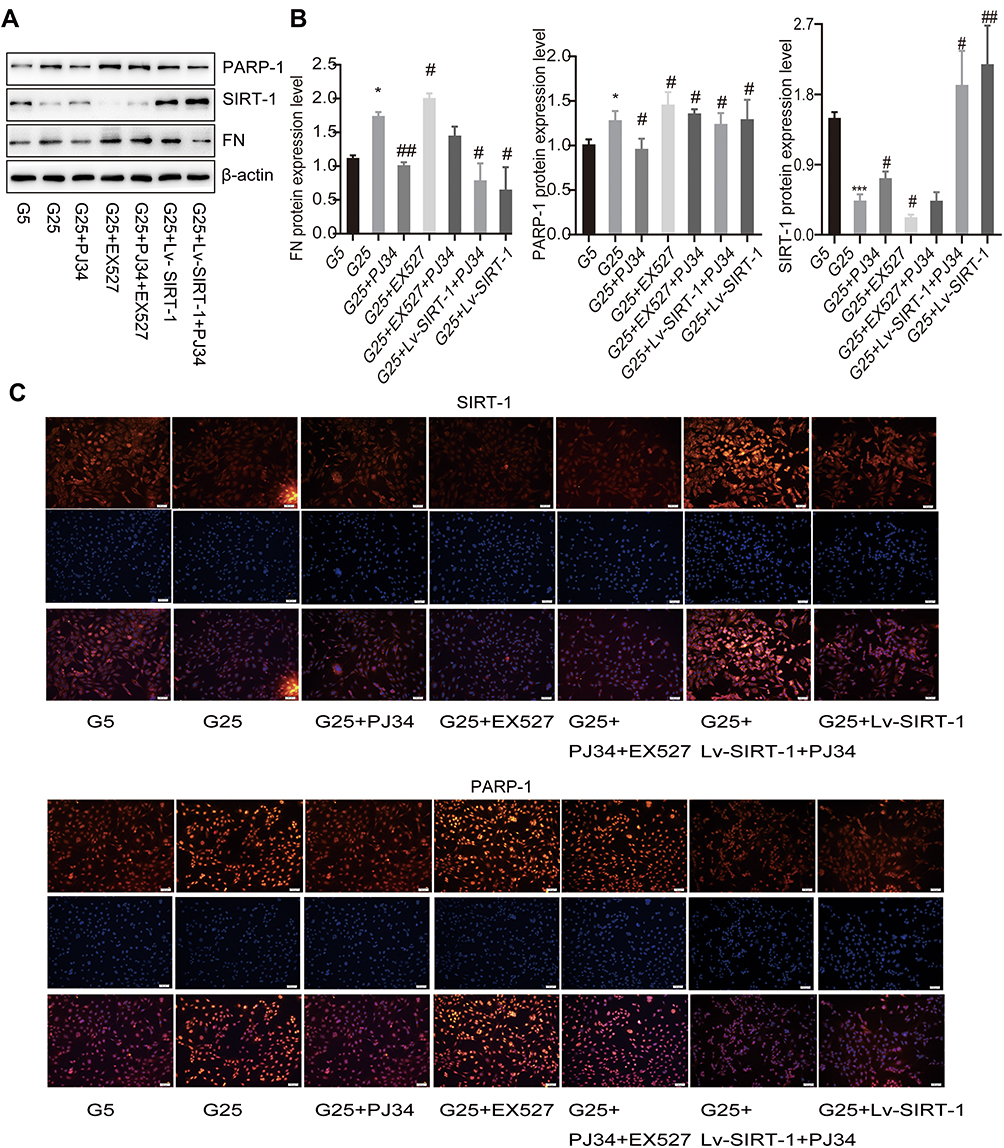 |
Figure 2 The effect of PARP-1 and SIRT-1 in MCs. (A and B) Proteins expression in MCs in high glucose condition or low glucose condition incubated with different reagents by Western blotting. β-Actin was used as control of protein loading. Bars represent mean ± SD of three independent experiments. *Compared with G5, P < 0.05; ***compared with G5, P < 0.001. #Compared with G25, P < 0.05; ##compared with G25, P < 0.01. (C) Immunofluorescence images showed the change of protein SIRT-1 and PARP-1 expression in different MCs groups. Blue, nuclear staining (DAPI); green, target protein staining. Images were analyzed by confocal microscopy under a 50-μm ruler. |
The Change of Cell Viability is Related to the Activity of PARP-1
In the meanwhile, we examined the viability of hyperglycemia induced-MCs by MTT assay in different groups (Figure 3B). Hyperglycemia can strongly elevate the viability of MCs compared to the low glucose (P < 0.01) and this promotion can be increased or declined by EX527 (p < 0.001) and PJ34 (p < 0.001), respectively. Overexpression SIRT-1 also cause a decline in cell viability (p < 0.001). In addition, when extra PJ34 and EX527 are added to high glucose to stimulate MCs, the changes in cell viability were between the PJ34 and EX527 groups (Figure 3B). The likely reason is that PARP-1 exposure to high glucose levels induces a promotion of cell viability in the MCs, while SIRT-1 possesses a reverse role. To further determine the effect of PARP-1 on hyperglycemia-induced MC activation, the activity of PARP-1 was observed (Figure 3A). The trend and outcomes were much the same between the PARP-1 activity and cell viability in each group, which further demonstrated PARP-1 and SIRT-1 had an opposite effect in diabetic kidney tissues.
