Back to Journals » International Journal of Nanomedicine » Volume 14
Oxidized carbon nanoparticles as an effective protein antigen delivery system targeting the cell-mediated immune response
Authors Sawutdeechaikul P, Jiangchareon B, Wanichwecharungruang S, Palaga T ![]()
Received 15 February 2019
Accepted for publication 15 May 2019
Published 4 July 2019 Volume 2019:14 Pages 4867—4880
DOI https://doi.org/10.2147/IJN.S204134
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Pritsana Sawutdeechaikul1,2, Banphot Jiangchareon,3–5 Supason Wanichwecharungruang,3–6 Tanapat Palaga1–3
1Graduate Program in Microbiology and Microbial Technology, Department of Microbiology, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand; 2Center of Excellence in Immune-mediated Diseases, Chulalongkorn University, Bangkok 10330, Thailand; 3Center of Excellence in Materials and Bio-Interfaces, Chulalongkorn University, Bangkok, 10330, Thailand; 4Nanotec-CU Center of Excellence on Food and Agriculture, Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand; 5Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand; 6Center of Excellence on Petrochemical and Materials Technology, Chulalongkorn University, Bangkok, 10330, Thailand
Background: The demand for an effective vaccine delivery system that drives a suitable immune response is increasing. The oxidized carbon nanosphere (OCN), a negatively charged carbon nanoparticle, has the potential to fulfill this requirement because it can efficiently deliver macromolecules into cells and allows endosomal leakage. However, fundamental insights into how OCNs are taken up by antigen-presenting cells, and the intracellular behavior of delivered molecules is lacking. Furthermore, how immune responses are stimulated by OCN-mediated delivery has not been investigated.
Purpose: In this study, the model protein antigen ovalbumin (OVA) was used to investigate the uptake mechanism and intracellular fate of OCN-mediated delivery of protein in macrophages. Moreover, the immune response triggered by OVA delivered by OCNs was characterized.
Methods: Bone-marrow-derived macrophages (BMDMs) from mice were used to study antigen uptake and intracellular trafficking. Mice were immunized using OCN–OVA combined with known adjuvants, and the specific immune response was measured.
Results: OCNs showed no cytotoxicity against BMDMs. OCN-mediated delivery of OVA into BMDMs was partially temperature independent process. Using specific inhibitors, it was revealed that intracellular delivery of OCN–OVA does not rely on phagocytosis or the clathrin- and lipid raft/caveolae-mediated pathways. Delivered OVA was found to colocalize with compartments containing MHC class I, but not with early endosomes, lysosomes, and autophagosomes. Immunization of OVA using OCNs in combination with the known adjuvant monophosphoryl lipid A specifically enhanced interferon gamma (IFNγ)- and granzyme B-producing cytotoxic T cells (CTLs).
Conclusion: OCNs effectively delivered protein antigens into macrophages that localized with compartments containing MHC class I partially by the temperature independent, but not clathrin- and lipid raft/caveolae-mediated pathways. Increased CD8+, T-cell activity was induced by OCN-delivered antigens, suggesting antigen processing toward antigen presentation for CTLs. Taken together, OCNs are a potential protein antigen delivery system that stimulates the cell-mediated immune response.
