Back to Journals » Journal of Inflammation Research » Volume 18
Parishin Protects Against Sepsis-Induced Intestinal Injury by Modulating the ACSL4/p-Smad3/PGC-1α Pathway: An Integrated Approach of Bioinformatics and Experimental Validation
Authors Bao D, Shao S, Wang Y, Chen X, Ren Y, Zhou C, Mao Q
Received 12 June 2025
Accepted for publication 8 October 2025
Published 15 October 2025 Volume 2025:18 Pages 14283—14305
DOI https://doi.org/10.2147/JIR.S546492
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Daiqin Bao,1,* Shifeng Shao,2,* Yingjie Wang,3,* Xian Chen,1 Yunqin Ren,1 Chaomin Zhou,1 Qingxiang Mao1
1Department of Anesthesiology, Daping Hospital, Army Medical University, Chongqing, 400042, People’s Republic of China; 2Department of ICU, Daping Hospital, Army Medical University, Chongqing, 400042, People’s Republic of China; 3Department of Post-Graduate School, Army Medical University, Chongqing, 400042, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qingxiang Mao, Department of Anesthesiology, Daping Hospital, Army Medical University, Chongqing, 400042, People’s Republic of China, Email [email protected]
Background: Sepsis is a major clinical challenge, with in-hospital mortality of 25%-40% in intensive care unit patients. The gastrointestinal tract is recognized as both the “initiating organ” of multiple organ dysfunction syndrome and the “central organ” in orchestrating the host stress response during critical illness. ACSL4, a regulator of lipid metabolism and ferroptosis, is a potential target for sepsis-induced intestinal injury, but its inhibitor parishin has not been evaluated in this context.
Methods: Key genes implicated in sepsis pathogenesis were identified through bioinformatic analysis of publicly available datasets from the GEO. Network pharmacology approaches were used to screen for small-molecule compounds with high binding affinity to the identified hub genes. Molecular docking, followed by in vivo and in vitro validation, was employed to evaluate the therapeutic efficacy and mechanistic impact of the top candidate compound in a murine sepsis model.
Results: Weighted Gene Co-expression Network Analysis identified five genes most significantly associated with sepsis diagnosis. Protein-protein interaction network analysis revealed 157 hub genes, among which ACSL4 was the sole gene shared across diagnostic and functional modules. Molecular docking analysis indicated that Parishin exhibited the strongest binding affinity to ACSL4 (docking score: − 17.701). In septic animal models, ACSL4 expression was markedly upregulated in both plasma monocytes and intestinal tissues (P < 0.05), accompanied by increased levels of inflammatory cytokines, lipid peroxidation (LPO), MDA, and Fe2+ (P < 0.05). Expression of ferroptosis-associated proteins was also evidently elevated (P < 0.05). Treatment with Parishin notably attenuated these pathological changes, reduced ferroptosis-related markers, and improved 72-hour survival rates in septic mice (P < 0.05).
Conclusion: Parishin ameliorates sepsis-induced intestinal injury by downregulating ACSL4 expression, thereby inhibiting Smad3 phosphorylation and suppressing ferroptosis. These findings suggest that ACSL4 is a promising therapeutic target for mitigating intestinal damage in sepsis.
Keywords: sepsis, parishin, acsl4, ferroptosis, mitochondrial function
Introduction
Sepsis is a life-threatening clinical syndrome that arises from a dysregulated host response to infection, leading to life-threatening organ dysfunction and high mortality. Globally, sepsis affects nearly 50 million individuals each year and is responsible for approximately 11 million deaths—accounting for nearly 20% of all global mortality.1 Despite advances in critical care, sepsis remains the leading cause of death among critically ill patients, with a reported 90-day mortality rate of 32.24%.2 The gastrointestinal (GI) tract is among the earliest and most severely affected organs in sepsis and plays a central role in the development of multiple organ dysfunction syndrome (MODS). The gut maintains a complex bidirectional relationship with other organs, particularly the lungs and liver. Disruption of the intestinal epithelial barrier allows the translocation of pathogens and toxins into the systemic circulation, which can activate immune cells in distant organs and disturb immune homeostasis. For example, bacterial translocation from the gut can trigger pulmonary immune responses, exacerbating lung injury.3 Similarly, increased intestinal permeability facilitates the passage of harmful substances to the liver, where they activate hepatic immune cells and impair metabolic function. Liver dysfunction, in turn, aggravates gut barrier damage, underscoring the critical gut-liver axis in sepsis pathophysiology.4 Elevated intestinal permeability is considered a key initiator of MODS in sepsis. Loss of gut barrier integrity allows microbial products and pro-inflammatory mediators to disseminate systemically, thereby activating immune responses across multiple organs and contributing to widespread tissue injury. Additionally, sepsis disrupts the balance between the gut microbiota and the host immune system, promoting the development and progression of gut-derived infections. As a result, the gastrointestinal tract is increasingly recognized as the initiating organ in sepsis-induced multi-organ dysfunction.5,6 The intestinal epithelium, composed of a single layer of cells, is essential for maintaining gut homeostasis. Beyond serving as a physical barrier, it acts as an immunological interface that mediates the interaction between commensal microbes and host immune cells.7 The unique structural and physiological characteristics of the gut position it as a pivotal player in the pathogenesis of sepsis.8,9 However, despite its central role, current clinical treatments for sepsis—such as antibiotic therapy, fluid resuscitation, and organ support—offer only symptomatic relief. No targeted therapies are currently available to effectively preserve or restore intestinal barrier function.10
Ferroptosis is a distinct, regulated form of cell death driven by iron-dependent lipid peroxidation and the accumulation of reactive oxygen species (ROS). It is tightly regulated by multiple metabolic pathways, including those governing iron homeostasis, lipid metabolism, and redox balance.11 While ferroptosis plays a physiological role in cellular homeostasis, aberrant activation under pathological conditions—both acute and chronic—can lead to tissue injury and disease progression.12–15 Emerging evidence has implicated ferroptosis in the pathogenesis of sepsis; however, its specific role in sepsis-induced intestinal injury remains insufficiently understood. To elucidate potential molecular targets, we performed a bioinformatic analysis comparing gene expression profiles between healthy individuals and sepsis patients. Among the differentially expressed genes, long-chain acyl-CoA synthetase 4 (ACSL4) emerged as a key candidate for both diagnostic and therapeutic targeting in sepsis. ACSL4, a member of the ACSL enzyme family, plays a central role in fatty acid metabolism and is particularly notable for its involvement in the formation of polyunsaturated fatty acid-containing phospholipids—crucial substrates for lipid peroxidation in ferroptosis. By facilitating iron-dependent lipid oxidation, ACSL4 contributes to the accumulation of lipid-derived ROS (L-ROS), ultimately leading to ferroptotic cell death.16
Our previous research demonstrated that ferroptosis, mediated by the Hippo/ACSL4 signaling axis, contributes to pericyte loss and vascular dysfunction in sepsis. Notably, modulation of this pathway was found to ameliorate vascular abnormalities.17 Building upon these findings, we aimed to further investigate the diagnostic and therapeutic relevance of ACSL4 in sepsis-associated intestinal injury. To this end, we applied network pharmacology and molecular docking approaches, which identified Parishin as a promising small-molecule compound targeting ACSL4. Parishin is a phenolic glucoside derived from Gastrodia elata, a traditional Chinese medicinal herb first recorded in the Shennong Bencao Jing. Among the major bioactive constituents of G. elata, Parishin has been shown in modern pharmacological studies to exert notable anti-inflammatory, antioxidant, and pro-angiogenic effects.18–20 For instance, Wang et al demonstrated that oxidative stress and inflammation are major contributors to cerebral ischemic injury, and that Parishin mitigates brain damage in ischemic rat models by regulating these pathological processes.21 Additional studies by Zhao and Gong revealed that Parishin also contributes to the restoration of intestinal microbiota balance and supports mucosal barrier integrity.22,23
Despite advances in supportive care, there remains a critical lack of reliable biomarkers and effective small-molecule therapeutics for sepsis-induced intestinal injury. Therefore, elucidating the underlying pathophysiological mechanisms, identifying novel molecular targets, and developing targeted treatment strategies are of urgent clinical importance. In this study, we demonstrate that Parishin significantly reduces inflammation, preserves intestinal barrier function, and improves survival in septic animal models. Moreover, we provide mechanistic insights into the role of ACSL4 in ferroptosis and its contribution to mitochondrial dysfunction and intestinal injury. These findings not only validate the therapeutic potential of Parishin in sepsis but also offer a foundation for the development of targeted interventions against sepsis-induced intestinal damage.
Materials and Methods
Data Collection and Processing
Gene expression profiles were retrieved from the NCBI Gene Expression Omnibus (GEO) database, including datasets GSE28750, GSE57065, GSE65682, GSE69528, GSE8121, and GSE95233, with the latter (GSE95233) designated as the validation set. Outlier samples and genes were filtered using the goodSamplesGenes function from the Weighted Gene Co-expression Network Analysis (WGCNA) package in R(version 4.1.2). Differentially expressed genes (DEGs) were identified and subjected to Gene Ontology (GO) enrichment analysis via the Database for Annotation, Visualization and Integrated Discovery (DAVID). Pathway enrichment analysis was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.24,25
WGCNA
WGCNA is a systems biology approach used to characterize correlation patterns among genes across different samples. This method facilitates the identification of gene modules with coordinated expression patterns and their associations with clinical phenotypes or disease states.26 Through the construction of gene co-expression networks, WGCNA clusters genes into modules based on expression similarity and correlates these modules with external traits, enabling the identification of candidate biomarkers and potential therapeutic targets. It has been widely applied in studies exploring genotype-phenotype relationships.27
Construction of the Protein-Protein Interaction (PPI) Network
Based on DEGs identified through WGCNA, a PPI network was constructed to explore functional associations. Genes with higher connectivity (ie, greater interaction degree) were considered more likely to represent key molecular targets. Hub genes significantly associated with sepsis were identified through topological network analysis. Cytoscape (version 3.9.1) was used to visualize the PPI network.28 In the visualization, node size and color intensity reflected gene connectivity—the higher the degree, the larger and lighter the node. Within each module, the top 30 genes with the highest degrees were designated as hub genes.
Receiver Operating Characteristic (ROC) Curve Analysis
To evaluate the diagnostic potential of candidate genes, ROC curve analysis was performed using the pROC package in R, a widely used tool for assessing biomarker performance. Sensitivity and specificity values were used to generate ROC curves, and the area under the curve (AUC) was calculated to quantify diagnostic accuracy.
Gene-Molecular Docking for Identification of Potential Therapeutic Compounds
Candidate therapeutic compounds were screened from the DrugBank database (https://go.drugbank.com/) based on Lipinski’s Rule of Five to ensure drug-likeness. A preliminary literature review guided the selection of ACSL4 as the target protein for virtual screening. Molecular docking was performed against the mouse ACSL4 protein using Schrödinger Maestro (version 12.8), while three-dimensional visualization was carried out with PyMol. High-throughput virtual screening was conducted via the Virtual Screening Workflow module, and docking was completed using the Glide module. Binding affinities and structural interactions were manually reviewed to evaluate compound-target compatibility. The top 500 compounds from the HY-L021P Natural Product Library Plus and HY-L0107V Life Chemicals Natural Product-like Compound Library were included in the screen. Docking scores were used to rank compounds and assess their potential binding strength to ACSL4.
Animal Preparation and Sepsis Model
Eight-week-old male C57BL/6 mice (20–22 g), free of specific pathogens, were used for all animal experiments. Mice were acclimated for 24 hours in a controlled environment (22 ± 2 °C, 40%–70% humidity) prior to procedures. Sepsis was induced via the cecal ligation and puncture (CLP) method, a well-established murine model of polymicrobial sepsis. Mice were anesthetized with intraperitoneal pentobarbital sodium (30 mg/kg). Following confirmation of anesthesia, animals were placed supine, and the abdominal region was disinfected using povidone-iodine. A midline laparotomy (~1 cm) was performed to expose the cecum. In the sham group, the abdomen was opened, but no ligation or puncture was performed. In the sepsis (Sep) and treatment (Parishin) groups, the cecum was ligated approximately one-quarter of the distance from the distal end toward the ileocecal junction using sterile 4–0 sutures. The cecum was then punctured once with a 22G needle to allow fecal extrusion (~0.2 mL). The cecum was repositioned into the peritoneal cavity, and the incision was closed in two layers with sterile 2–0 sutures. Age- and sex-matched (8–12 weeks) controls were used in each experiment.
