Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Parkinson’s disease-associated melanin steal
Authors Hinz M, Stein A, Cole T
Received 26 September 2014
Accepted for publication 17 October 2014
Published 10 December 2014 Volume 2014:10 Pages 2331—2337
DOI https://doi.org/10.2147/NDT.S74952
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
This paper has been retracted.
Marty Hinz, 1 Alvin Stein, 2 Ted Cole 3
1Clinical Research, NeuroResearch Clinics, Inc., Cape Coral, FL, USA; 2Stein Orthopedic Associates, Plantation, FL, USA; 3Cole Center for Healing, Cincinnati, OH, USA
Abstract: Urinary dopamine fluctuations in the competitive inhibition state were first documented in 2009. At that time, it was noted that progressively higher daily dosing values of L-tyrosine decreased the magnitude of these fluctuations. While extensive statistical analysis has been performed by the authors since 2004, it was not until 2012 that a plausible explanation was formulated. In the process, correlations with L-tyrosine administration and the on/off effect of Parkinson’s disease were defined. This paper documents the current knowledge with regard to the management of retrograde phase 1 dopamine fluctuations and investigates the hypothesis that they are caused by a melanin steal phenomenon.
Keywords: fluctuations, L-dopa, dopamine, melanocyte
Expression of Concern for this paper has been published
Introduction
In 2004, during interpretation of urinary serotonin and dopamine amino acid load testing, retrograde phase 1 urinary dopamine fluctuations were defined. By 2005, it was noted that increasing the daily L-tyrosine consumption significantly decreased fluctuations. The phenomenon was formally documented in 2009:
[…] the fluctuations in dopamine excretion were smaller in samples obtained from patients ingesting incrementally greater amounts of tyrosine; and this difference for the combined phases 1, 2, and 3 reached a high level of statistical significance (p < 0.0001) when compared to phase 0 samples.1
A potential etiology of dopamine fluctuations was not defined until 2012. This paper documents the current knowledge regarding this phenomenon as associated with the treatment of Parkinson’s disease. The primary hypothesis is that if significant retrograde phase 1 urinary dopamine fluctuations exist in the competitive inhibition state, then the primary force causing this phenomenon is melanin steal, which causes dopaquinone to preferentially utilize L-tyrosine and L-3,4-dihydroxyphenylalanine (L-dopa), leading to an inconsistency of dopamine synthesis.
The three-phase response
L-tryptophan is metabolized to 5-hydroxytryptophan (5-HTP), which in turn is metabolized by aromatic L-amino acid decarboxylase (AADC) to serotonin. L-tyrosine is metabolized to L-dopa, which in turn is metabolized by AADC to dopamine.2–8 Competitive inhibition between serotonin and dopamine exists in transport, synthesis, and metabolism when precursors of both are administered simultaneously in levels that are high enough and properly balanced.5,6 Objective verification of the competitive inhibition state is under the apical regulatory super system (APRESS) model. Under APRESS, changes in only serotonin concentrations will affect changes in dopamine concentrations in a predictable manner. The inverse is also true: changes to only dopamine concentrations will affect serotonin concentrations in a predictable manner. Functions exclusively controlled by manipulation of serotonin in the endogenous state may be regulated by dopamine and/or serotonin manipulation in the competitive inhibition state. Functions exclusively controlled by manipulation of dopamine in the endogenous state may be regulated by dopamine and/or serotonin manipulation in the competitive inhibition state.5,6