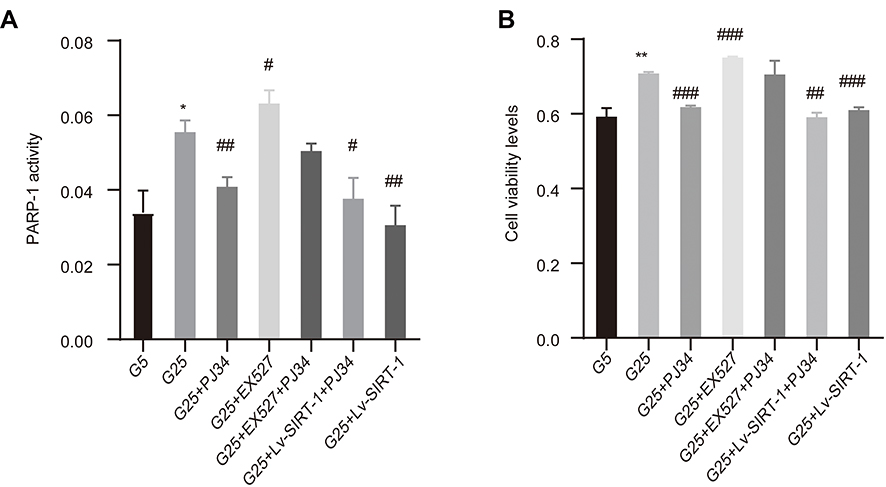 |
Figure 3 The result of PARP-1 activity and cell viability. (A) PARP-1 activity was measured by PARP-1 activity kit. (B) Cell viability measured by MTT assay upon 48 h treatment of different reagents. Data of three experiments are presented as mean ± SD. *Compared with G5, P < 0.05; **compared with G5, P < 0.01. #Compared with G25, P < 0.05; ##compared with G25, P < 0.01; ###compared with G25, P < 0.001. |
PARP-1-Induced SIRT-1 Down-Regulation is Mediated by the SIRT-1-AMPK-PGC-1α Signaling Pathway
Both PARP-1 and SIRT-1 affect chromatin remodeling and thus modulation of gene expression affecting inflammation and diabetic related diseases.54 In addition, the interaction of PARP-1 and SIRT-1 plays a role in the regulation of oxidative stress and diabetic related diseases.35,55 To clarify the molecular mechanism of how PARP-1 regulates SIRT-1 expression, we screened a variety of target proteins of which SIRT-1 can participate in the deacetylation, such as PGC-1α and p-AMPK (Thr172) (Figure 4A and B). We determined that an increase in SIRT-1 expression was associated with elevation of protein PGC-1α (p < 0.05) and p-AMPK (p < 0.05) expression in the MCs, accompanied by a drop while inhibiting the level of protein SIRT-1 (p < 0.05) and reversed by an inhibition of PARP-1 (Figure 4A and B). In conclusion, these results suggest a direct relationship between SIRT-1 and PGC-1α protecting renal tissues against oxidative stress and accumulation ECM which participate in the pathogenesis of DN disease. And inhibiting PARP-1 leading to an up-expression of SIRT-1 can be considered as an effective therapeutic target for DN (summarized in Figure 4C).