Postoperatively, mice were housed individually and monitored. Twelve hours after CLP, mean arterial pressure (MAP) was measured via carotid artery cannulation; a ≥30% reduction from baseline was considered indicative of successful sepsis induction. Mice in the Sep group received no further intervention. Mice in the Parishin group were administered an intraperitoneal injection of Parishin (50 mg/kg) one hour prior to surgery. All animals were sacrificed 24 hours post-CLP for tissue collection and analysis. All procedures involving animals were granted by the Ethics Committee of the Army Medical University (Approval No. AMUWEC20237064) and conducted at the Experimental Animal Center of the Army Specialty Medical Center, Chinese PLA Army Medical University.
Cell Culture and Reagents
IEC-6 Cell Culture
IEC-6 cells (obtained from ATCC, USA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) enriched with 10% fetal bovine serum (FBS) and seeded into sterile culture dishes. Cells were maintained at 37 °C in a humidified incubator with 5% CO2. Upon reaching 75–80% confluence, cells were used for subsequent experiments. Experimental groups were defined as follows: Control group (CTL): Cells were incubated with serum-free DMEM for 12 hours. Sepsis group (LPS): Cells were treated with 10 μg/mL lipopolysaccharide (LPS) in serum-free DMEM. Treatment group (Parishin): Following exposure to 10 μg/mL LPS, cells were further treated with Parishin at concentrations of 20, 50, 100, and 200 μmol/L for 12 hours.
Monocyte Isolation
Peripheral blood monocytes were isolated using a PBMC isolation kit (Solarbio, China). Briefly, 2 mL of fresh EDTA-anticoagulated whole blood was mixed with an equal volume of normal saline and gently layered over 3 mL of lymphocyte separation medium. After centrifugation at 400 g for 25–35 minutes, the mononuclear cell layer—appearing as a milky white ring—was collected. Cells were washed with 10 mL saline, centrifuged at 250 g for 10 minutes, and washed twice before being used in downstream assays.
Main Reagents
Parishin and Ferrostatin-1 were procured from MedChemExpress (MCE, USA). LPS, Evans Blue, and Type I collagenase were obtained from Sigma-Aldrich (USA). DMEM and FBS were sourced from Hyclone (USA). ELISA kits for diamine oxidase (DAO), D-lactate (D-LA), TNF-α, IL-6, and IL-1β were obtained from Elabscience (China). Lipid peroxidation (LPO) assay kits were brought from Nanjing Jiancheng Bioengineering Institute (China). Mitochondrial membrane potential (JC-1) and ROS detection kits were obtained from Beyotime Biotechnology (China). Primary antibodies againstβ-Actin(ab170325), GPX4(ab252833), ACSL4(A6826), phosphorylated Smad3 (P-Smad3,ab52903), total Smad3(ab208182), SLC7A11(ab216876), PGC-1α(ab313559), COX2(ab179800), ZO-1(ab276131), and β-actin, as well as HRP-conjugated goat anti-rabbit secondary antibodies, were sourced from Abcam (USA). The Seahorse XF Cell Mito Stress Test Kit was procured from Agilent Technologies (USA). The BODIPY 581/591 C11 lipid peroxidation probe was purchased from Thermo Fisher Scientific (USA), and the FerroOrange probe and Fe2+ detection kit were acquired from Dojindo Laboratories (Japan).
Histopathological Examination of Intestinal Tissue (Hematoxylin and Eosin Staining, HE)
Histological analysis of intestinal tissues was performed following euthanasia via intraperitoneal injection of a lethal dose of pentobarbital. A ~10 cm segment of the small intestine proximal to the stomach was excised, thoroughly rinsed with cold PBS to remove luminal contents, and fixed in 4% paraformaldehyde for 48–72 hours. Fixed tissues were trimmed into 2–3 mm sections and processed using standard histological techniques, including dehydration, clearing, paraffin embedding, sectioning, and staining. HE staining was conducted, and histopathological changes were examined under a light microscope.
Transmission Electron Microscopy (TEM)
Ultrastructural analysis of intestinal tissues was performed using transmission electron microscopy. After euthanasia, a 10 cm segment of the proximal small intestine was isolated and flushed alternately with PBS and sodium citrate to remove residual contents. The ends of the intestinal segment were ligated, and the lumen was infused with 2.5% glutaraldehyde. After 20 minutes, the tissue was sectioned into 1–2 mm fragments and fixed again in 2.5% glutaraldehyde for 30 minutes at room temperature. Samples were subsequently immersed in sucrose solution and incubated at 4 °C for 2 hours, followed by fixation in osmium tetroxide for 2 hours in the dark. Following fixation, tissues were dehydrated through a graded acetone series and embedded in a 1:1 mixture of acetone and resin. Samples were pre-cured at room temperature for 1.5 hours, then polymerized at 37 °C for 24 hours and at 60 °C for an additional 48 hours. After cooling to room temperature, ultra-thin sections were cut and stained with lead citrate and uranyl acetate. Ultrastructural features were examined using a TEM (JEM-1400, JEOL, Japan).
Western Blotting
Total proteins were extracted from cells or tissues using RIPA lysis buffer supplemented with protease inhibitors, and concentrations were quantified using a BCA protein assay kit. Equal amounts of protein were resolved by SDS-PAGE and transferred onto PVDF membranes. Membranes were blocked with 5% non-fat milk for 2 hours at room temperature and incubated overnight at 4 °C with primary antibodies at the following dilutions: GPX4, COX2, SLC7A11, PGC-1α, and ZO-1 (1:1000); ACSL4, total Smad3, and phosphorylated Smad3 (P-Smad3) (1:2000); and β-actin (1:5000). After washing, membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies (1:20,000) for 1 hour at room temperature. Protein bands were visualized using the Odyssey CLx infrared imaging system, and signal intensities were quantified using ImageJ software.
Immunofluorescence Staining
Following treatment, IEC-6 cells were fixed in 4% paraformaldehyde for 10 minutes and washed three times with PBS (5 minutes each). Cells were then permeabilized with 0.3% Triton X-100 for 1 minute at room temperature. After blocking with 1% BSA for 1 hour, cells were incubated overnight at 4 °C with ZO-1 primary antibody (1:200) on a shaker. The following day, cells were washed with PBS and incubated with a fluorescent secondary antibody (1:200) in the dark for 1 hour at room temperature. Nuclei were counterstained with DAPI, and fluorescence images were captured using a laser scanning confocal microscope.
Detection of Lipid ROS in IECs
Lipid ROS in IECs were assessed by flow cytometry. Cells were prepared as single-cell suspensions and stained according to the antibody manufacturer’s protocol. Fluorescent probes—BODIPYTM 581/591 C11, JC-1, and MitoSOXTM—were diluted 1:1000 in serum-free medium and added to culture plates. Cells were incubated at 37 °C in a 5% CO2 atmosphere for 30 minutes. After incubation, the staining solution was removed, and cells were washed with PBS. Following trypsinization, cells were resuspended in DMEM-containing serum, transferred to flow cytometry tubes, and subsequently washed with PBS. Lipid ROS levels were detected using flow cytometry (excitation: 488 nm; emission: 501–563 nm).
Seahorse Analysis of Mitochondrial Respiratory Function
Mitochondrial respiration in IECs was assessed using the Seahorse XF Cell Mito Stress Test Kit (Agilent, USA). When cell confluence reached approximately 50%, cells were seeded into XF24-well microplates following standard Seahorse protocols. After adherence, 150 μL of DMEM was added to each well, and plates were allowed to sit at room temperature for 1 hour to promote even cell settling. On the day of the assay, XF calibrant (1 mL/well) was added to the sensor cartridge and incubated overnight at 37 °C. Before measurement, cells were washed twice and incubated with 500 μL of Seahorse assay medium at 37 °C for 1 hour. Mitochondrial morphology and ROS fluorescence were visualized using a Leica TCS SP5 confocal microscope (excitation: 488 nm; emission: 501–563 nm).
Animal Survival Analysis
For survival analysis, 16 C57BL/6 mice per group were randomly selected using a random number table Starting 12 hours after CLP surgery, survival rates and times were monitored and recorded over a 72-hour period.
Statistical Analysis
All statistical analyses were conducted using R software (version 4.0.2; https://www.r-project.org/). For normally distributed continuous variables, data are presented as mean±standard deviation. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparison test. For non-normally distributed data, the Mann–Whitney U-test (Wilcoxon rank-sum test) was applied. ROC curves were generated using the pROC package to evaluate classification performance. Survival rates were analyzed by the Kaplan-Meier estimator and compared using a log-rank hazard ratio. Double-blind method was adopted for intestinal pathological scoring and image quantitative analysis. All statistical tests were two-sided, and p < 0.05 was considered statistically significant.
Results
Identification of DEGs Between Sepsis and Control Samples
Five gene expression datasets were obtained from public databases. Differential expression analysis was performed on each dataset, and the results were visualized using volcano plots. The number of DEGs identified in the individual datasets was 826, 709, 1049, 1,310, and 543, respectively. Integration of these results using a Venn diagram (Figure 1A) revealed 159 overlapping DEGs that were consistently dysregulated across all five datasets (Figure 1B).
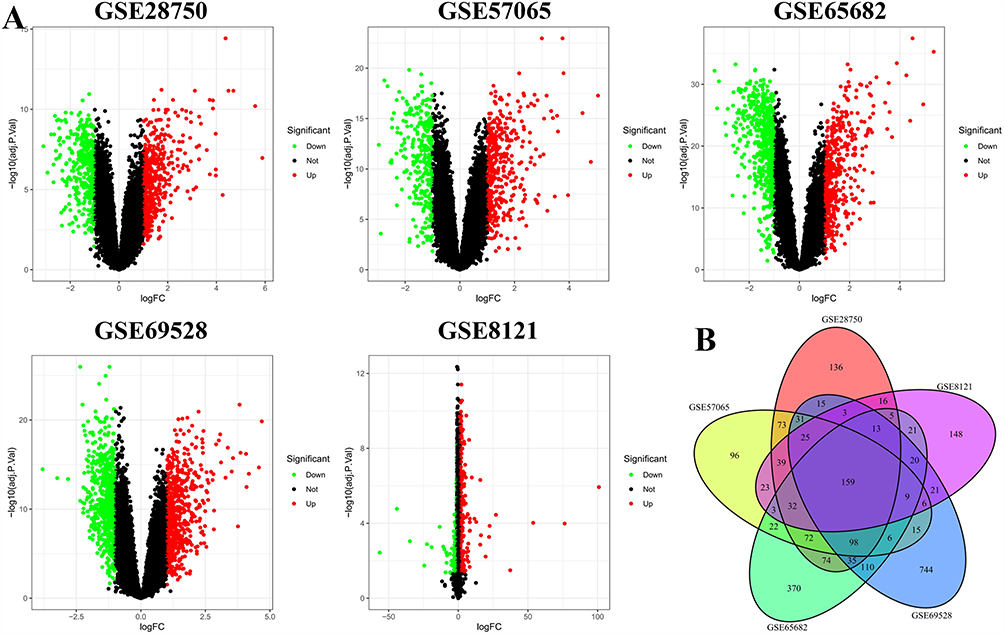 |
Figure 1 Identification of DEGs. (A) Volcano plots displaying the distribution of upregulated and downregulated genes in each dataset. (B) Venn diagram illustrating the intersection of DEGs among all five datasets, yielding 159 common DEGs. Each colored area represents one dataset. |
GO and KEGG Pathway Enrichment Analysis
To explore the biological functions of the identified DEGs, GO enrichment analysis was conducted using the DAVID platform. The results highlighted autophagy, phagocytic vesicle assembly, and cysteine-type peptidase activity as the most significantly enriched terms within the biological process (BP), cellular component (CC), and molecular function (MF) categories, respectively (Figure 2A). Notably, genes upregulated in sepsis were primarily involved in pathways related to phagosome formation and autophagy, whereas downregulated genes were enriched in processes related to ubiquitin-like protein modifications, inflammasome activity, and enzymatic regulation—highlighting a disruption of immune-related pathways in sepsis. A GO circle plot further displays the top five enriched GO terms (Figure 2B), and a chord diagram visualizes the relationships between representative DEGs and these top GO categories (Figure 2C).