Figure 1 illustrates the three phases of correlation between urinary serotonin or dopamine with the total daily dosing values of serotonin and dopamine precursors in competitive inhibition: inverse (phase 1), no (phase 2), and direct (phase 3) correlation.1 These responses are generated by the basolateral organic cation transporters type-2 (OCT-2) of the proximal convoluted renal tubule cells’ (PCT) S3 segment. Under normal conditions, serotonin and dopamine filtered at the glomerulus are transported into the PCT and then metabolized, and are not found in the final urine. The serotonin and dopamine (which are centrally acting monoamines) in the final urine represent monoamines that are newly synthesized by the kidneys. The monoamines newly synthesized in the PCT are preferentially transported by serotonin transporters (SERT) and OCT-2 across the S3 basolateral border of the PCT to the peripheral circulation via the renal vein. SERT is responsible for bulk transport, while OCT-2 is responsible for fine tuning the exact final amount of monoamines transported to the system. The monoamines not transported to the peripheral system are carried across the apical PCT S3 surface by organic cation transporters novel type-2 (OCT-N2), then onto the final urine as waste.2,9 The importance of renal OCT-2 functional status analysis to the central nervous system is documented:
The current knowledge of the distribution and functional properties […] of cation transport measured in intact plasma membranes is used to postulate identical or homologous transporters in intestine, liver, kidney, and brain.10
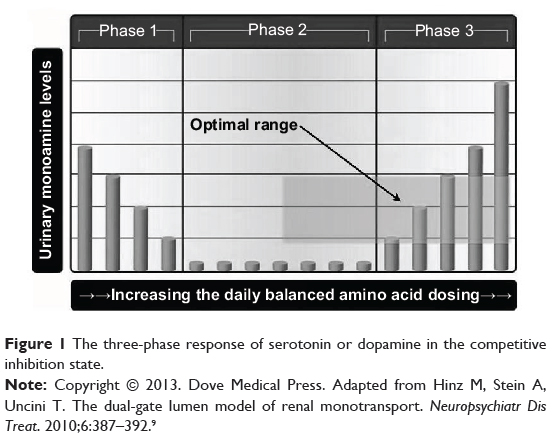
|
Figure 1 The three-phase response of serotonin or dopamine in the competitive inhibition state. |
Serotonin and dopamine both need to be conceptualized independent of each other, each with its own three-phase model. The phases illustrated in Figure 1 correlate with specific configurations which, in the competitive inhibition state, define the functional status of the OCT-2 in the transport of serotonin and dopamine. Phase 1 correlates with the transporter entrance gate inhibiting full monoamine access to the unsaturated transporter lumen. Entrance gate restriction of monoamine access to the transporter lumen dissipates as the sum total of serotonin and dopamine presenting at the gate increases. This causes increasing amounts of monoamine to be transported to the peripheral system, while decreasing concentrations observed in the final urine. Phase 2 correlates with full monoamine access to the unsaturated lumen as the effects of the entrance gate restriction are no longer present. Phase 3 correlates with full transporter lumen access as the entrance gate effects are no longer a factor while the lumen transporter is saturated. This leads to the phenomenon whereby increases in serotonin or dopamine concentrations presenting at the saturated transporter lead to increased excretion of these monoamines in the final urine.2,5,6,9
Fluctuations
It takes 5 days to achieve equilibrium when the daily dosing value of a serotonin or dopamine amino acid precursor is started or changed.5,6 Urinary dopamine levels that are higher than can be achieved in phase 3 at the equilibrium of a specific amino acid dosing value are definitive evidence that urinary dopamine fluctuations are present. For example, while ingesting 120 mg of L-dopa per day, the phase 3 urinary dopamine response limit is defined as 1,500 μg of dopamine per gram of creatinine (μg/g cr). Urinary levels higher than 1,500 μg/g cr are not a phase 3 response, but represent retrograde phase 1 dopamine fluctuations, as illustrated in Figure 2.