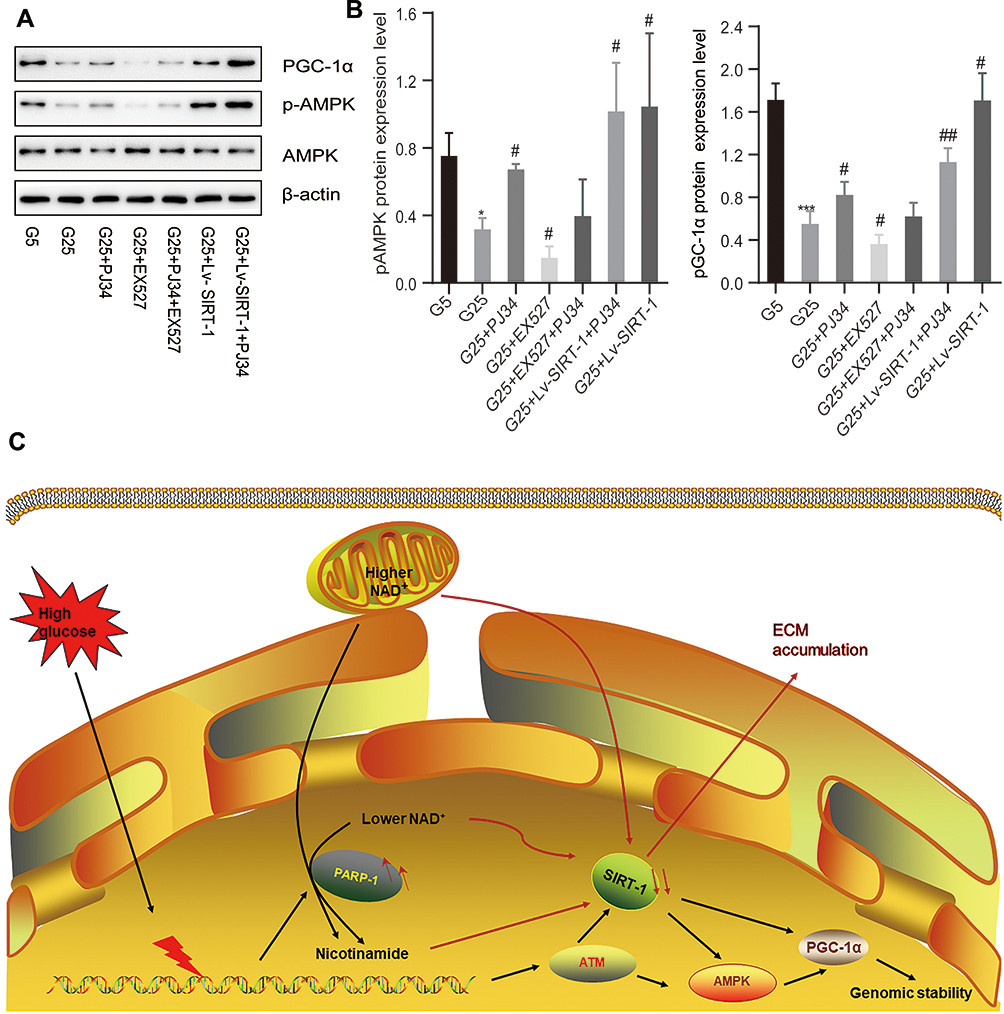 |
Figure 4 PARP-1 may have a crosslink with SIRT-1 via PGC-1α axis. (A and B) The expression of pAMPK and PGC-1α in MCs in high glucose condition or low glucose condition incubated with different reagents by Western blotting. Data of three experiments are presented as mean ± SD. *Compared with G5, P < 0.05; ***compared with G5, P < 0.001. #Compared with G25, P < 0.05, ##compared with G25, P < 0.01. (C) The possible pathway of the accumulation of ECM in DN patients: PARP-1-NAD+-SIRT-1-PGC-1α axis. High glucose can cause a strong stress to DNA replication, which then be transduced by DNA damage response sensors, such as PARP-1, ataxia telangiectasia mutated (ATM). Then SIRT-1 and AMPK propagate the signal via posttranslational modifications of PGC-1α and other proteins to maintain the integrity of genome. However, the overactivation of PARP-1 can reverse this protectable role, which subsequently contributes to the subcellular difference in NAD+ content between the nucleus and mitochondria and leads to the down-expression of SIRT-1 and finally causes the activation of ECM. The red arrow refers to the pathological phenomenon caused by excessive activation of PARP-1. |
Discussion
The accumulation of ECM plays a critical role in the pathophysiologic of glomerulosclerosis in DN, which can be triggered by various pathological conditions such as oxidative stress and autophagy.4,56 A plethora of evidences have shown a crucial role of SIRT-1 in retarding the ECM accumulation during MCs injury.57,58 In addition, Chen et al59 determined a significant connection of PARP-1 and ECM in the effect of fluorofenidone attenuating experimental cyclosporine A-induced kidney injuries through a variety of molecular biology experiments. And Xu et al60 also illustrated that hyperhexosemia and diabetes cause an upregulation of ECM proteins at the transcriptional level in the kidney via a signaling pathway mediated by PARP, which can be subsequently attenuated by PARP blockade. These findings suggest that PARP-1 attributes to the ECM accumulation in kidney. Moreover, previous studies indicated that there was a direct effect of SIRT-1 on PARP-1 regulation through the deacetylation of PARP-1 in many other kidney-related diseases.20,61 However, the precise connection between SIRT-1 and PARP-1 in ECM accumulation in DN remains unknown.