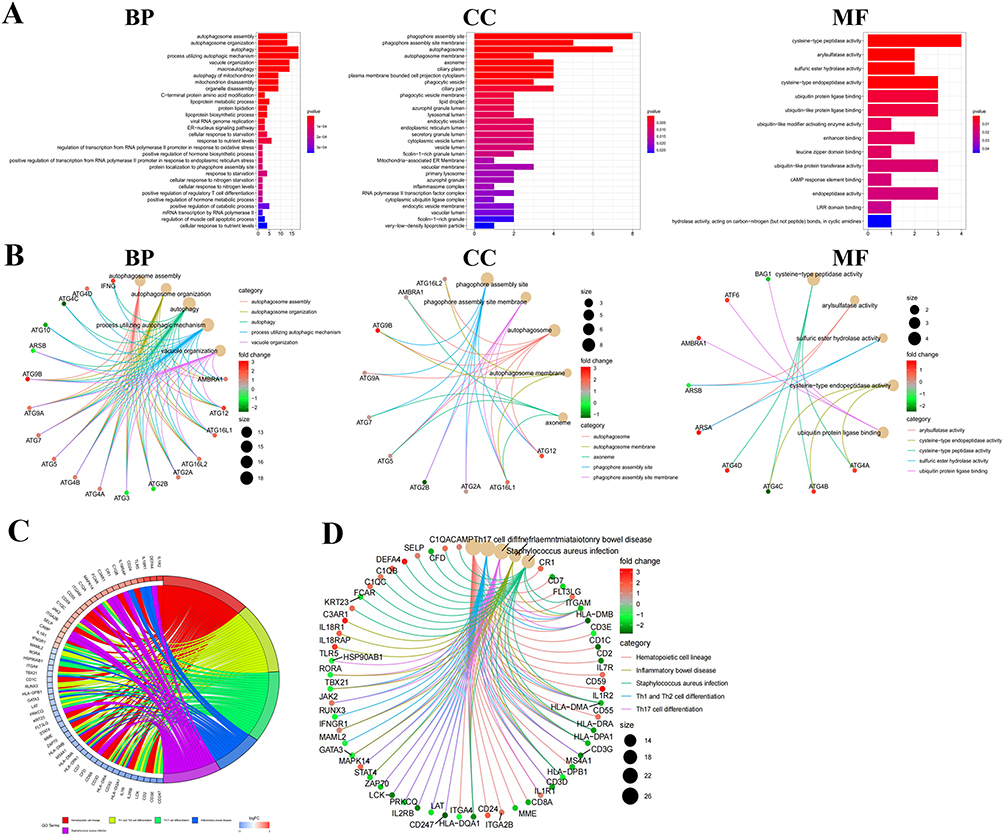 |
Figure 2 GO and KEGG Enrichment Analyses. (A) GO analysis results are categorized into three domains: biological processes, cellular components, and molecular functions. (B) GOCircle plot showing the top five significantly enriched GO terms and associated DEGs, with gene color indicating fold-change magnitude. (C) Chord plot illustrating the connections between common DEGs and top GO terms. (D) KEGG pathway enrichment analysis highlighting the top five significantly enriched pathways associated with immune and neutrophil-related processes. |
To further understand the signaling pathways involved, pathway enrichment analysis was performed using KOBAS 3.0, referencing multiple databases, including KEGG, Reactome, BioCyc, and PANTHER. The analysis revealed that common DEGs were significantly enriched in immune-related pathways, particularly those associated with neutrophil activation and dysfunction (Figure 2D).
WGCNA
WGCNA was employed to identify gene modules with highly correlated expression patterns, summarize each module using its eigengene or representative hub genes, and relate these modules to clinical traits such as sepsis. This network-based approach facilitates the discovery of biomarkers and prioritization of therapeutic targets across complex biological conditions.
Network construction: Gene expression matrices from five independent datasets—GSE28750, GSE57065, GSE65682, GSE69528, and GSE8121—were analyzed. Hierarchical clustering was performed using Pearson correlation coefficients to detect outlier samples. Sample dendrograms indicated no significant outliers across the datasets (Figure 3A).
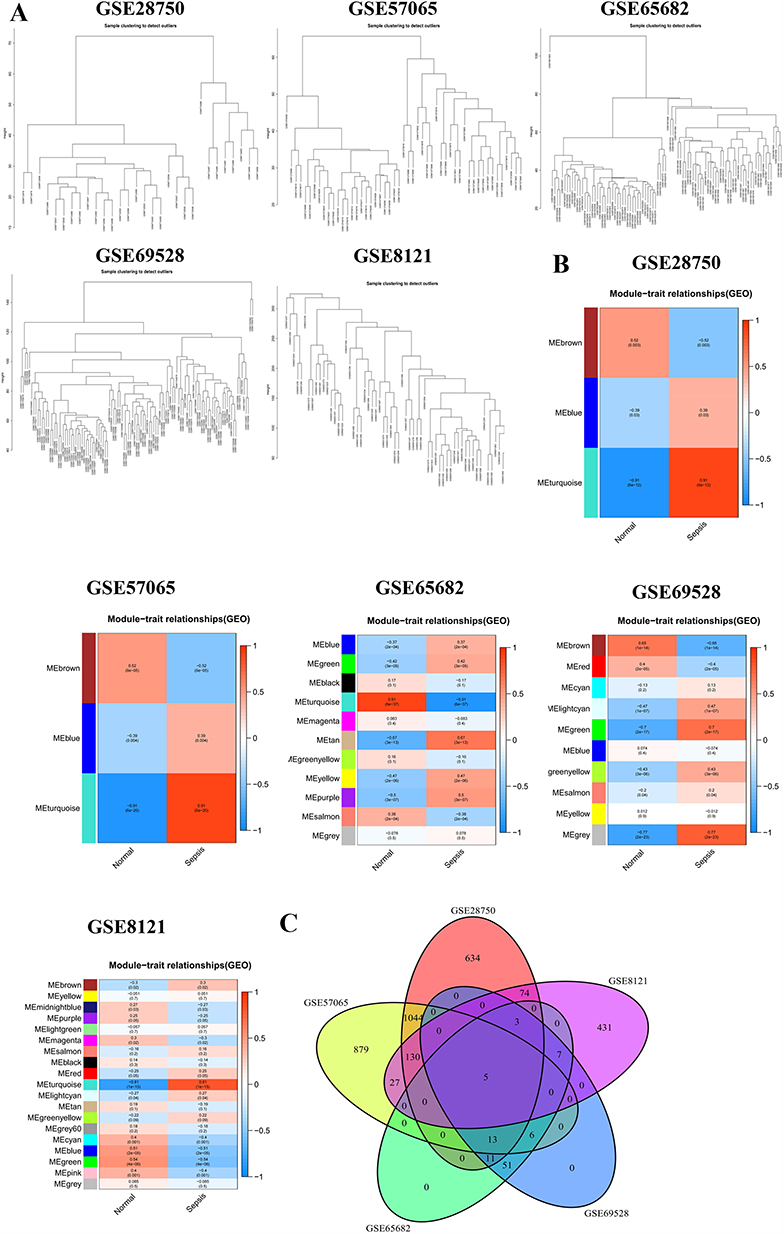 |
Figure 3 WGCNA results. (A) Sample clustering dendrogram for outlier detection. Samples from five datasets (GSE28750, GSE57065, GSE57066, GSE65682, and GSE69528) were clustered, with most disease-status-matched samples grouped together. (B) Heatmap of module-trait relationships. Correlations between each module and sepsis across the five datasets are shown. Each row represents a module eigengene, and each column represents a trait (sepsis or healthy control). The values within the cells indicate the correlation coefficient and corresponding p-value. Color intensity reflects the strength of the correlation. (C) Venn diagram identifying five overlapping hub genes across sepsis-associated modules from all five datasets. |
Module detection and trait association: Genes exhibiting similar expression profiles were grouped into modules using dynamic tree cutting. These modules were largely independent at the transcriptomic level. Module-trait relationships were assessed by correlating module eigengenes with the clinical trait (sepsis vs healthy control). In each dataset, a specific module—indicated by a distinct color—exhibited the strongest and most statistically significant association with sepsis (Figure 3B).
Gene significance (GS) and hub selection. Within each sepsis-associated module, GS was calculated to quantify the correlation between individual gene expression and the sepsis phenotype. Several genes with high GS scores, including CD177 and CLEC4D, have been previously implicated in sepsis pathogenesis, supporting the biological relevance of the identified modules.
Cross-dataset integration: To identify robust candidate genes, sepsis-related modules from all five datasets were compared. A Venn diagram revealed five overlapping hub genes consistently present across datasets: ACSL4, IL13RA1, MARCKS, NUMB, and RNF19B (Figure 3C). Among these, ACSL4 displayed the highest GS. Collectively, these results highlight ACSL4 and four additional hub genes as key regulators consistently associated with sepsis across multiple independent cohorts.
Construction of PPI Network and Identification of Hub Genes
To further explore key genes associated with sepsis, we constructed a PPI network based on the 159 DEGs identified through integrative analysis. Using the genes most strongly correlated with sepsis from the WGCNA modules, we established a PPI network to assess protein-level interactions (Figure 4A). From this network, the top 30 hub genes were ranked by connectivity, with node color intensity (red to yellow) indicating descending levels of connectivity (Figure 4B). To refine the list of candidate genes, we intersected the PPI-derived hub genes with the 159 DEGs previously identified. Notably, only one gene, ACSL4, overlapped between these two gene sets (Figure 4C).
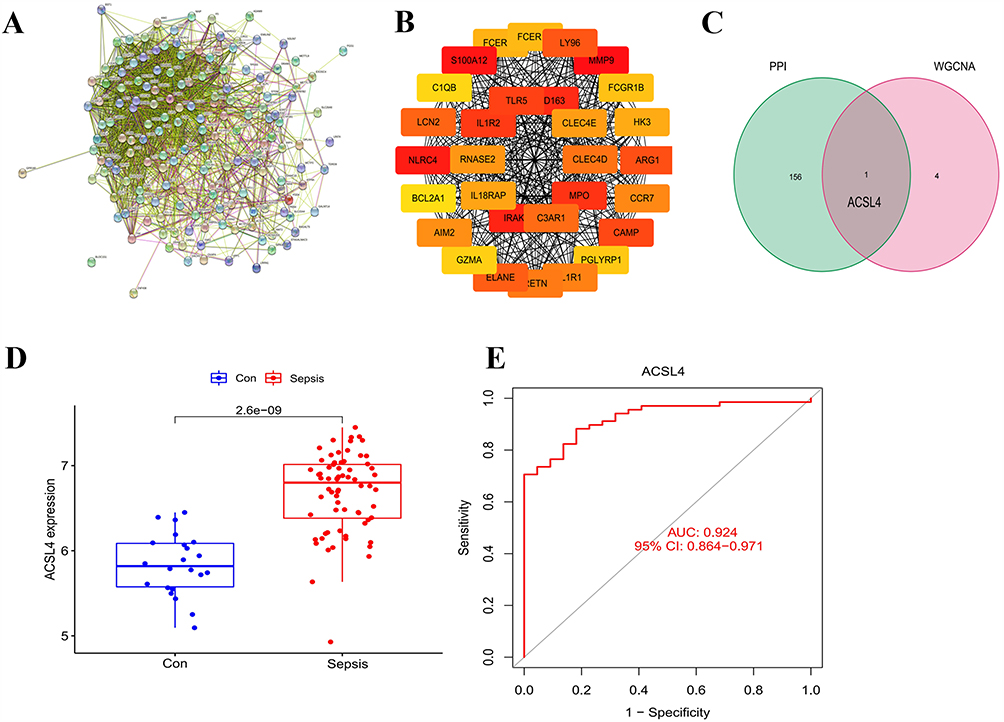 |
Figure 4 Construction of the PPI Network and Identification of ACSL4 as a Core Gene. (A) A PPI network was constructed based on 159 common DEGs. (B) The top 30 hub genes were identified based on network connectivity, with nodes colored from red to yellow indicating descending degrees of connectivity. (C) Key genes were selected based on the overlap between hub genes identified in the PPI network and genes enriched in the co-expression network. (D) Box plot showing significantly increased ACSL4 expression in sepsis patients compared to healthy controls. Blue indicates the healthy control group; red represents the sepsis group. (E) ROC curve demonstrating the diagnostic performance of ACSL4, with an AUC of 0.924, indicating high sensitivity and specificity. |
Validation of ACSL4 Expression in an Independent Dataset
To validate the upregulation and diagnostic relevance of ACSL4, we performed a single-gene expression analysis using the independent GSE95233 dataset. As a candidate biomarker, ACSL4 should exhibit high sensitivity and specificity, typically evaluated via ROC curve analysis. Consistent with previous findings, ACSL4 expression was significantly elevated in sepsis patients compared to healthy controls (Figure 4D). ROC analysis yielded an area under the curve (AUC) value of 0.924, indicating excellent diagnostic performance in distinguishing sepsis from non-septic individuals (Figure 4E). These results reinforce the robustness of ACSL4 as a potential biomarker for sepsis and support its relevance across multiple datasets.