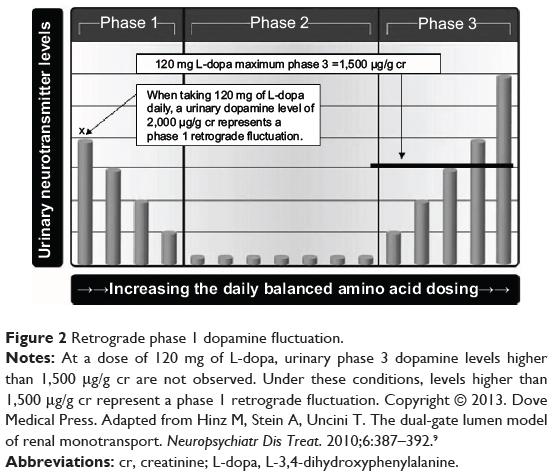
|
Figure 2 Retrograde phase 1 dopamine fluctuation. |
These fluctuating retrograde phase 1 urinary dopamine assay results are not reproducible. They are also revealed with repeat assays on different days with the same L-dopa dosing values. In studying this phenomenon, it is apparent that there is a force variably compromising dopamine synthesis, pulling dopamine back into phase 1, which then fluctuates. These variations are a random occurrence in the laboratory assay. The urinary dopamine assay is a single snapshot of the fluctuating urinary dopamine level in the competitive inhibition state. Serotonin levels vacillate in response to dopamine fluctuations, a phenomenon that is explained by the APRESS model.5
Methodology
Table 1 is based on statistical analysis of urinary serotonin and dopamine amino acid precursor load testing performed on Parkinson’s disease patients by DBS Laboratory Services, Inc. (Duluth, MN, USA) between January 1, 2014 and September 4, 2014.
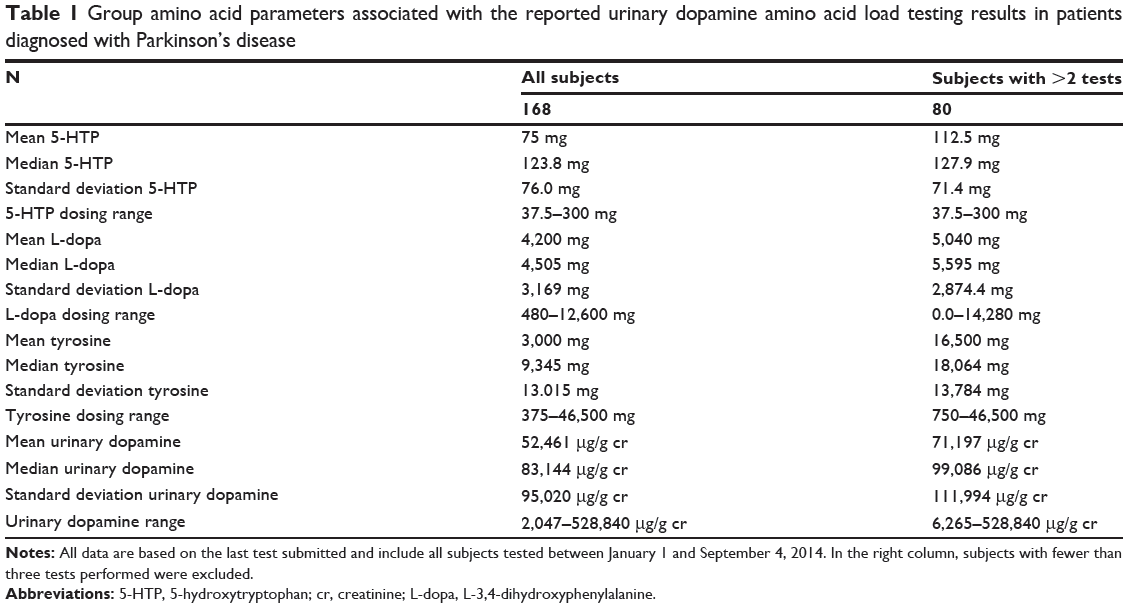
|
Table 1 Group amino acid parameters associated with the reported urinary dopamine amino acid load testing results in patients diagnosed with Parkinson’s disease |
Laboratory sample processing was as follows. After 1 week on a specific amino acid dosing, a urinary sample was obtained 6 hours prior to onset of the sleep cycle; 4 pm was the most frequent time of collection. After stabilization with 6 N HCl for preservation of the monoamines, the samples were shipped to DBS Laboratory Services, Inc. Commercial kits for radioimmunoassay were used (3 CAT PIA IC88501 and IB89527; Immuno Biological Laboratories, Inc, Minneapolis, MN, USA). DBS Laboratory Services, Inc. performs high-complexity laboratory testing that is accredited by Clinical Laboratory Improvement Amendments (CLIA).