To elucidate this point, we measured the expression of PARP-1 and SIRT-1, as well as FN in kidney tissue of db/db mice and wild-type mice. The increase in PARP-1 activity and the reduction expression of SIRT-1 were detected, coupled with increased FN expression in db/db mice kidney compared with wildtype mice kidney. PARP-1 inhibitor treatment by PJ34 reversed these changes, suggesting that there may exist a potential relationship between PARP-1 and SIRT-1 and down-expression of PARP-1 could exert a vital protective role in the diabetic mice.
We further tested the respective effects of an overexpression or decline of SIRT-1 on PARP-1 and an inhibitor of PARP-1 on SIRT-1 in hyperglycemia-activated MCs. Hyperglycemia induced the accumulation of ECM in MCs and increased the expression of FN and PARP-1, followed by the down-expression of SIRT-1. The reduction in PARP-1 activity resulting from PARP-1 inhibition by PJ34 in the hyperglycemia mice was followed by increasing SIRT-1 and FN expression in MCs. And PARP-1 levels were significantly declined in MCs treated with overexpression SIRT-1 or PJ34 and increased after down-expression SIRT-1 via the treatment of EX257. These findings indicate that SIRT-1 and PARP-1 have an interplay in hyperglycemia-activated MCs.
In addition, PARP-1 can be modulated by PGC-1α signaling.14,39,40 Wadman et al40 reported that activation of SIRT-1-PGC-1α signaling pathway can protect cardiomyocytes from suffering diabetes through downregulating PARP-1. In this study, we found that hyperglycemia-induced MC activation suppressed SIRT-1 activity along with declining PGC-1α signaling, both of which were reversed by PARP-1 inhibitor PJ34. The possible mechanism employed by PJ34 is the inhibition of AMPK-PGC-1α signaling by declining SIRT-1 deacetylation. To prove this hypothesis, MCs were pretreated with the Lv-SIRT-1 or inhibitor EX257. MCs treated with Lv-SIRT-1 showed enhanced PGC-1α and higher p-AMPK levels compared to PJ34 group, while a completely opposite effect was seen with inhibitor EX527 treatment. Therefore, PARP-1 acts as an SIRT-1 activity opponent to inhibit the deacetylation of PGC-1α and enhances DNA breaks in hyperglycemia-activated MCs.
To date, the role of PARP-1 and SIRT-1 in DN remains still controversial. In this experiment, an obvious increase of PARP-1 and a dramatic decrease of SIRT-1 were observed in hyperglycemia-activated MCs and db/db mice followed by the imbalance of AMPK/PGC-1α pathway, suggesting it reasonable to consider PARP-1 and SIRT-1 as adaptive targets to combat kidney damage caused by hyperglycemia in the future. The treatment of regulating AMPK/PGC-1α pathway could protect the kidney from damage caused by hyperglycemia and possibly prevents stress-induced PARP-1 activation.