Molecular Docking and Targeted Drug Screening
To identify potential therapeutic compounds targeting ACSL4, molecular docking was conducted using the top 500 candidates from two natural product libraries: the HY-L021P Natural Product Library Plus and the HY-L0107V Life Chemicals Natural Product-like Compound Library . In molecular docking, a more negative docking score indicates a stronger predicted binding affinity between the ligand and the target protein.
The top-ranked compound, designated HY-N2031, was derived from the HY-L021P Natural Product Library Plus and exhibited a docking score of –17.701, the highest among all screened compounds. Structural analysis revealed that HY-N2031 binds specifically and strongly to the mouse ACSL4 protein (Figure 5A and B), suggesting its potential as a lead compound for therapeutic targeting in sepsis. HY-N2031 forms a total of 12 hydrogen bonds and one π–cation interaction with ACSL4. Specifically, hydroxyl groups of the compound form hydrogen bonds with amino acid residues GLY465, LEU446, GLN464, LYS690, ASP573, ARG570, TYR582, GLY505, and GLN566. An additional hydrogen bond is formed between an oxygen atom on the compound ring and LEU446. A π–cation interaction occurs between the phenyl ring of the compound and LYS587 (Figure 5C and D).
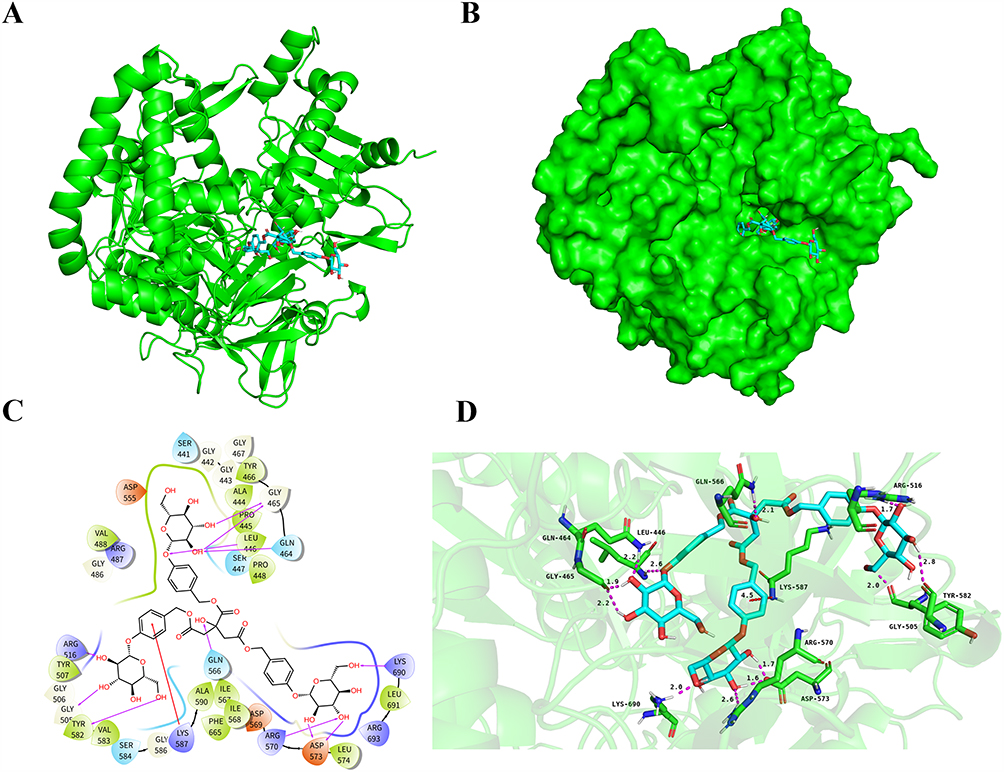 |
Figure 5 Molecular Docking and Binding Affinity Analysis of HY-N2031 (Parishin) with Established Ferroptosis-Associated Sepsis Genes. (A and B) The docking model shows the mouse ACSL4 protein backbone in green. (C and D) Nitrogen atoms are depicted in blue, oxygen atoms in red, hydrogen atoms in white, and HY-N2031 is shown as light blue sticks. Red dashed lines represent π–cation interactions, while purple dashed lines denote hydrogen bonds (with longer lines indicating weaker interactions). |
Targeted Therapy with Parishin Attenuates Inflammatory Responses and Improves Intestinal Function and Prognosis in Septic Mice by Modulating ACSL4
To validate the therapeutic potential of Parishin and its regulatory effect on ferroptosis-related genes in sepsis, we conducted both in vivo and in vitro experiments using murine and cellular models. Our results showed that, compared with the untreated sepsis group, Parishin significantly downregulated the mRNA expression of ACSL4 in monocytes and small intestinal epithelial cells. Specifically, the expression level of ACSL4 decreased from 17.97±4.2 to 9.23±0.95 in monocytes, and from 8.57±1.3 to 4.53±0.78 in small intestinal epithelial cells (P < 0.05, Figure 6A and Supplementary Table 1).
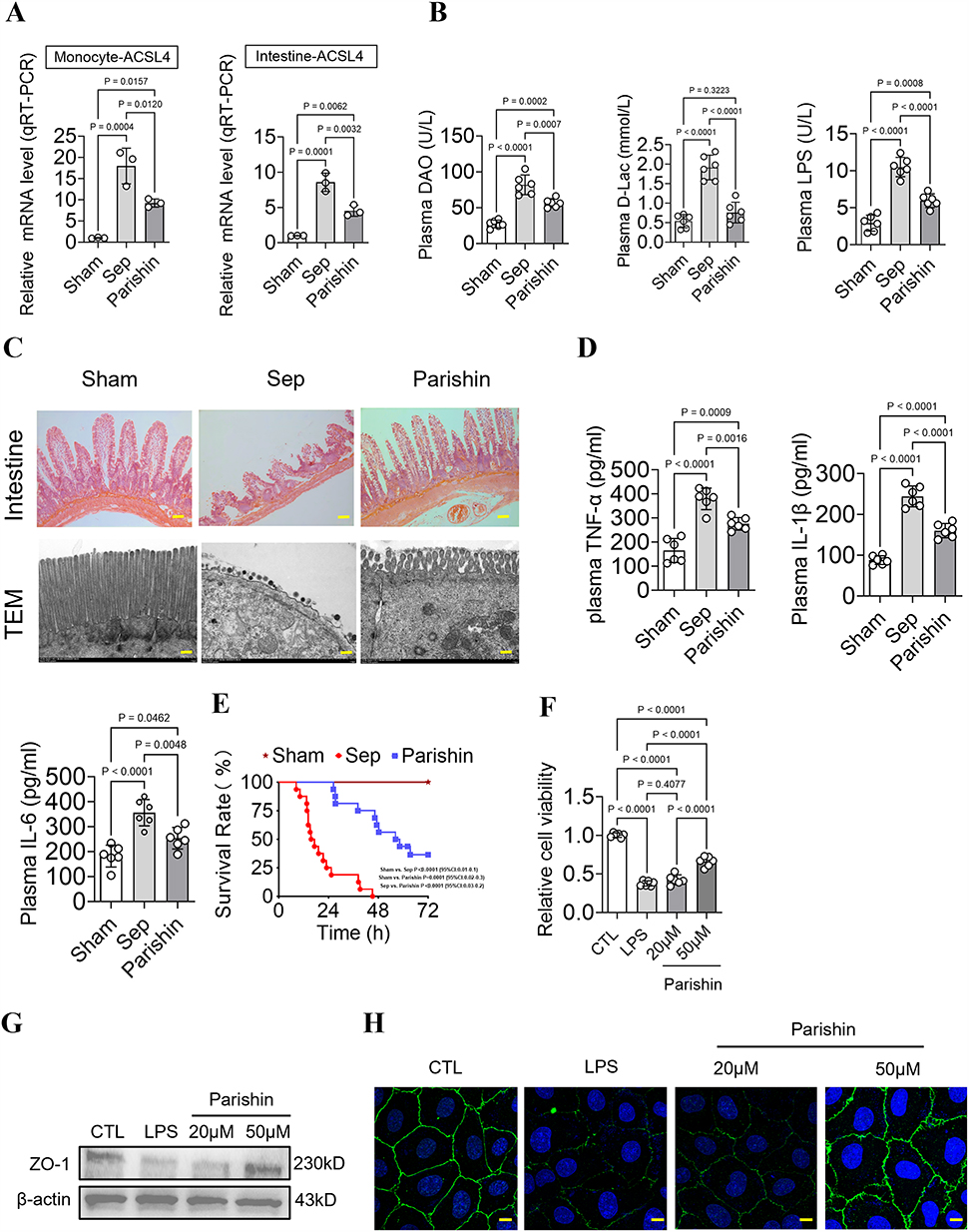 |
Figure 6 Therapeutic Potential of Parishin in Alleviating Sepsis-Induced Intestinal Injury. (A) mRNA expression of ferroptosis-related genes in septic mice following Parishin treatment (n=3). (B) Levels of plasma diamine oxidase (DAO), D-lactate (D-Lac), and lipopolysaccharide (LPS) in septic mice (n=6). (C) Histological evaluation of intestinal mucosa in mice (scale bar,50um), and transmission electron microscopy (TEM) images of intestinal villi (scale bar,3um). (D) Levels of plasma inflammatory cytokines (TNF-α, IL-1β, IL-6) in septic mice (n=6). (E) Survival analysis of septic mice following Parishin treatment (Log-Rank (Mantel-Cox) Test) (n=16). (F) Cell viability on IEC-6 cells (n=6). (G) Expression levels of ZO-1 protein in intestinal tissues (n=3). (H) Laser confocal microscopy analysis of IEC6 cells (scale bar, 50 um) (n=3). |
To assess intestinal barrier integrity, we measured plasma levels of DAO, D-lactate, and LPS. In the Sham group, the levels of these markers were 27.43±5.33, 0.54±0.17, and 2.92±1.08. Compared with the untreated sepsis group, the levels of these markers in septic mice treated with Parishin were significantly decreased from 81.88±13.71 to 56.60±6.04, 1.91±0.32 to 0.76±0.27, and 10.50±1.31 to 5.94±0.92, respectively (P<0.05). Parishin treatment distinctly reduced all three markers compared to the sepsis group, they have decreased by 30.87%, 60.27% and 43.36%.This result indicates that Parishin alleviates intestinal permeability damage (P < 0.05, Figure 6B).
Histopathological examination via HE staining revealed that intestinal tissues from control mice exhibited normal mucosal architecture, including intact epithelial alignment, well-formed crypts, and an absence of inflammatory infiltration. In contrast, septic mice displayed extensive epithelial damage, crypt destruction, crypt abscess formation, and prominent infiltration of lymphocytes and neutrophils. Treatment with Parishin led to notable improvements in mucosal structure and reduced inflammatory changes.TEM further supported these findings. In control mice, intestinal epithelial cells displayed well-organized microvilli, intact tight junctions, and mitochondria with clearly defined cristae. Septic mice, by comparison, showed shortened and disorganized microvilli, disrupted tight junctions, and swollen, cristae-depleted mitochondria. Parishin treatment partially restored mitochondrial morphology and epithelial integrity (Figure 6C).