Keywords: oxidized carbon nanosphere, macrophages, adjuvant, cell-mediated immune response
Introduction
Nanomaterials have gained more attraction in recent years for medical applications because they show promising results in the diagnosis, prevention, and treatment of diseases. There are various properties of nanomaterials that make them suitable for medical applications, such as their size that comes with a large surface area, shape, and chemical composition, which can be modified.1
Vaccines are powerful tools for public health interventions to prevent and/or treat infectious and noninfectious diseases. Because safety issues are one of the main concerns for vaccine development, a subunit vaccine using recombinant proteins has gained more attention for novel vaccine development.2 The inherent nature of this type of vaccine results in low immunogenicity, and there is a need for effective delivery and adjuvants to enhance their efficacy.3–5 Currently, there are only a handful of adjuvants and vaccine delivery systems approved for use in humans, and the new generation of such system is expected to help facilitate the development of new vaccines. Nanoparticles for application in vaccine development are one of the fastest growing areas of research.3,6
The oxidized carbon nanosphere (OCN) is a new type of carbon nanomaterial that shows promising performance as a cellular delivery system. This nanoparticle is stable with a spherical shape of approximately 130 nm in diameter, is water dispersible, has a negatively charged surface and is an oxidized sp2-hybridized carbon nanoparticle.7,8 Previously, our group reported that OCNs can be used to deliver macromolecules into cells efficiently, such as curcumin into human embryonic kidney (HEK293T) cells, human monoclonal antibodies (150 kDa) into Vero cells, and the microparticles into HepG2 cells.7,9,10 Moreover, peptide nucleic acid (PNA), designed to specifically bind to the NF-ĸB-binding motif of the il6 promoter, was successfully delivered by OCNs into the RAW264.7 macrophage cell line, resulting in suppression of the expression of the targeted gene.8 Based on the results from the synthetic cell-sized liposome study, the ability to escape from endosomes into the cytosol by generating transient pores at the lipid bilayer was reported.8
Macrophages play an essential role in the innate immune response and function as antigen-presenting cells (APCs).6,11 Therefore, macrophages are one of the target cells for vaccine delivery that can initiate a desirable adaptive immune response.12 In general, when APCs uptake antigen, exogenous antigen is processed through the endocytic pathway, and the resulting peptides are presented to CD4+ T cells via MHC class II.13,14 Meanwhile, endogenous antigen or cytosolic antigen is processed by the proteasome and loaded onto MHC class I, leading to CD8+ T-cell activation. However, the exogenous antigen is also presented to CD8+T cells via MHC class I by the process called cross-presentation.15,16 Based on our previous findings regarding OCNs, we hypothesized that protein antigens delivered by OCNs into APCs, such as macrophages, would end up in the cytosol by leaking out of vesicles such as endosomes and likely be processed for MHC class I presentation, which is effective for cytotoxic T-cell activation.
In this study, we tested this hypothesis using ovalbumin (OVA) as a model protein antigen. The cellular uptake and intracellular fate of delivered OVA in macrophage cell lines and bone marrow-derived macrophages were investigated. Finally, the effect of OCN-delivered OVA on the cell-mediated immune response was also determined in an in vivo study.
Materials and methods
Animals
Eight-week-old BALB/c female mice were purchased from Nomura Siam International (Thailand). All experiments involving animals were approved by the Chulalongkorn University Institutional Animal Care and Use Committee (CU-IACUC) (No.1673005). All procedures were carried out according to the guidelines and regulations issued by CU-IACUC.
Materials
Dulbecco’s Modified Eagle Medium (DMEM), sodium pyruvate, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and penicillin/streptomycin were purchased from GE Healthcare Life Sciences (Pittsburgh, PA, USA). Fetal bovine serum (FBS) was purchased from Life Technologies (Carlsbad, CA, USA). Horse serum was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Poly(I:C) and MPL were all vaccine grade and purchased from InvivoGen (San Diego, CA, USA). OVA was purchased from Sigma-Aldrich (St. Louis, MO, USA). Endotoxin-free water was purchased from Merck Millipore (Darmstadt, Germany).
OCN–OVA complex preparation
OCNs in water were sterilized by autoclaving at 121°C for 15 mins and sonicated for 5 mins. The mixture of OCNs and OVA was prepared by mixing at various weight ratios (w/w) such as 1:1 and 3:1 and incubated overnight at 4°C. The mixture was added to cells cultured in complete DMEM (DMEM supplemented with 10% FBS (v/v) 1% sodium pyruvate (w/v) 1% HEPES (w/v) penicillin (100 U/ml) and streptomycin (0.25 mg/ml)) at a final concentration of 2 or 6 µg/mL for OCN and 2 µg/mL for OVA.