Parkinson’s disease melanin steal model
With the induction of suboptimal L-tyrosine and/or L-dopa concentrations, retrograde phase 1 fluctuations can and will occur.1,5,6 Dopamine fluctuations may be intermittent. It is common to observe initial assays wherein the dopamine and serotonin are following the three-phase model prior to the occurrence of retrograde phase 1 dopamine fluctuations. It has been previously documented that, in the competitive inhibition state, increasing L-tyrosine daily dosing decreases dopamine fluctuations.1 Prior research has revealed that administration of progressively higher amounts of L-tyrosine can move the urinary dopamine out of phase 1, through phase 2, and into phase 3. This research further revealed that L-tyrosine alone will not establish urinary dopamine concentrations higher than 475 μg/g cr going into phase 3.5,6
The prototype for understanding the etiology of dopamine fluctuations is the exaggerated response seen in Parkinson’s disease. Retrograde phase 1 urinary dopamine levels of 20,000 to 200,000 μg/g cr are common, while levels above 2 million μg/g cr are occasionally observed. Exacerbation of dopamine fluctuations may occur as the serotonin and dopamine daily amino acid precursor dosing levels (not including L-tyrosine) increase and/or as time passes on a static dose.
While carbidopa/L-dopa combinations are the current standard in medicine, 5-HTP, as documented in Table 1, was administered in place of carbidopa based on the following considerations. Carbidopa has no efficacy in the treatment of Parkinson’s disease symptoms. Its only indication is management of the L-dopa-induced side effect nausea.11 It is documented that carbidopa irreversibly binds to and permanently deactivates the active form of vitamin B6 (pyridoxal 5′-phosphate [PLP]), PLP-dependent enzymes, and depletes B6 reserves.12 Depletion of B6 by carbidopa and benserazide adversely affects over 300 enzymes and proteins that depend on B6 for their function.13 Both are effective in controlling L-dopa-induced nausea by the same mechanism of action, AADC inhibition.5,6,11 The inhibition caused by carbidopa is irreversible, while 5-HTP inhibition is reversible.5,6,12 A primary advantage of 5-HTP over carbidopa is that it does not irreversibly bind to nor permanently deactivate or deplete PLP, PLP-dependent enzymes, and PLP reserves, thereby avoiding system-wide nutritional deficiency and collapse of B6.12 Both Parkinson’s disease and administration of L-dopa are associated with serotonin depletion.4,14–17 5-HTP is freely metabolized to serotonin without biochemical feedback regulation. This means that it is the most powerful precursor available for serotonin synthesis in compensating for the known serotonin depletion associated with Parkinson’s disease and its treatment.5,6
L-tyrosine administration has not been previously documented at the daily dosing levels found in Table 1. It is asserted that daily L-tyrosine dosing, greater than 5,000 mg, needs to be started or increased only in response to a confirmed laboratory indication in the competitive inhibition state. Previous research by the authors has revealed that, when the laboratory indications exist, virtually all patients tolerate L-tyrosine administered at the individualized levels reflected in Table 1. The hypothesis is that if there is a tolerance to the nutrients being administered, then there was a need in the body for these nutrients.