We next measured plasma concentrations of inflammatory cytokines. Compared to the normal Sham group, TNF-α, IL-1β, and IL-6 levels increased from164.8±49.65 to 379.2±44.45, 87.83±10.72 to 244±25.4, and 182.2±43.92 to 355.8±52.8, respectively (P < 0.05). Treatment with Parishin markedly reduced these proinflammatory cytokines relative to the untreated sepsis group, they have decreased to 275.5±26.15, 160.2±17.39 and 253.5±43.11 (P < 0.05, Figure 6D). Importantly, 72h survival analysis showed high early mortality in septic mice, with no surviving beyond 72 hours (mean survival, 16.4h). Treatment with Parishin significantly improved outcomes, with six mice surviving beyond 72 hours (mean survival, 57.3h). Parishin significantly improved the 72-hour survival rate of septic mice (P < 0.05, Figure 6E and Table 1).
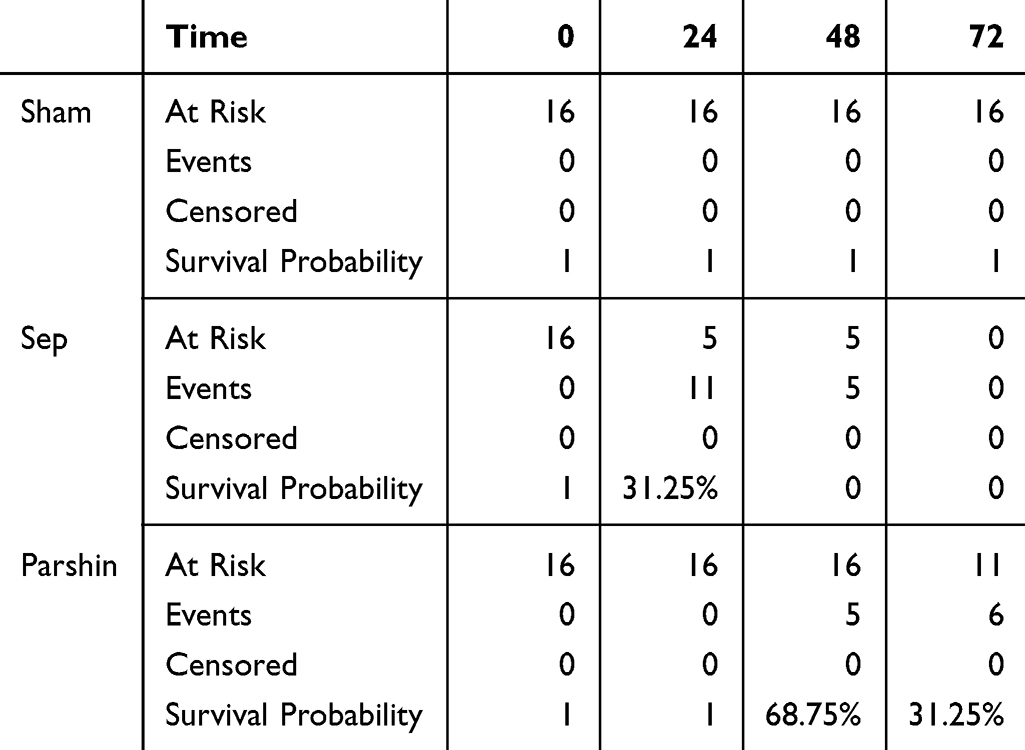 |
Table 1 Kaplan–Meier Table |
At the cellular level, compared with the control group (1±0.03), cell proliferation capacity was decreased in the sepsis group (0.38±0.03), while it was upregulated by 75.32% in the Parishin (50um,0.66±0.07) group (P < 0.05, Figure 6F). At the protein level, ZO-1 expression in intestinal tissues was markedly reduced in septic mice, consistent with compromised barrier function. Parishin treatment significantly restored ZO-1 levels (P < 0.05, Figure 6G). These findings were further corroborated by immunofluorescence imaging, which showed improved mucosal barrier integrity following Parishin treatment (Figure 6H).
Parishin Reduces Ferroptosis in Intestinal and Circulating Mononuclear Cells by Targeting Sepsis-Specific Ferroptosis-Related Genes
To elucidate the effects of Parishin on ferroptosis during sepsis, we evaluated ferroptosis-associated markers in both intestinal tissues and circulating mononuclear cells. Prussian blue staining of intestinal sections demonstrated a marked reduction in iron deposition following Parishin treatment, indicating that Parishin effectively mitigates cell death by suppressing ferroptosis (Figure 7A). In vitro, LPS was utilized to simulate septic conditions in intestinal epithelial cells and monocytes. LPS exposure distinctly increased intracellular iron levels. It is specifically manifested as in intestinal epithelial cells, MDA and Fe²⁺ levels increased from 3.14±1.1 to 17.32±2.8 and 15.05±5.97 to 49.4±8.85. In monocytes, MDA and Fe²⁺ levels increased from 5±1.73 to 22.22±8.44 and 32.05±7.22 to 80.97±9.59 (P < 0.05), whereas treatment with Parishin notably diminished ferrous ion fluorescence intensity. It is specifically manifested as in intestinal epithelial cells, MDA and Fe²⁺ levels decreased to 7.61±1.08 and 30.12±7.3. In monocytes, MDA and Fe²⁺ levels decreased to 11.53±1.62 and 47.79±5.41, reflecting reduced iron accumulation (P < 0.05, Figure 7B, Supplementary Table 2). A similar reduction in iron overload was observed in Mean Fluorescence Intensity (MFI) from septic mice treated with Parishin.Compared with the untreated sepsis group, the MFI of Fe and LPO in septic mice treated with Parishin were significantly decreased from 10086±1839.14 to 6446±694.77 and 2531±932.34 to 730.6±87.84 (P < 0.05, Figure 7C). Furthermore, GPX4 and SLC7A11, both of which are central to ferroptosis inhibition, were significantly downregulated in septic intestinal tissues and PBMCs, as well as in LPS-stimulated monocytes and IEC-6 cells. Parishin treatment restored the expression levels of both proteins. In contrast, COX2, a pro-ferroptotic marker typically upregulated during iron-induced lipid peroxidation, was significantly increased under septic conditions and was markedly reduced following Parishin intervention. Compared to the sepsis group, treatment with Parishin is specifically manifested as in intestine, GPX and SLC7A11 levels increased from 0.35±0.12 to 0.67±0.05 and 0.39±0.09 to 0.69±0.04, respectively, while COX2 decreased from 1.57±0.03 to 1.25±0.03. In monocytes, GPX and SLC7A11 levels increased from 0.46±0.08 to 0.69±0.04 and 0.44±0.09 to 0.7±0.06, respectively, while COX2 decreased from 1.74±0.06 to 1.36±0.08. In IEC-6 cell, GPX and SLC7A11 levels increased from 0.53±0.1 to 0.74±0.1 and 0.49±0.11 to 0.7±0.08, respectively, while COX2 decreased from 1.73±0.04 to 1.41±0.07 (P < 0.05, Figure 7D). Collectively, these results demonstrate that Parishin alleviates sepsis-induced ferroptosis in both intestinal and circulating mononuclear cells, likely by modulating sepsis-specific ferroptosis-related genes.
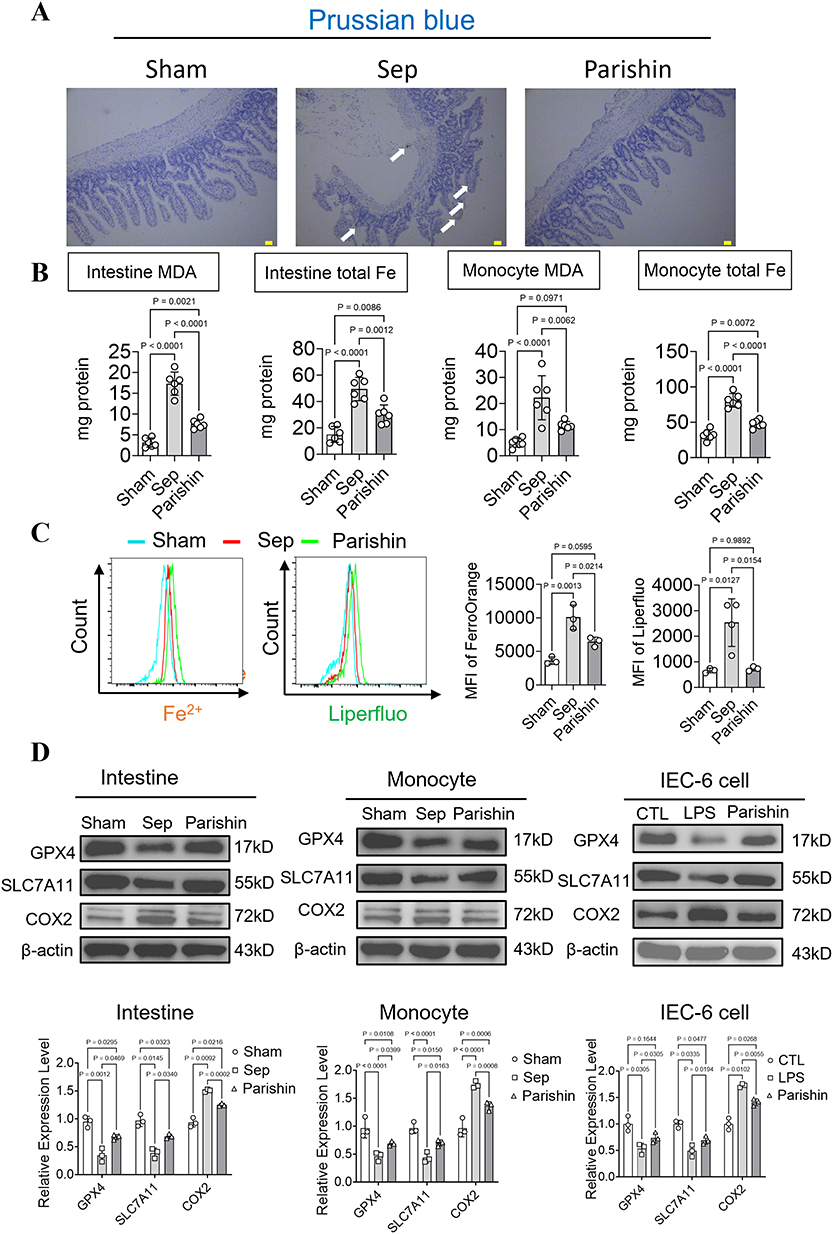 |
Figure 7 Regulatory Effects of Parishin on Iron Overload and Ferroptosis During Sepsis. (A) Representative Prussian blue staining images showing ferrous iron accumulation in intestinal tissue (scale bar, 50 μm). (B) Measurement of malondialdehyde (MDA) levels and total iron content in intestinal epithelial cells and monocytes under LPS stimulation (n=6). (C) Effects of Parishin on iron accumulation and lipid peroxidation (LPO) in septic monocytes (n=3). (D) Protein expression levels of GPX4, SLC7A11, and COX2 (n=3). |
Parishin Improves Mitochondrial Function and Reduces Ferroptosis by Enhancing Mitochondrial Apoptosis
To determine whether the intestinal protective effects of Parishin in sepsis are linked to mitochondrial function, we examined the expression of key mitochondrial regulatory proteins via Western blot analysis. In septic mononuclear cells and IEC-6, the expression of PGC-1α was significantly decreased from 1±0.15 to 0.53±0.04 and 0.96±0.08 to 0.46±0.1, while ACSL4 and phosphorylated Smad3 (p-Smad3) levels were markedly increased from 1±0.14 to 1.54±0.08, 1±0.07 to 1.54±0.11 and 0.97±0.12 to 1.35±0.08, 0.99±0.11 to 1.37±0.09. Treatment with Parishin reversed these trends, upregulating PGC-1α (0.75±0.07 and 0.69±0.12) while downregulating ACSL4 (0.67±0.05 and 1.15±0.03) and p-Smad3 (0.69±0.04 and 1.14±0.04) (Figure 8A, Supplementary Figure 1A and Supplementary Table 3). Moreover, Silencing ACSL4 led to reduced expression of both ACSL4 and p-Smad3, manifests itself specifically as decreased from 1±0.15 to 0.41±0.07 and 1±0.1 to 0.43±0.06, and concomitantly increased levels of PGC-1α from 1±0.08 to 1.5±0.07 (Figure 8B and Supplementary Figure 1B). These molecular findings were validated through confocal microscopy, which revealed improved mitochondrial membrane potential and reduced mitochondrial ROS levels in both IEC-6 and mononuclear cells treated with Parishin (Figure 8C). In addition, Seahorse metabolic analysis showed that Parishin significantly improved both basal and maximal mitochondrial respiration in LPS-stimulated intestinal epithelial cells and monocytes, compared to the sepsis group, in IEC-6 and mononuclear cells its level increased by 1.56-fold, 2.2-fold, and 0.68-fold, 0.61-fold, respectively (P < 0.05, Figure 8D). These findings confirm that Parishin enhances mitochondrial metabolic activity, possibly by inhibiting Smad3 phosphorylation and ACSL4-mediated ferroptotic signaling.