Conclusion
In summary, we elucidated the possibility of a potential relationship between PARP-1 and SIRT-1 at the animal and cell levels, respectively. Our study suggests that PARP-1 involves in the downregulation of SIRT-1 impeding renal function in db/db mice. And the reduction of PARP-1 could protect MCs from hyperglycemia-induced MC activation ameliorating renal ECM accumulation via SIRT-1-AMPK-PGC-1α pathway. These findings provide the experimental basis to further investigate the therapeutic target to DN through this intricate connection between PARP-1 and SIRT-1.
Abbreviations
DN, diabetic nephropathy; ECM, extracellular matrix; PARP-1, poly (ADP-ribose) polymerase 1; SIRT-1, sirtuin 1; AMPK, AMP-activated protein kinase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1- α; HG, high glucose; FN, fibronectin; DKD, diabetic kidney disease; ESRD, end-stage renal disease; NAD+, nicotinamide adenine dinucleotide; PAR, ADP-ribose polymers; CR, caloric restriction; NF-κB, nuclear factor kappa B; DMEM, Dulbecco’s Modified Eagle Medium; FBS, fetal bovine serum; MOI, multiplicity of infection.
Data Sharing Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All animal experiments were guided in accordance with the guidelines of the National Institutes of Health, eighth edition, 2011. All experiments involving ethics have been ethically approved by the Department of Science and Technology of the First Affiliated Hospital of Nanchang University, and the ethics number is 2020 Medical Research Ethics No. 116.
Acknowledgments
The authors thank all of the members in their lab. Thanks to the Shenzhen Science and Technology Innovation Commission and the Jiangxi Natural Sciences Foundation Commission for providing financial support for us.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. Hengmei Zhu and Zhi Fang contributed to the work equally. Everyone participated in the final approval of the manuscript.
Funding
This work was supported by the Basic Research Project of Shenzhen Science and Technology Innovation Commission (JCYJ20160429181842402, JCYJ20190809112003711) and Jiangxi Natural Sciences Youth Science Foundation-Youth Fund Project (20202BAB216007).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Breyer MD, Susztak K. The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov. 2016;15(8):568–588.
2. Papadopoulou-Marketou N, Kanaka-Gantenbein C, Marketos N, Chrousos GP, Papassotiriou I. Biomarkers of diabetic nephropathy: a 2017 update. Crit Rev Clin Lab Sci. 2017;54(5):326–342. doi:10.1080/10408363.2017.1377682
3. Gheith O, Farouk N, Nampoory N, Halim MA, Al-Otaibi T. Diabetic kidney disease: world wide difference of prevalence and risk factors. J Nephropharmacol. 2016;5(1):49–56.
4. Liu HF, Liu H, Lv LL, et al. CCN3 suppresses TGF-beta1-induced extracellular matrix accumulation in human mesangial cells in vitro. Acta Pharmacol Sin. 2018;39(2):222–229. doi:10.1038/aps.2017.87
5. Abboud HE. Mesangial cell biology. Exp Cell Res. 2012;318(9):979–985. doi:10.1016/j.yexcr.2012.02.025
6. Liu J, Wang C, Liu F, Lu Y, Cheng J. Metabonomics revealed xanthine oxidase-induced oxidative stress and inflammation in the pathogenesis of diabetic nephropathy. Anal Bioanal Chem. 2015;407(9):2569–2579. doi:10.1007/s00216-015-8481-0
7. Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond). 2013;124(3):139–152. doi:10.1042/CS20120198
8. Sifuentes-Franco S, Padilla-Tejeda DE, Carrillo-Ibarra S, Miranda-Diaz AG. Oxidative stress, apoptosis, and mitochondrial function in diabetic nephropathy. Int J Endocrinol. 2018;2018:1875870. doi:10.1155/2018/1875870
9. Kitada M, Kume S, Takeda-Watanabe A, Kanasaki K, Koya D. Sirtuins and renal diseases: relationship with aging and diabetic nephropathy. Clin Sci (Lond). 2013;124(3):153–164. doi:10.1042/CS20120190