Table 2 presents real-life data from one subject. While the daily dosing value of L-dopa was decreased by 33.3% between the first and second assays, the urinary dopamine levels increased more than 16-fold. L-cysteine is administered to compensate for the ability of L-dopa to deplete thiols.5,6 The next recommended step in treatment would be to start L-tyrosine 3,750 mg twice a day for control of dopamine fluctuations and to continue adjusting the L-dopa daily dosing with pill stops consistent with previous documentation.8
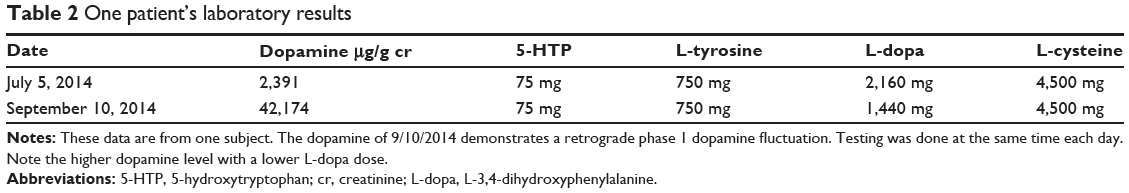
|
Table 2 One patient’s laboratory results |
L-tyrosine is an amino acid synthesized from phenylalanine. The recommended dietary allowance (RDA) for phenylalanine is 3,000 to 5,000 mg per day.18 In humans, L-tyrosine is metabolized to one of five metabolites: tyramine, 3-iodo-L-tyrosine, 4-hydroxyphenylpyruvate, dopaquinone, or L-dopa.19 Fluctuations of tyramine, 3-iodo-L-tyrosine, and 4-hydroxyphenylpyruvate have not been documented. By default, this leaves the melanin system, with its precursor dopaquinone, as the prime candidate for generating L-dopa/dopamine fluctuations in the competitive inhibition state. Both melanin and dopamine fluctuations have been documented.1,4,20–25 The inverse association between the increasing daily L-dopa dosing value with the magnitude of dopamine fluctuations in the competitive inhibition state has also been documented.1,4 Not documented until now are the interactions of L-tyrosine, L-dopa, and dopaquinone, with regard to the previously documented dopamine variances.1
As noted in Figure 3, two enzymes catalyze metabolism of L-tyrosine to L-dopa: tyrosinase and tyrosine hydroxylase. Tyrosinase also catalyzes metabolism of both L-tyrosine and L-dopa to dopaquinone, the critical precursor of melanin synthesis (Figure 3).19,26 Tyrosinase is known to have higher efficacy in the conversion of dopamine to dopaquinone than in the conversion of L-tyrosine to L-dopa.27 In Parkinson’s disease, there is a 50% to 90% loss of tyrosine hydroxylase-containing cells and a 33% to 80% loss of neuromelanin-containing neurons in the substantia nigra.28 As a result, the system attempts to synthesize more melanin by increasing concentrations of β-melanocyte-stimulating hormone (MSH).29 An increase in MSH induces increased activity in tyrosinase. Increases in tyrosinase activity, up to 90-fold, secondary to MSH stimulation have been reported.30 Melanin fluctuations have been documented in vivo and in vitro.20–22 The hypothesis is that if decreasing tyrosine hydroxylase activity and increasing tyrosinase activity is more effective in the synthesis of dopaquinone than dopamine, then this leaves melanin synthesis and fluctuations in a more primary position, while the melanin-induced fluctuations of L-tyrosine and L-dopa are reflected as dopamine fluctuations on laboratory assay.
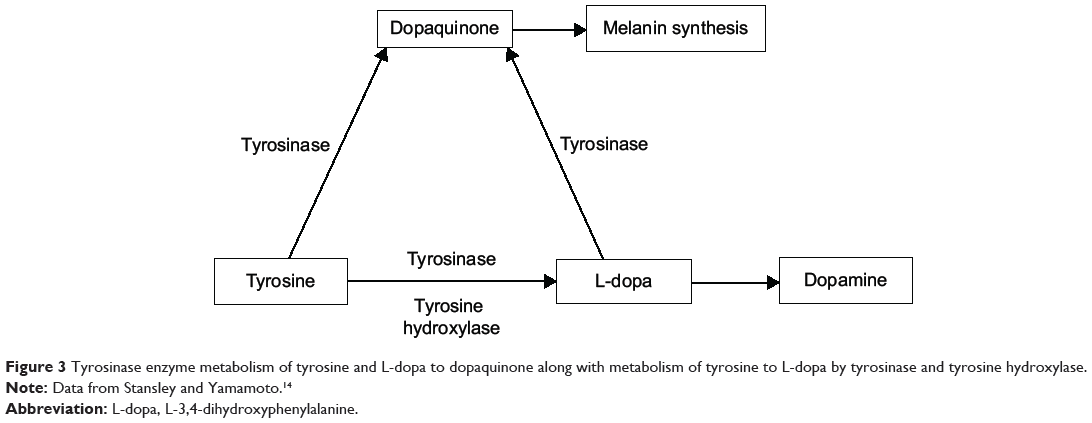
|
Figure 3 Tyrosinase enzyme metabolism of tyrosine and L-dopa to dopaquinone along with metabolism of tyrosine to L-dopa by tyrosinase and tyrosine hydroxylase. |
Implications
The on/off effect is defined as symptoms of Parkinson’s disease that wax and wane and are not fully controlled with L-dopa. With classic on/off effect, Parkinson’s disease patients have better control early in the day, decreased control 6 to 12 hours after rising, then better control prior to onset of sleep.31,32 The results reported herein adhered to the retrograde fluctuation model and L-tyrosine was only administered for the control of fluctuations when urinary dopamine levels exceeded 40,000 μg/g cr. This paper is based on accumulated data, which reveal control of the on/off effect in over 800 Parkinson’s disease patients treated with L-tyrosine, administered only when indicated, with no refractory cases reported. Curiously, low-protein diets have been reported to have a positive effect in the management of the on/off effect, yet this laboratory-guided amino acid administration approach ameliorates the phenomenon completely.33