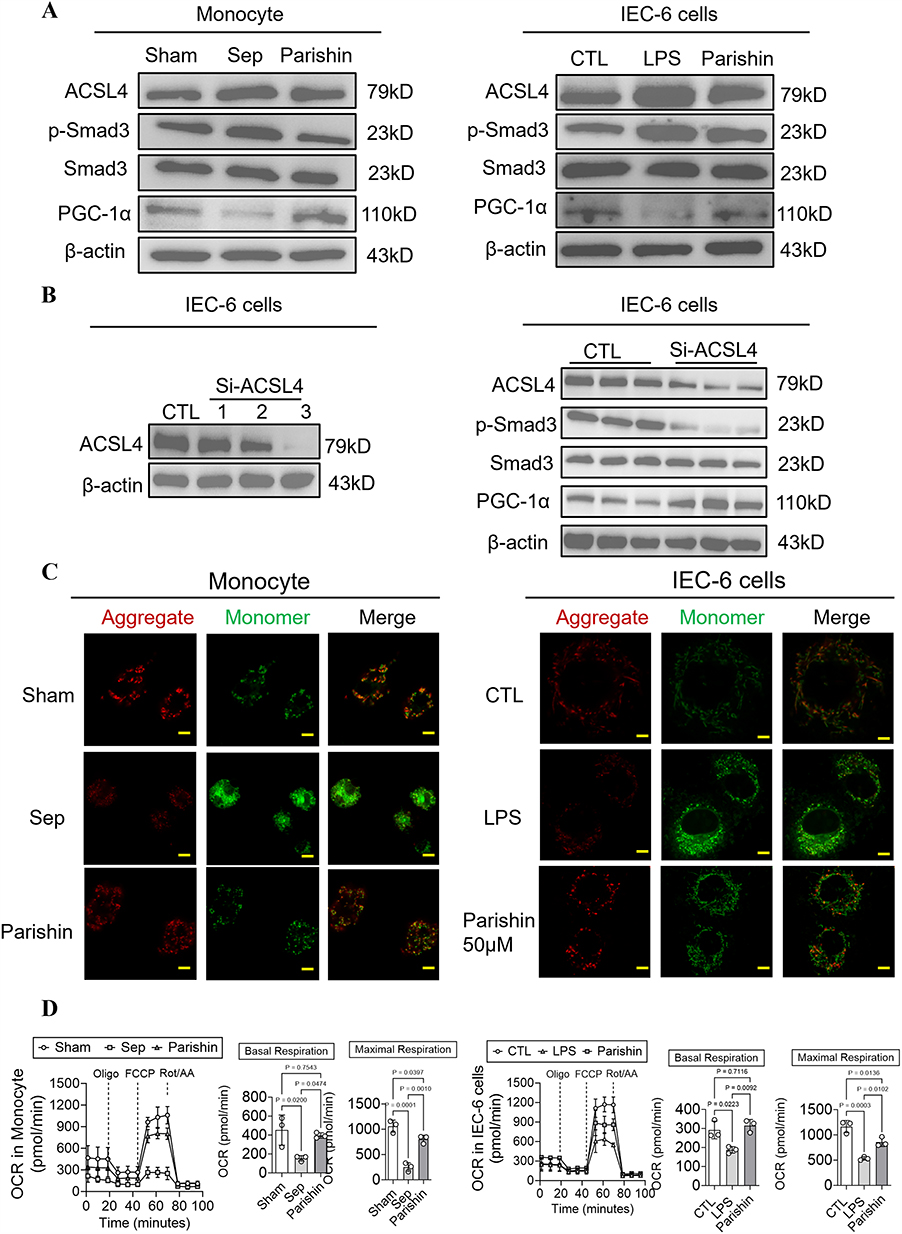 |
Figure 8 Parishin Improves Mitochondrial Function in LPS-Stimulated Intestinal Epithelial Cells and Monocytes. (A) Western blot analysis showing the effects of Parishin on the expression of ACSL4, Smad3, phosphorylated Smad3 (p-Smad3), and PGC-1α in LPS-stimulated intestinal epithelial cells and monocytes (n=3). (B) The relative expression level of ACSL4 after treatment in Si-ACSL4 of IEC-6 cells (n=3). (C) Confocal microscopy images of mitochondrial ROS and mitochondrial membrane potential (MMP) in intestinal epithelial cells (scale bar, 20 μm) and monocytes (scale bar: 40 μm) (n=3). (D) Effects of Parishin on mitochondrial respiration in intestinal epithelial cells and monocytes (n=3). |
These results suggest that Parishin can reduce the occurrence of ferroptosis in intestinal and blood leukocytes after sepsis, which may be closely related to Parishin improving mitochondrial quality by regulating ACSL4 (Figure 9).
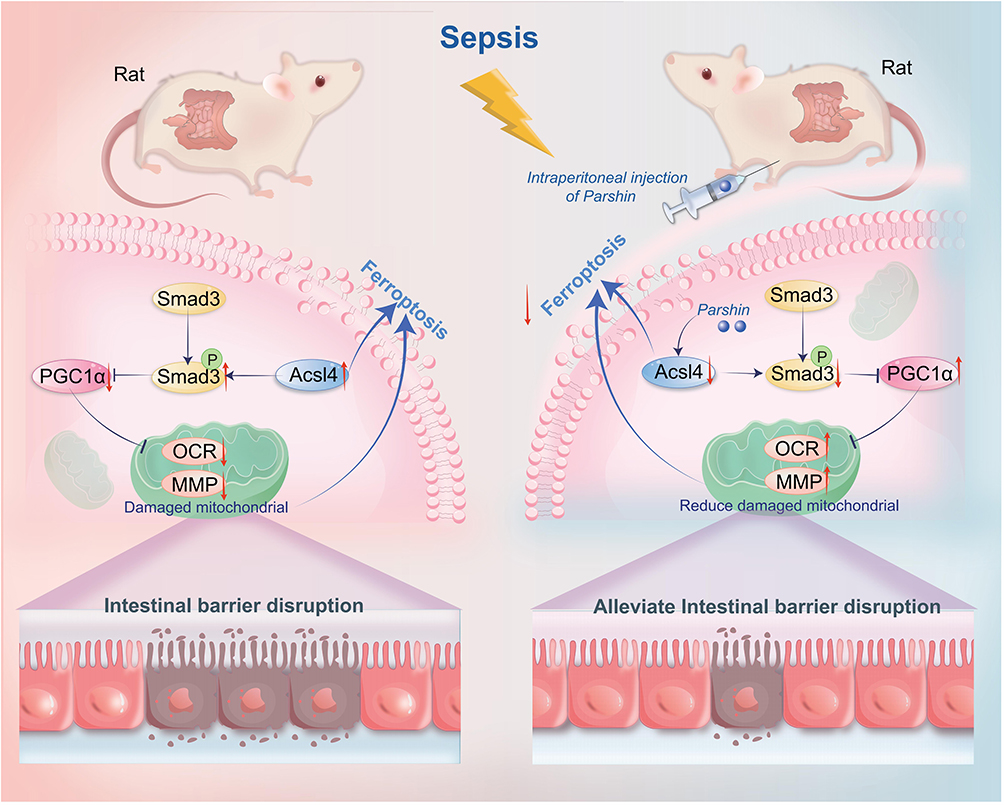 |
Figure 9 The mechanism diagram of the protective effects of Parishin on sepsis-induced intestinal injury. The red arrow pointing upward represents promotion, and the red arrow pointing downward represents inhibition. |
Discussion
Sepsis is a life-threatening syndrome characterized by acute organ dysfunction resulting from a dysregulated host response to infection. It poses a significant global health burden, with wide heterogeneity in infection sources, causative pathogens, and affected organ systems. This variability complicates both timely diagnosis and the development of effective, targeted treatments.29 A hallmark of sepsis pathophysiology is profound immune dysregulation. Current clinical management strategies—centered on antimicrobial therapy, source control, hemodynamic stabilization, and organ support—are largely supportive. However, the absence of specific biomarkers to assess immune dysfunction continues to limit diagnostic precision and therapeutic targeting. There is therefore an urgent need to identify reliable biomarkers and novel therapeutic agents capable of altering the disease trajectory and improving patient outcomes.29,30
Our study addressed this gap by identifying ACSL4 as a core gene involved in sepsis pathogenesis through integrated analysis of GEO transcriptomic datasets. Using network pharmacology and molecular docking, we further identified Parishin, a natural compound from G. elata, as a potential therapeutic agent targeting ACSL4. Both in vivo and in vitro studies confirmed Parishin’s protective role against sepsis-induced intestinal injury, advancing our understanding of the mechanistic link between ACSL4 and ferroptosis. These findings suggest that pharmacological inhibition of ACSL4 may represent a novel strategy for mitigating intestinal damage in sepsis.
To address this challenge, we employed an integrated bioinformatics approach using five independent transcriptomic datasets. Differential expression analysis and WGCNA were used to identify genes associated with sepsis pathogenesis. ACSL4 emerged as a robust, consistently upregulated gene across all datasets. Its expression was further validated in the independent GSE95233 cohort, confirming its relevance as a potential biomarker for sepsis.
ACSL4 is known to regulate lipid metabolism and plays a pivotal role in mediating ferroptosis. Recent studies have highlighted its contribution to organ injury in various models of sepsis, suggesting that ACSL4 may be a promising therapeutic target.17,31–34 For instance, Wu et al demonstrated that histone lactylation enhances METTL3-mediated m6A modification of ACSL4 mRNA, promoting ferroptosis and exacerbating sepsis-induced lung injury. Pharmacological inhibition of METTL3 or ACSL4 significantly improved pulmonary outcomes.32 Similarly, Xiao et al showed that the NFIL3–ACSL4 axis regulates both ferroptosis and inflammation in septic acute kidney injury, with inhibition of this pathway improving renal function.35 Sun et al reported that exogenous histone exposure induced both pyroptosis and ferroptosis in sepsis-associated encephalopathy, a process partially mitigated by vitamin D supplementation.36 In parallel, Huang et al developed small-molecule ACSL4 inhibitors that effectively reduced ferroptotic cell death and associated tissue damage in preclinical models.31
Ferroptosis is a distinct form of iron-dependent, non-apoptotic cell death, increasingly recognized as a critical contributor to the pathogenesis of diverse conditions, including cancer, ischemia-reperfusion injury, neurodegeneration, and organ dysfunction.37 Geng et al demonstrated that activation of the Nrf2 pathway suppressed ferroptosis in macrophages, thereby alleviating sepsis-induced acute lung injury.33 Similarly, Li et al reported that the Nrf2/GPX4 axis inhibited ferroptosis and offered neuroprotection in sepsis-associated encephalopathy.38 Zhang et al found that downregulation of Smad3 reduced ferroptosis-mediated cardiac damage during sepsis.39 Despite advancements in identifying endogenous regulators that protect against ferroptosis by limiting lipid peroxidation, certain cell types remain resistant to these mechanisms.40 ACSL4, a key enzyme involved in polyunsaturated fatty acid metabolism, plays a pivotal role in sensitizing cells to ferroptosis by promoting the incorporation of oxidizable fatty acids into membrane phospholipids. While ACSL4 has been established as a potential therapeutic target, no clinically approved compound specifically targeting ACSL4 is currently available. Moreover, its precise role in sepsis-induced intestinal injury, a critical yet underexplored aspect of sepsis pathology, remains insufficiently characterized.