10. Hao CM, Haase VH. Sirtuins and their relevance to the kidney. J Am Soc Nephrol. 2010;21(10):1620–1627. doi:10.1681/ASN.2010010046
11. Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4(5):421–440. doi:10.1038/nrd1718
12. Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13(13):3046–3082. doi:10.2741/2909
13. Bai P. Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol Cell. 2015;58(6):947–958. doi:10.1016/j.molcel.2015.01.034
14. Lu P, Hogan-Cann AD, Kamboj A, et al. Poly(ADP-ribose) polymerase-1 inhibits mitochondrial respiration by suppressing PGC-1alpha activity in neurons. Neuropharmacology. 2019;160:107755. doi:10.1016/j.neuropharm.2019.107755
15. Farivar AS, McCourtie AS, MacKinnon-Patterson BC, et al. Poly (ADP) ribose polymerase inhibition improves rat cardiac allograft survival. Ann Thorac Surg. 2005;80(3):950–956. doi:10.1016/j.athoracsur.2005.02.035
16. Mormile R, Vittori G, De Michele M, Squarcia U, Quaini F. Is a deceptive role of IGF-1 in Sirt1-PARP1 interactions the primary step of postnatal regression of hypertrophic cardiomyopathy in infants of diabetic mothers? Int J Cardiol. 2012;154(1):87–88. doi:10.1016/j.ijcard.2011.10.072
17. Eros K, Magyar K, Deres L, et al. Chronic PARP-1 inhibition reduces carotid vessel remodeling and oxidative damage of the dorsal hippocampus in spontaneously hypertensive rats. PLoS One. 2017;12(3):e0174401. doi:10.1371/journal.pone.0174401
18. Li X, Ling Y, Cao Z. Targeting intestinal epithelial cell-programmed necrosis alleviates tissue injury after intestinal ischemia/reperfusion in rats. J Surg Res. 2018;225:108–117. doi:10.1016/j.jss.2018.01.007
19. Meng YY, Wu CW, Yu B, Li H, Chen M, Qi GX. PARP-1 involvement in autophagy and their roles in apoptosis of vascular smooth muscle cells under oxidative stress. Folia Biol (Praha). 2018;64(3):103–111.
20. Mohamed JS, Wilson JC, Myers MJ, Sisson KJ, Alway SE. Dysregulation of SIRT-1 in aging mice increases skeletal muscle fatigue by a PARP-1-dependent mechanism. Aging (Albany NY). 2014;6(10):820–834. doi:10.18632/aging.100696
21. Del Moral RM, Gomez-Morales M, Hernandez-Cortes P, et al. PARP inhibition attenuates histopathological lesion in ischemia/reperfusion renal mouse model after cold prolonged ischemia. ScientificWorldJournal. 2013;2013:486574. doi:10.1155/2013/486574
22. Minchenko AG, Stevens MJ, White L, et al. Diabetes-induced overexpression of endothelin-1 and endothelin receptors in the rat renal cortex is mediated via poly(ADP-ribose) polymerase activation. FASEB J. 2003;17(11):1514–1516. doi:10.1096/fj.03-0013fje
23. Szabo C, Biser A, Benko R, Bottinger E, Susztak K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes. 2006;55(11):3004–3012. doi:10.2337/db06-0147
24. Chow BSM, Allen TJ. Mouse models for studying diabetic nephropathy. Curr Protoc Mouse Biol. 2015;5(2):85–94. doi:10.1002/9780470942390.mo140192
25. Peixoto EB, Papadimitriou A, Lopes de Faria JM, Lopes de Faria JB. Tempol reduces podocyte apoptosis via PARP signaling pathway in experimental diabetes mellitus. Nephron Exp Nephrol. 2012;120(2):e81–90. doi:10.1159/000337364
26. Guarente L, Franklin H. Epstein lecture: sirtuins, aging, and medicine. N Engl J Med. 2011;364(23):2235–2244. doi:10.1056/NEJMra1100831
27. Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149–159. doi:10.1016/S0092-8674(01)00527-X
28. Gagarina V, Gabay O, Dvir-Ginzberg M, et al. SirT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 2010;62(5):1383–1392. doi:10.1002/art.27369
29. Zhang Z, Lin J, Nisar M, et al. The Sirt1/P53 axis in diabetic intervertebral disc degeneration pathogenesis and therapeutics. Oxid Med Cell Longev. 2019;2019:7959573.