Cell line and culture conditions
J774A.1, a murine macrophage cell line (ATCC®TIB-67™) and MH-S, a murine alveolar macrophage cell line (ATCC® CRL-2019™), were purchased from ATCC (Manassas, VA, USA). Cells were cultured in complete DMEM and incubated at 37°C in a humidified atmosphere containing 5% CO2.
Bone marrow-derived macrophages (BMDMs)
Bone marrow cells were isolated from tibias and femurs of BALB/c female mice. Harvested cells were incubated in BMDM differentiation media (DMEM supplemented with 10% (v/v) FBS, 1% (w/v) sodium pyruvate, 1% (w/v) HEPES, 100 U/mL penicillin, 0.25 mg/mL streptomycin, 20% (v/v) L929 cell-conditioned medium, and 5% (v/v) horse serum) for a week at 37°C in a humidified atmosphere containing 5% CO2.17
Cell viability assay
Cell viability was tested by the MTT assay. BMDMs were seeded at 2×104 cells/well in 96 well plates overnight and treated with OCNs at the indicated concentrations (0.78, 1.56, 3.13, 6.25, 12.5, 25, 50, and 100 µg/mL) for 24 hrs. In addition, the cytotoxicity of the mixture of OCN and OVA with or without adjuvant (Poly(I:C) and MPL) was also tested. The mixture of OCN and OVA with two weight ratios, 1:1 and 3:1, were incubated at 4°C overnight before addition of MPL or Poly(I:C) to obtain the final concentrations of 2 or 6 µg/mL for OCN, 2 µg/mL for OVA, 2 µg/mL for MPL, and 10 µg/mL for Poly(I:C). The mixtures were incubated with BMDMs for 24 hrs. The blank control was medium and OCNs without cells. In the last 4 hrs of incubation, MTT solution (Alfa Aesar, UK) was added to all wells before addition of DMSO (Merck, Darmstadt, Germany) to dissolve the insoluble formazan. Absorbance was measured at 540 nm with a microplate reader (Anthos 2010, Biochrom, UK). The percentage of cell viability was calculated using the following formula.

Measurement of OVA loading on OCN
OCN and OVA mixture at indicated weight ratios were prepared by mixing 10, 20, and 30 µg of OCN with 10 µg of OVA in water in the total volume of 50 µL. The mixtures were incubated at 4°C for overnight before adjusting the volume to 1 mL by water. Clear supernatant was harvested after centrifugation of the mixtures at 13,000 rpm at 4°C for 20 mins. Free OVA in the supernatant was measured by indirect ELISA using a rabbit polyclonal antibody against OVA (Sigma Aldrich). The OVA- loading capacity and the OVA adsorption efficiency on OCN were determined by the following formulas.

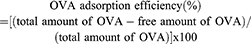
Uptake mechanism study by flow cytometry
The J774A.1 cell line was seeded at 5×105 cells/well in a 12-well plate (Thermo Scientific) and incubated at 37°C overnight. Fresh complete DMEM was added the next day. Cells were pre-treated for 30 mins with cytochalasin D (Sigma-Aldrich) and methyl-β-cyclodextrin (mβcd) (Sigma-Aldrich) at indicated concentrations. Pretreatment of 15 mins was performed in the case of chlorpromazine (Selleckchem, Houston, TX, USA) and nystatin (Sigma-Aldrich). After inhibitor pretreatment, the mixture of OCN+OVA at 1:1 (w/w) was added to cells. For cytochalasin D treatment, cells were incubated with OCN+OVA in the presence of cytochalasin D. For mβcd, chlorpromazine and nystatin treatment, the inhibitor was removed before the addition of OCN+OVA. After 1 hr of incubation, the cells were collected and divided into two sets for surface OVA staining and total OVA staining. The Fc receptor was blocked with anti-mouse CD16/CD32 antibody. Cells were subsequently stained for surface OVA by rabbit polyclonal anti-OVA antibody and followed by anti-rabbit IgG conjugated with Alexa Fluor® 488 (Cell Signaling Technology). For total OVA staining, the cells were fixed and permeabilized by absolute methanol (Merck Millipore, Darmstadt, Germany) and stained for intracellular OVA using anti-OVA antibody described above. Samples were subjected to analysis by flow cytometry (FC500, Beckman Coulter, Brea, CA, USA). Data were analyzed by FlowJo software (Tree Star, USA). The uptake was calculated by subtracting surface staining from the staining of the total OVA.