Existing clinical interventions for sepsis predominantly emphasize infection control, fluid resuscitation, and hemodynamic support. However, these strategies inadequately address GI involvement, despite the GI tract being among the earliest and most severely affected organs in sepsis.41,42 Impaired visceral perfusion can lead to intestinal ischemia and epithelial injury, which in turn triggers the release of gut-derived inflammatory mediators. Compounding this, systemic inflammation disrupts mucosal barrier integrity, facilitating the translocation of intestinal microbiota and endotoxins into the systemic circulation—thereby intensifying the inflammatory cascade.43 The GI tract has thus been termed the “motor” of MODS in sepsis, functioning as a central hub of immune activation and systemic inflammation.41,44 Therefore, therapeutic strategies that focus on mitigating intestinal injury, restoring epithelial barrier integrity, and interrupting the cycle of immune dysregulation are urgently needed to improve sepsis outcomes.45
Emerging evidence documents that ferroptosis is crucial in GI dysfunction during sepsis. In thalassemic mice with iron overload, sepsis severity is significantly exacerbated due to iron-induced mucosal injury, leading to elevated serum levels of LPS and (1,3)-β-D-glucan. Iron appears to amplify the inflammatory response initiated by these pathogen-associated molecular patterns.46 To explore targeted interventions aimed at modulating ACSL4 and ferroptosis in sepsis-induced intestinal injury, we employed a network pharmacology approach combined with molecular docking using the DrugBank database. Among the screened compounds, Parishin—a small-molecule polyphenolic glycoside primarily derived from G. elata, a traditional Chinese medicinal herb—emerged as a promising ACSL4-targeting agent. Historically documented in classical texts such as the Shennong Bencao Jing, Parishin has been used in traditional medicine for over two millennia and is noted for its favorable pharmacological safety profile.47,48 Modern pharmacological studies have shown that Parishin delays vascular and cardiac aging in murine models.20,23,49 In the current study, Parishin notably reduced systemic inflammatory cytokine levels, improved organ function, enhanced survival in septic mice, suppressed ferroptosis, and preserved mitochondrial integrity. These effects align with findings from Wang et al, who reported that Parishin attenuated LPS-induced inflammation,18 highlighting its anti-inflammatory potential in septic conditions. Importantly, Parishin demonstrated therapeutic efficacy at doses well below its known clinical safety threshold (220–500 mg/kg). Pharmacokinetic studies have shown that Parishin exhibits rapid absorption and clearance, with peak plasma concentrations occurring within 3 hours and near-complete elimination by 8 hours. Its pharmacokinetics follow first-order kinetics, with a dose-proportional AUC, supporting its predictable systemic behavior.48,50–55
To further elucidate the protective mechanism of Parishin in sepsis-induced intestinal injury, we reviewed recent studies linking ACSL4 to ferroptosis in septic models.33,56,57 Liangming Liu et al have showed that ACSL4 can disrupt pericyte mitochondrial function by downregulating PGC1α in pericytes, thereby inducing pericyte ferroptosis, which leads to the dysfunction of the pulmonary vascular barrier after sepsis.17 Yang Y et al also have demonstrated that knockdown of ACSL4 can inhibit the phosphorylation of Smad3, and promote PGC-1α expression in a Smad3-dependent manner. ACSL4 may regulate fatty acid oxidation through the TGF-β1/Smad3/PGC1α axis, thereby regulating ferroptosis.58
Therefore, in this study, we observed the expression levels of ACSL4, p-Smad3, and PGC-1α in monocytes and intestinal epithelial cells after sepsis and after treatment with parishin. Our study found that both monocytes and intestinal epithelial cells exhibited significantly upregulated expression levels of ACSL4 and p-Smad3, as well as significantly downregulated expression of PGC-1α, leading to severe impairment of mitochondrial function after sepsis. Parishin recovered these changes. We further knocked down ACSL4 in intestinal epithelial cells and found that after knocking down ACSL4, the expression level of p-Smad3 in intestinal epithelial cells significantly decreased, while the expression level of PGC-1α significantly increased. Based on the above results, our study pointed out that Parishin Protects Against Sepsis-Induced Intestinal Injury by Modulating the ACSL4/p-Smad3/PGC-1α Pathway.
Consistent with our findings, ACSL4 upregulation in sepsis correlates with mitochondrial dysfunction. Our results demonstrated that Parishin inhibited ACSL4 expression and modulated the Smad3 pathway. Specifically, Parishin reduced Smad3 phosphorylation, which in turn relieved suppression of PGC-1α, a key regulator of mitochondrial biogenesis and function. This regulatory effect contributes to the preservation of mitochondrial integrity and supports intestinal barrier protection during sepsis.
While our study highlights the role of the ACSL4/p-Smad3/PGC-1α axis in parishin-mediated protection against sepsis-induced intestinal injury, we acknowledge that other mechanisms may also contribute. Parishin has been reported to exert antioxidant effects by activating the Nrf2/HO-1 signaling pathway, thereby reducing oxidative stress and lipid peroxidation independently of ACSL4.59 Additionally, parishin possesses anti-inflammatory properties, such as inhibition of NF-κB activation and downstream pro-inflammatory cytokines,18,21,60 which could alleviate intestinal epithelial damage regardless of ACSL4 expression. Furthermore, parishin may enhance mitochondrial function through SIRT1-mediated deacetylation of PGC-1α, promoting mitochondrial biogenesis and cellular energy homeostasis via an ACSL4-independent route.61 These potential pathways suggest that parishin’s protective effects are likely multifactorial, involving both ACSL4-dependent and -independent mechanisms. Future studies using ACSL4 knockout or overexpression models will help further clarify the relative contribution of these pathways.
In patients with sepsis, disruption of the intestinal epithelial barrier facilitates the translocation of bacteria and endotoxins, leading to the activation of mucosal immunity and the release of pro-inflammatory cytokines by both epithelial and immune cells.62 These responses are further amplified by the systemic dissemination of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through the portal circulation and mesenteric lymphatics, contributing to distant organ dysfunction.63
In the later stages of sepsis, immunosuppression becomes a predominant clinical concern. MDSCs are key in this phase due to their immunoregulatory and suppressive functions.64 A study by Xue et al showed that ferumoxytol (FMT) an FDA-approved iron supplement—can alleviate MDSC-mediated immunosuppression by inhibiting arginase-1 activity and ROS production, suggesting potential for therapeutic use in late-stage sepsis.65 Despite the recognized clinical relevance of gut dysfunction in sepsis pathophysiology, it remains inadequately addressed in current treatment protocols.66,67
However, this study has several limitations. First, although the intestinal protective effects of Parishin were clearly demonstrated, but does not reproduce the clinical scenario in which therapy should be initiated after sepsis is recognized, and its potential impact on other sepsis-affected organs. Accordingly, our data only establish biological plausibility. Second, while our data support the anti-ferroptotic mechanism of Parishin, additional anti-inflammatory or antioxidant pathways may also contribute to its therapeutic efficacy and warrant further investigation. Thus, while parishin shows clear mechanistic promise against sepsis-induced intestinal injury, its clinical potential remains speculative until post-exposure efficacy, systemic safety and long-term benefit are rigorously established.
Conclusion
This study identifies ACSL4 as a multifunctional biomarker with diagnostic and prognostic value for sepsis. Our findings demonstrate that Parishin exerts protective effects by improving mitochondrial function, inhibiting ferroptosis, reducing systemic inflammation, and preserving intestinal barrier integrity through modulation of ACSL4. These results provide important insights into the molecular mechanisms underlying sepsis-induced intestinal injury and support the development of innovative diagnostic and therapeutic strategies. Moreover, our work highlights Parishin as a promising candidate derived from traditional Chinese medicine with potential clinical utility in sepsis management. Further investigation is warranted to explore the broader therapeutic applications of ACSL4 inhibition and to validate Parishin’s effects in clinical settings.
Ethics Statement
The data used in this study were obtained from the NCBI Gene database (https://www.ncbi.nlm.nih.gov/gene/), a publicly available repository of gene-specific information that complies with international data sharing standards. All data in NCBI Gene have been de-identified and processed in accordance with ethical guidelines for public genetic data, and no individual-level identifiable information was accessed or used in this study.Since this research solely relied on publicly accessible secondary data and did not involve the collection, use, or interaction with human participants, animals, or biological specimens, no ethical approval or informed consent was required in accordance with the regulations of the Declaration of Helsinki. According to item 3 of Article 32 of the “Notice on Issuing the Measures for Ethical Review of Human-Related Life Science and Medical Research” (Guo Wei Ke Jiao Fa 〔2023〕 No. 4), research conducted using anonymized information and data may be exempted from ethical review.
The animal study was sourced from the Animal Experimentation Center of the Army Specialty Medical Center (Animal Production License No. SCXK (Yu) 2022-0018 and an Animal Use License No. SYXK (Yu) 2022-0018). All experimental procedures were conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals established by the Animal Ethics Committee of the Army Medical University (Ethics Approval No. AMUWE20237064). The care and handling of the animals were conducted in strict accordance with the guidelines outlined in the “Guide for the Care and Use of Laboratory Animals” (https://www.nature.com/srep/journal-policies/editorial-policies#experimental-subjects). All methods were performed in accordance with the relevant guidelines and regulations. The study was carried out in compliance with the ARRIVE guidelines (https://arriveguidelines.org).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Chongqing Natural Science Foundation project (cstc2019jcyj-msxmX0018).
Disclosure
There are no conflicts in this work to declare within the author reports.
References
1. Rudd KE, Johnson SC, Agesa KM. et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–211. doi:10.1016/S0140-6736(19)32989-7
2. Bauer M, Gerlach H, Vogelmann T, et al. Mortality in sepsis and septic shock in Europe, North America and Australia between 2009 and 2019— results from a systematic review and meta-analysis. Critical Care. 2020;24(1). doi:10.1186/s13054-020-02950-2.
3. Zhou X, Liao Y. Gut-Lung Crosstalk in Sepsis-Induced Acute Lung Injury. Front Microbiol. 2021;12:779620. doi:10.3389/fmicb.2021.779620
4. Sun J, Zhang J, Wang X, et al. Gut-liver crosstalk in sepsis-induced liver injury. Critical Care. 2020;24(1):614. doi:10.1186/s13054-020-03327-1
5. Zhou Q, Verne GN. Intestinal hyperpermeability: a gateway to multi-organ failure? J Clin Invest. 2018;128(11):4764–4766. doi:10.1172/JCI124366
6. Wang C, Li Q, Ren J. Microbiota-Immune Interaction in the Pathogenesis of Gut-Derived Infection. Front Immunol. 2019;10:1873. doi:10.3389/fimmu.2019.01873
7. Allaire JM, Crowley SM, Law HT, et al. The Intestinal Epithelium: central Coordinator of Mucosal Immunity. Trends Immunol. 2018;39(9):677–696. doi:10.1016/j.it.2018.04.002
8. Xie B, Wang M, Zhang X, et al. Gut-derived memory γδ T17 cells exacerbate sepsis-induced acute lung injury in mice. Nat Commun. 2024;15(1). doi:10.1038/s41467-024-51209-9.
9. Yan Y, Li B, Gao Q, et al. Intestine‐Decipher Engineered Capsules Protect Against Sepsis‐induced Intestinal Injury via Broad‐spectrum Anti‐inflammation and Parthanatos Inhibition. Adv Sci. 2025;12(10). doi:10.1002/advs.202412799.