30. Daenthanasanmak A, Iamsawat S, Chakraborty P, et al. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting treg stability in mice. Blood. 2019;133(3):266–279. doi:10.1182/blood-2018-07-863233
31. Wang Y, Luo W, Wang Y. PARP-1 and its associated nucleases in DNA damage response. DNA Repair (Amst). 2019;81:102651. doi:10.1016/j.dnarep.2019.102651
32. Wang W, Sun W, Cheng Y, Xu Z, Cai L. Role of sirtuin-1 in diabetic nephropathy. J Mol Med (Berl). 2019;97(3):291–309. doi:10.1007/s00109-019-01743-7
33. de Kreutzenberg SV, Ceolotto G, Papparella I, et al. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes. 2010;59(4):1006–1015. doi:10.2337/db09-1187
34. Chen Z, Gong L, Zhang P, et al. Epigenetic down-regulation of sirt 1 via DNA methylation and oxidative stress signaling contributes to the gestational diabetes mellitus-induced fetal programming of heart ischemia-sensitive phenotype in late life. Int J Biol Sci. 2019;15(6):1240–1251. doi:10.7150/ijbs.33044
35. Papadimitriou A, Silva KC, Peixoto EB, Borges CM, Lopes de Faria JM, Lopes de Faria JB. Theobromine increases NAD+ /Sirt-1 activity and protects the kidney under diabetic conditions. Am J Physiol Renal Physiol. 2015;308(3):F209–225. doi:10.1152/ajprenal.00252.2014
36. Kitada M, Kume S, Imaizumi N, Koya D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes. 2011;60(2):634–643. doi:10.2337/db10-0386
37. Karbasforooshan H, Karimi G. The role of SIRT1 in diabetic cardiomyopathy. Biomed Pharmacother. 2017;90:386–392. doi:10.1016/j.biopha.2017.03.056
38. Wang W, Sun W, Cheng Y, Xu Z, Cai L. Management of diabetic nephropathy: the role of sirtuin-1. Future Med Chem. 2019;11(17):2241–2245. doi:10.4155/fmc-2019-0153
39. Lu P, Kamboj A, Gibson SB, Anderson CM. Poly(ADP-ribose) polymerase-1 causes mitochondrial damage and neuron death mediated by Bnip3. J Neurosci. 2014;34(48):15975–15987. doi:10.1523/JNEUROSCI.2499-14.2014
40. Waldman M, Nudelman V, Shainberg A, et al. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1alpha axis. Exp Cell Res. 2018;373(1–2):112–118. doi:10.1016/j.yexcr.2018.10.003
41. Eo H, Park JE, Jeon YJ, Lim Y. Ameliorative effect of ecklonia cava polyphenol extract on renal inflammation associated with aberrant energy metabolism and oxidative stress in high fat diet-induced obese mice. J Agric Food Chem. 2017;65(19):3811–3818. doi:10.1021/acs.jafc.7b00357
42. Szrejder M, Piwkowska A. AMPK signalling: implications for podocyte biology in diabetic nephropathy. Biol Cell. 2019;111(5):109–120. doi:10.1111/boc.201800077
43. Ma T, Zheng Z, Guo H, et al. 4-O-methylhonokiol ameliorates type 2 diabetes-induced nephropathy in mice likely by activation of AMPK-mediated fatty acid oxidation and Nrf2-mediated anti-oxidative stress. Toxicol Appl Pharmacol. 2019;370:93–105. doi:10.1016/j.taap.2019.03.007
44. Hong YA, Lim JH, Kim MY, et al. Extracellular superoxide dismutase attenuates renal oxidative stress through the activation of adenosine monophosphate-activated protein kinase in diabetic nephropathy. Antioxid Redox Signal. 2018;28(17):1543–1561. doi:10.1089/ars.2017.7207
45. Liao Z, Zhang J, Wang J, et al. The anti-nephritic activity of a polysaccharide from okra (Abelmoschus esculentus (L.) Moench) via modulation of AMPK-Sirt1-PGC-1alpha signaling axis mediated anti-oxidative in type 2 diabetes model mice. Int J Biol Macromol. 2019;140:568–576. doi:10.1016/j.ijbiomac.2019.08.149