In Parkinson’s disease patients, the correlation between motor fluctuations and dopamine fluctuations has been documented for several years.23–25 Until this novel approach, there was no objective, laboratory-based method for addressing the problem of dopamine-driven motor fluctuations. Observations also support the assertion that patients not exhibiting the on/off effect who are more difficult to control with L-dopa may benefit from administration of L-tyrosine in response to laboratory indications. Once enough L-tyrosine is administered to compensate for and meet dopaquinone synthesis needs, further administration of L-tyrosine optimizes dopamine synthesis by meeting some of the dopamine precursor needs (Figure 3).
Discussion
The need for higher levels of L-tyrosine is not absolute. Administration needs to be guided by laboratory indication. When laboratory indication exists, there is a tolerance of the higher dosing of L-tyrosine. Bearing in mind that the melanin system has the ability to preferentially steal the precursors of dopamine, the following question was posed: “What is the melanin system involved with that is so important that it has the ability to assume priority in the metabolism of amino acid precursors over the catecholamine system?” Melanin regulates cytokines. If optimization of melanin synthesis occurs using this approach, then this approach may be the foundation for regulating and optimizing cytokine function.34,35 No evidence has been found to support the concept that cytokine function is important enough to preferentially steal the precursors required for catecholamine synthesis and optimal function. A more compelling argument is that the various forms of melanin are DNA protective. Ultraviolet (UV) radiation may cause melanoma and other deadly events. Melanin has the ability to establish a protective barrier around DNA, protecting it from toxin damage.36,37 It is documented that “[…] melanin binds directly to DNA, it acts as a direct photosensitizer of mtDNA damage during UVA irradiation.”38 The hypothesis is that if the momentary load of toxins or UV protection fluctuates, then melanin needs will also fluctuate.
Conclusion
Urinary dopamine fluctuations have been recognized since 2004, but were first documented in 2009.1 Melanin concentrations are known to fluctuate under normal conditions. In Parkinson’s disease patients, there is a defect of the postsynaptic dopamine neurons found in the substantia nigra of the brain. The dark color of the substantia nigra is from neuromelanin-rich cells. As the disease progresses, the substantia nigra turns progressive shades of lighter gray with the loss of neuromelanin. There is diminished tyrosine hydroxylase activity and an increase in MSH which increases tyrosinase activity. The enhanced tyrosinase activity is more effective at synthesizing dopaquinone than L-dopa. The model is that dopaquinone/melanin synthesis has priority in utilization of L-tyrosine and L-dopa at the expense of stable dopamine synthesis and concentrations. As a result, dopamine synthesis fluctuates as the needs of melanin synthesis are preferentially met.
The diagnosis of dopamine fluctuations in the competitive inhibition state is a novel approach for determining whether there is adequate L-tyrosine supplementation in the stabilization of Parkinson’s disease patients and may be a powerful tool for the management of the on/off effect and for the control of cytokines.