10. Hou Q, Dou Z, Zhu L, et al. Shielding the Gut: ghrelin and Ferrostatin-1’s Protective Role Against Sepsis-Induced Intestinal Ferroptosis. Biomedicines. 2024;13(1):77. doi:10.3390/biomedicines13010077
11. Xl L, Gy Z, G R, et al. Ferroptosis in sepsis: the mechanism, the role and the therapeutic potential. Front Immunol. 2022;13:956361. doi:10.3389/fimmu.2022.956361
12. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi:10.1038/nature14344
13. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401–2421. doi:10.1016/j.cell.2022.06.003
14. Zheng J, Conrad M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020;32(6):920–937. doi:10.1016/j.cmet.2020.10.011
15. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424–442. doi:10.1038/s41580-024-00703-5
16. Zhang Y, Li S, Li F, et al. High-fat diet impairs ferroptosis and promotes cancer invasiveness via downregulating tumor suppressor ACSL4 in lung adenocarcinoma. Biology Direct. 2021;16(1):10. doi:10.1186/s13062-021-00294-7
17. Liu Y, Bao D, She H, et al. Role of Hippo/ACSL4 axis in ferroptosis-induced pericyte loss and vascular dysfunction in sepsis. Redox Biol. 2024;78:103353. doi:10.1016/j.redox.2024.103353
18. Ma T, Chen P, Dong H, et al. Identification of key anti-neuroinflammatory components in Gastrodiae Rhizoma based on spectrum-effect relationships and its mechanism exploration. J Pharma Biomed Anal. 2024;248:116266. doi:10.1016/j.jpba.2024.116266
19. Shi Y, Chen W, Yang R, et al. Unravelling pharmacological mechanisms and effects of Tianma Siwu Decoction-derived compounds on ischemic stroke by multidimensional network pharmacological analysis. J Ethnopharmacol. 2025;337(Pt 3):118979. doi:10.1016/j.jep.2024.118979
20. Zhao X, Zhou S, Liu Y, et al. Parishin alleviates vascular ageing in mice by upregulation of Klotho. J Cell & Mol Med. 2023;27(10):1398–1409. doi:10.1111/jcmm.17740
21. Wang T, Chen H, Xia S, et al. Ameliorative Effect of Parishin C Against Cerebral Ischemia-Induced Brain Tissue Injury by Reducing Oxidative Stress and Inflammatory Responses in Rat Model. Neuropsychiatr Dis Treat. 2021;17:1811–1823. doi:10.2147/NDT.S309065
22. Zhao X, Zhou S, Yan R, et al. Parishin From Gastrodia Elata Ameliorates Aging Phenotype in Mice in a Gut Microbiota-Related Manner. Front Microbiol. 2022;13:877099. doi:10.3389/fmicb.2022.877099
23. Gong CX, Ma C, Irge DD, et al. Gastrodia elata and parishin ameliorate aging induced ‘leaky gut’ in mice: correlation with gut microbiota. Biomedical Journal. 2023;46(4):100547. doi:10.1016/j.bj.2022.07.001
24. Huang Da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
25. Liu W, Li L, Ye H, et al. Weighted gene co-expression network analysis in biomedicine research]. Sheng wu gong cheng xue bao =. Chin J Biotechnol. 2017;33(11):1791–1801. doi:10.13345/j.cjb.170006
26. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559. doi:10.1186/1471-2105-9-559
27. Gao XM, Zhou XH, Jia MW, et al. Identification of key genes in sepsis by WGCNA. Preventive Med. 2023;172:107540. doi:10.1016/j.ypmed.2023.107540
28. Zhang JX, Xu WH, Xing XH, et al. ARG1 as a promising biomarker for sepsis diagnosis and prognosis: evidence from WGCNA and PPI network. Hereditas. 2022;159(1):27. doi:10.1186/s41065-022-00240-1
29. Meyer NJ, Prescott HC. Sepsis and Septic Shock. New Engl J Med. 2024;391(22):2133–2146. doi:10.1056/NEJMra2403213
30. Cecconi M, Evans L, Levy M, et al. Sepsis and septic shock. Lancet. 2018;392(10141):75–87. doi:10.1016/S0140-6736(18)30696-2
31. Huang Q, Ru Y, Luo Y, et al. Identification of a targeted ACSL4 inhibitor to treat ferroptosis-related diseases. Sci Adv. 2024;10(13):eadk1200. doi:10.1126/sciadv.adk1200
32. Wu D, Spencer CB, Ortoga L, et al. Histone lactylation-regulated METTL3 promotes ferroptosis via m6A-modification on ACSL4 in sepsis-associated lung injury. Redox Biol. 2024;74:103194. doi:10.1016/j.redox.2024.103194
33. Lai K, Song C, Gao M, et al. Uridine Alleviates Sepsis-Induced Acute Lung Injury by Inhibiting Ferroptosis of Macrophage. Int J Mol Sci. 2023;24(6):5093. doi:10.3390/ijms24065093
34. Ding K, Liu C, Li L, et al. Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism. Chinese Med J. 2023;136(21):2521–2537. doi:10.1097/CM9.0000000000002533
35. Xiao Z, Zhang J, Qiu Z, et al. Ferroptosis and inflammation are modulated by the NFIL3-ACSL4 axis in sepsis associated-acute kidney injury. Cell Death Discovery. 2024;10(1):349. doi:10.1038/s41420-024-02113-0
36. Sun Y, Pu Z, Zhao H, et al. Vitamin D can mitigate sepsis-associated neurodegeneration by inhibiting exogenous histone-induced pyroptosis and ferroptosis: implications for brain protection and cognitive preservation. Brain Behav Immun. 2025;124:40–54. doi:10.1016/j.bbi.2024.11.019
37. Mishima E, Nakamura T, Doll S, et al. Recommendations for robust and reproducible research on ferroptosis. Nat Rev Mol Cell Biol. 2025;26(8):615–630. doi:10.1038/s41580-025-00843-2
38. Wang J, Zhu Q, Wang Y, et al. Irisin protects against sepsis-associated encephalopathy by suppressing ferroptosis via activation of the Nrf2/GPX4 signal axis. Free Radic Biol Med. 2022;187:171–184. doi:10.1016/j.freeradbiomed.2022.05.023
39. Fang X, Fu W, Zou B, et al. Tectorigenin relieved sepsis-induced myocardial ferroptosis by inhibiting the expression of Smad3. Toxicol Res. 2023;12(3):520–526. doi:10.1093/toxres/tfad038
40. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi:10.1038/nchembio.2239
41. Appiah MG, Park EJ, Darkwah S, et al. Intestinal Epithelium-Derived Luminally Released Extracellular Vesicles in Sepsis Exhibit the Ability to Suppress TNF-a and IL-17A Expression in Mucosal Inflammation. Int J Mol Sci. 2020;21(22):8445. doi:10.3390/ijms21228445
42. Izadparast F, Riahi-Zajani B, Yarmohammadi F, et al. Protective effect of berberine against LPS-induced injury in the intestine: a review. Cell Cycle. 2022;21(22):2365–2378. doi:10.1080/15384101.2022.2100682
43. Sonnier DI, Bailey SR, Schuster RM, et al. Proinflammatory chemokines in the intestinal lumen contribute to intestinal dysfunction during endotoxemia. Shock. 2012;37(1):63–69. doi:10.1097/SHK.0b013e31823cbff1
44. Chen GY, Chen X, King S, et al. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nature Biotechnol. 2011;29(5):428–435. doi:10.1038/nbt.1846
45. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. 2017;14(1):9–21. doi:10.1038/nrgastro.2016.169
46. Sae-Khow K, Charoensappakit A, Visitchanakun P, et al. Pathogen-Associated Molecules from Gut Translocation Enhance Severity of Cecal Ligation and Puncture Sepsis in Iron-Overload β-Thalassemia Mice. J Inflamm Res. 2020;13:719–735. doi:10.2147/JIR.S273329
47. Kim DW, Kim JK, Gebru YA, et al. Identification of Novel Parishin Compounds from the Twig of Maclura tricuspidata and Comparative Analysis of Parishin Derivatives in Different Parts. Molecules. 2022;28(1):7. doi:10.3390/molecules28010007
48. Wu J, Wu B, Tang C, et al. Analytical Techniques and Pharmacokinetics of Gastrodia elata Blume and Its Constituents. Molecules. 2017;22(7):1137. doi:10.3390/molecules22071137
49. Zhou S, Zhao X, Wu L, et al. Parishin treatment alleviates cardiac aging in naturally aged mice. Heliyon. 2023;9(12):e22970. doi:10.1016/j.heliyon.2023.e22970
50. Tang C, Wang L, Liu X, et al. Comparative pharmacokinetics of gastrodin in rats after intragastric administration of free gastrodin, parishin and Gastrodia elata extract. J Ethnopharmacol. 2015;176:49–54. doi:10.1016/j.jep.2015.10.007
51. Tang C, Wang L, Liu X, et al. Pharmacokinetic study of Gastrodia elata in rats. Anal BioanalChem. 2015;407(29):8903–8910. doi:10.1007/s00216-015-9054-y
52. Zhao Y, Gong X-J, Zhou X, et al. Relative bioavailability of gastrodin and parishin from extract and powder of Gastrodiae rhizoma in rat. J Pharma Biomed Anal. 2014;100:309–315. doi:10.1016/j.jpba.2014.08.017
53. Dai Y, Ban W, Yang Z. Gastrodin, a Promising Natural Small Molecule for the Treatment of Central Nervous System Disorders, and Its Recent Progress in Synthesis. Pharmacol Pharmacokinet Int J Mol Sci. 2024;25(17).
54. Tang C, Wang L, Cheng M, et al. Rapid and sensitive analysis of parishin and its metabolites in rat plasma using ultra high performance liquid chromatography-fluorescence detection. J Chromatogr B Anal Technol Biomed Life Sci. 2014;973:104–109. doi:10.1016/j.jchromb.2014.08.020
55. Bing W, Yan-Tao S, Zhi-Dong P, et al. Pharmacokinetic and tissue distributions study of adenosine, 4-hydroxybenzyl alcohol and Parishin C from Gastrodia elata extract in rats. Pak J Pharm Sci. 2018;31(5(Supplementary):2053–2060.
56. Li FJ, Hu H, Wu L, et al. Ablation of mitophagy receptor FUNDC1 accentuates septic cardiomyopathy through ACSL4-dependent regulation of ferroptosis and mitochondrial integrity. Free Radic Biol Med. 2024;225:75–86. doi:10.1016/j.freeradbiomed.2024.09.039
57. Zhao N, Tao W, Ouyang X, et al. Nicotinamide mononucleotide mitigates hyperoxia-aggravated septic lung injury via the GPx4-mediated anti-ferroptosis signaling pathway in alveolar epithelial cells. Free Radic Biol Med. 2025;234:86–99. doi:10.1016/j.freeradbiomed.2025.04.021
58. Duan J, Wang Z, Duan R, et al. Therapeutic targeting of hepatic ACSL4 ameliorates NASH in mice. Hepatology. 2022;75(1):140–153. doi:10.1002/hep.32148
59. Wang Y, Wu W, Wu X, et al. Parishin C Attenuates Oxidative Stress and Inflammation in HT22 Hippocampal Neurons and BV2 Microglia Through Nrf2 Signaling Pathway. Int J Mol Sci. 2025;26(15):7263. doi:10.3390/ijms26157263
60. Shu G, Yang T, Wang C, et al. Gastrodin stimulates anticancer immune response and represses transplanted H22 hepatic ascitic tumor cell growth: involvement of NF-κB signaling activation in CD4+ T cells. Toxicol Appl Pharmacol. 2013;269(3):270–279. doi:10.1016/j.taap.2013.02.019
61. Zhao X, Zhou S, Sheng Z, et al. Parishin Alleviates Pulmonary Fibrosis by Reducing CD38 Levels in Naturally Aging Mice. Rejuvenation Res. 2025;28(1):25–32. doi:10.1089/rej.2024.0042
62. Otani S, Coopersmith CM. Gut integrity in critical illness. J Intensive Care. 2019;7(1):17. doi:10.1186/s40560-019-0372-6
63. Yoseph BP, Klingensmith NJ, Liang Z, et al. Mechanisms of Intestinal Barrier Dysfunction in Sepsis. Shock Augusta Ga. 2016;46(1):52–59. doi:10.1097/SHK.0000000000000565
64. Landoni VI, Martire-Greco D, Rodriguez-Rodrigues N, et al. Immature myeloid Gr-1+ CD11b+ cells from lipopolysaccharide-immunosuppressed mice acquire inhibitory activity in the bone marrow and migrate to lymph nodes to exert their suppressive function. Clin Sci. 2016;130(4):259–271. doi:10.1042/CS20150653
65. Xue Y, Xu Y, Liu X, et al. Ferumoxytol Attenuates the Function of MDSCs to Ameliorate LPS-Induced Immunosuppression in Sepsis. Nanoscale Res Lett. 2019;14(1):379. doi:10.1186/s11671-019-3209-2
66. Kobritz M, Nofi C, Sfakianos M, et al. Targeting sting to reduce sepsis-induced acute intestinal injury. Surgery. 2023;174(4):1071–1077. doi:10.1016/j.surg.2023.06.032
67. Zhang X, Ning W, Gao G, et al. Bazedoxifene attenuates intestinal injury in sepsis by suppressing the NF-κB/NLRP3 signaling pathways. Eur J Pharmacol. 2023;947:175681. doi:10.1016/j.ejphar.2023.175681
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.