46. Wang X, Gao Y, Tian N, et al. Astragaloside IV represses high glucose-induced mesangial cells activation by enhancing autophagy via SIRT1 deacetylation of NF-kappaB p65 subunit. Drug Des Devel Ther. 2018;12:2971–2980. doi:10.2147/DDDT.S174058
47. Lei P, Jiang Z, Zhu H, Li X, Su N, Yu X. Poly(ADP-ribose) polymerase-1 in high glucose-induced epithelial-mesenchymal transition during peritoneal fibrosis. Int J Mol Med. 2012;29(3):472–478. doi:10.3892/ijmm.2011.859
48. Fakouri NB, Durhuus JA, Regnell CE, et al. Rev1 contributes to proper mitochondrial function via the PARP-NAD(+)-SIRT1-PGC1alpha axis. Sci Rep. 2017;7(1):12480. doi:10.1038/s41598-017-12662-3
49. Jiang Y, Zhang Z, Cha L, et al. Resveratrol plays a protective role against premature ovarian failure and prompts female germline stem cell survival. Int J Mol Sci. 2019;20(14):3605. doi:10.3390/ijms20143605
50. Fan C, Ma Q, Xu M, et al. Ginsenoside Rb1 attenuates high glucose-induced oxidative injury via the NAD-PARP-SIRT axis in rat retinal capillary endothelial cells. Int J Mol Sci. 2019;20(19):4936. doi:10.3390/ijms20194936
51. Horvath EM, Zsengeller ZK, Szabo C. Quantification of PARP activity in human tissues: ex vivo assays in blood cells and immunohistochemistry in human biopsies. Methods Mol Biol. 2011;780:267–275.
52. Horvath EM, Zsengeller ZK, Szabo C. Quantification of PARP activity in human tissues: ex vivo assays in blood cells and immunohistochemistry in human biopsies. Methods Mol Biol. 2017;1608:19–26.
53. Pascua-Maestro R, Corraliza-Gomez M, Diez-Hermano S, Perez-Segurado C, Ganfornina MD, Sanchez D. The MTT-formazan assay: complementary technical approaches and in vivo validation in drosophila larvae. Acta Histochem. 2018;120(3):179–186. doi:10.1016/j.acthis.2018.01.006
54. Ciccarone F, Zampieri M, Caiafa P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin Cell Dev Biol. 2017;63:123–134. doi:10.1016/j.semcdb.2016.11.010
55. Duarte DA, Rosales MA, Papadimitriou A, et al. Polyphenol-enriched cocoa protects the diabetic retina from glial reaction through the sirtuin pathway. J Nutr Biochem. 2015;26(1):64–74. doi:10.1016/j.jnutbio.2014.09.003
56. Shemesh II, Rozen-Zvi B, Kalechman Y, Gafter U, Sredni B. AS101 prevents diabetic nephropathy progression and mesangial cell dysfunction: regulation of the AKT downstream pathway. PLoS One. 2014;9(12):e114287. doi:10.1371/journal.pone.0114287
57. Shang G, Gao P, Zhao Z, et al. 3,5-Diiodo-l-thyronine ameliorates diabetic nephropathy in streptozotocin-induced diabetic rats. Biochim Biophys Acta. 2013;1832(5):674–684. doi:10.1016/j.bbadis.2013.01.023
58. Huang XZ, Wen D, Zhang M, et al. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-beta/Smad3 pathway. J Cell Biochem. 2014;115(5):996–1005. doi:10.1002/jcb.24748
59. Chen Y, Wang N, Yuan Q, et al. The protective effect of fluorofenidone against cyclosporine a-induced nephrotoxicity. Kidney Blood Press Res. 2019;44(4):656–668. doi:10.1159/000500924
60. Xu B, Chiu J, Feng B, Chen S, Chakrabarti S. PARP activation and the alteration of vasoactive factors and extracellular matrix protein in retina and kidney in diabetes. Diabetes Metab Res Rev. 2008;24(5):404–412. doi:10.1002/dmrr.842
61. Rajamohan SB, Pillai VB, Gupta M, et al. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29(15):4116–4129. doi:10.1128/MCB.00121-09
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.