Disclosure
MH discloses his relationship with DBS Laboratory Services, Inc. and NeuroResearch Clinics, Inc. MH also discloses his relationship with West Duluth Distribution Company, which ended in June 2011. The authors report no other conflicts of interest in this work.
References
Trachte GJ, Uncini T, Hinz M. Both stimulatory and inhibitory effects of dietary 5-hydroxytryptophan and tyrosine are found on urinary excretion of serotonin and dopamine in a large human population. Neuropsychiatr Dis Treat. 2009;5:227–235. | ||
Stein A, Hinz M, Uncini T. Amino acid-responsive Crohn’s disease: a case study. Clin Exp Gastroenterol. 2010;3:171–177. | ||
Hinz M, Stein A, Neff R, Weinberg R, Uncini T. Treatment of attention deficit hyperactivity disorder with monoamine amino acid precursors and organic cation transporter assay interpretation. Neuropsychiatr Dis Treat. 2011;7:31–38. | ||
Hinz M, Stein A, Uncini T. Amino acid management of Parkinson’s disease: a case study. Int J Gen Med. 2011;4:165–174. | ||
Hinz M, Stein A, Uncini T. APRESS: apical regulatory super system, serotonin, and dopamine interaction. Neuropsychiatr Dis Treat. 2011;7:457–463. | ||
Hinz M, Stein A, Uncini T. Relative nutritional deficiencies associated with centrally acting monoamines. Int J Gen Med. 2012;5:413–430. | ||
Hinz M, Stein A, Uncini T. 5-HTP efficacy and contraindications. Neuropsychiatr Dis Treat. 2012;8:323–328. | ||
Hinz M, Stein A, Cole T. Management of L-dopa overdose in the competitive inhibition state. Drug Healthc Patient Saf. 2014;6:93–99. | ||
Hinz M, Stein A, Uncini T. The dual-gate lumen model of renal monoamine transport. Neuropsychiatr Dis Treat. 2010;6:387–392. | ||
Koepsell H. Organic cation transporters in intestine, kidney, liver, and brain. Annu Rev Physiol. 1998;60:243–266. | ||
SINEMET® (carbidopa levodopa) tablets [prescribing information]. Whitehouse Station, NJ: Merck & Co., Inc.; 2014. Available from: https://www.merck.com/product/usa/pi_circulars/s/sinemet/sinemet_pi.pdf. Accessed September 24, 2014. | ||
Daidone F, Montioli R, Paiardini A, et al. Identification by virtual screening and in vitro testing of human DOPA decarboxylase inhibitors. PLoS One. 2012;7(2):e31610. | ||
UniProtKB: pyridoxal AND organism: “Homo sapiens (Human) [9606]” [webpage on the Internet]. UniProt Consortium. Available from: http://www.uniprot.org/uniprot/?query=pyridoxal+AND+organism%3A%22Homo+sapiens+%5B9606%5D%22&sort=score. Accessed September 24, 2014. | ||
Stansley BJ, Yamamoto BK. Chronic l-dopa decreases serotonin neurons in a subregion of the dorsal raphe nucleus. J Pharmacol Exp Ther. 2014;351(2):440–447. | ||
Eskow Jaunarajs KL, George JA, Bishop C. L-DOPA-induced dysregulation of extrastriatal dopamine and serotonin and affective symptoms in a bilateral rat model of Parkinson’s disease. Neuroscience. 2012;30(218):243–256. | ||
García NH, Berndt TJ, Tyce GM, Knox FG. Chronic oral L-dopa increases dopamine and decreases serotonin excretions. Am J Physiol. 1999;277(5 Pt 2):R1476–R1480. | ||
Borah A, Mohanakumar KP. Long term L-DOPA treatment causes indiscriminate increases in dopamine levels at the cost of serotonin synthesis in discrete brain regions of rats. Cell Mol Neurobiol. 2007;27:985–996. | ||
Phenylalanine [webpage on the Internet]. Bethesda, MD: National Center for Biotechnology Information, U.S. National Library of Medicine. Available from: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=6140. Accessed September 24, 2014. | ||
Tyrosine metabolism – Homo sapiens (human) [webpage on the Internet]. Kanehisa Laboratories; 2014. Available from: http://www.genome.jp/kegg-bin/show_pathway?org_name=hsa&mapno=00350&mapscale=&show_description=hide. Accessed September 24, 2014. | ||
Kim O, McMurdy J, Jay G, Lines C, Crawford G, Alber M. Combined reflectance spectroscopy and stochastic modeling approach for noninvasive hemoglobin determination via palpebral conjunctiva. Physiol Rep. 2014;2(1):e00192. | ||
Baquié M, Kasraee B. Discrimination between cutaneous pigmentation and erythema: comparison of the skin colorimeters Dermacatch and Mexameter. Skin Res Technol. 2014;20(2):218–227. | ||
McMurdy JW, Jay GD, Suner S, Crawford G. Noninvasive optical, electrical, and acoustic methods of total hemoglobin determination. Clin Chem. 2008;54(2):264–272. | ||
Chase TN, Mouradian MM, Engber TM. Motor response complications and the function of striatal efferent systems. Neurology. 1993;43(12 Suppl 6):S23–S27. | ||
Obeso JA, Luquin MR, Martínez-Lage JM. Lisuride infusion pump: a device for the treatment of motor fluctuations in Parkinson’s disease. Lancet. 1986;1(8479):467–470. | ||
de la Fuente-Fernández R, Lu JQ, Sossi V, et al. Biochemical variations in the synaptic level of dopamine precede motor fluctuations in Parkinson’s disease: PET evidence of increased dopamine turnover. Ann Neurol. 2001;49(3):298–303. | ||
Slominski A, Zmijewski MA, Pawelek J. L-tyrosine and L-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment Cell Melanoma Res. 2012;25:14–27. | ||
Redding K, Masterman D, Randall J, Collins M. Advanced Biology with Vernier. Beaverton, OR: Vernier. 2008:15–2. | ||
Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136:2419–2431. | ||
Rainero I, Kaye JA, May C, et al. Alpha-melanocyte-stimulating hormonelike immunoreactivity is increased in cerebrospinal fluid of patients with Parkinson’s disease. Arch Neurol. 1988;45(11):1224–1227. | ||
Fuller BB, Lunsford JB, Iman DS. Alpha-melanocyte-stimulating hormone regulation of tyrosinase in Cloudman S-91 mouse melanoma cell cultures. J Biol Chem. 1987;262(9):4024–4033. | ||
Marsden CD, Parkes JD. “On-off” effects in patients with Parkinson’s disease on chronic levodopa therapy. Lancet. 1976;1(7954):292–296. | ||
Hardie RJ, Lees AJ, Stern GM. On-off fluctuations in Parkinson’s disease. A clinical and neuropharmacological study. Brain. 1984;107(Pt 2):487–506. | ||
Sweet RD, McDowell FH. Plasma dopa concentrations and the “on-off” effect after chronic treatment of Parkinson’s disease. Neurology. 1974;24(10):953–956. | ||
Mohagheghpour N, Waleh N, Garger SJ, Dousman L, Grill LK, Tusé D. Synthetic melanin suppresses production of proinflammatory cytokines. Cell Immunol. 2000;199(1):25–36. | ||
Morelli JG, Norris DA. Influence of inflammatory mediators and cytokines on human melanocyte function. J Invest Dermatol. 1993;100:191S–195S. | ||
Mouret S, Forestier A, Douki T. The specificity of UVA-induced DNA damage in human melanocytes. Photochem Photobiol Sci. 2012;11:155–162. | ||
Page S, Chandhoke V, Baranova A. Melanin and melanogenesis in adipose tissue: possible mechanisms for abating oxidative stress and inflammation? Obes Rev. 2011;12:e21–e31. | ||
Swalwell H, Latimer J, Haywood RM, Birch-Machin MA. Investigating the roll of melanin in UVA/UVB- and hydrogen peroxide-induced cellular and mitochondrial ROS production and mitochondrial DNA damage in human melanoma cells. Free Radic Biol Med. 2012;52(3):626–634. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.