Immunofluorescen ce staining
BMDMs were cultured at 1×105 cells/well in 8-well chamber slides (Thermo Fisher Scientific) in BMDM differentiation media overnight. On the next day, the media was changed to complete DMEM, and the cells were further incubated overnight. After removing the culture supernatant, OCN+OVA mixtures in DMEM media were added to cells, and the cells were incubated at 37°C and 5% CO2 for the indicated times. Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) and permeabilized with 0.2% Triton-X 100 (Amersham Biosciences, Little Chalfont, UK). For MHC-I staining, cells were stained for surface MHC-I before being fixed and permeabilized and stained again for intracellular MHC-I. OVA and cellular compartments were stained with specific dye or antibodies, ie, Hoechst (Invitrogen, Waltham, MA, USA) for nuclei staining, rabbit anti-EEA1 antibody (Cell Signaling Technology, Danvers, MA, USA) for early endosomes, rat anti-LAMP1 antibody (BioLegend, San Diego, CA, USA, USA) for lysosomes, rabbit polyclonal anti-LC3B antibody (Cell Signaling Technology) for autophagosomes, biotin conjugated anti-mouse H2Dd (BioLegend) for MHC-I and rabbit polyclonal anti-OVA antibody (Sigma-Aldrich) for OVA. Moreover, fluorescein-conjugated OVA (Molecular Probes, Eugene, OR, USA) was also used. For detection, fluorescent dye-conjugated secondary antibodies were used, including anti-rabbit IgG antibody conjugated with Alexa Fluor® 488 (Cell Signaling Technology), anti-rabbit IgG antibody conjugated with Alexa Fluor® 555 (Cell Signaling Technology), anti-rat IgG antibody conjugated with Alexa Fluor® 555 (BioLegend) and Dylight 488 conjugated streptavidin (BioLegend). The images were acquired with a FLUOVIEW confocal microscope (Olympus, Tokyo, Japan).
OCN+OVA preparation for immunization
The OCN+OVA mixtures that contained 10 µg of OCN and 10 µg of OVA ( total volumn was 50 µL) were prepared as described above. After overnight incubation, adjuvants, Poly(I:C) (50 µg) or MPL (10 µg) or endotoxin-free water (InvivoGen) were added to OCN+OVA mixture to bring the final volume to 100 µL. Each mouse received 100 µL of the indicated mixture solution. Eight-week-old BALB/c female mice (n=3 per group) were used for immunization. Before immunization, preimmunized blood was collected from the facial vein. Each vaccine formula, ie, OCN+OVA+Poly(I:C), OVA+Poly(I:C), OCN+OVA+MPL, and OVA+MPL, was administered subcutaneously three times at 2-week intervals (Figure 7A). Weight gain was monitored during the experimental period, and no difference was observed among these groups. On day 35, mice were sacrificed, and blood and spleens were collected. OVA-specific antibody titers were measured by indirect ELISA, and the endpoint titer was calculated.
Splenocyte restimulation assay and intracellular cytokine staining
Spleens were homogenized and strained through a 100 µm cell strainer. After lysis of red blood cells, splenocytes were seeded at 4×106 cells/well in 24 well plates (Thermo Scientific). OVA (200 µg/mL) was added, and the cells were incubated for the indicated times. For intracellular granzyme B detection, cells were incubated for 72 hrs. Golgi plug (Brefeldin A) (BD Biosciences, San Jose, CA, USA) was added to the culture 4 hr before harvesting. For intracellular IFN-γ detection, cells were incubated for 48 hrs in the presence of PMA (10 ng/mL) and ionomycin (1 µg/mL). The Golgi plug was added 4 hrs before harvesting. For intracellular staining, the cells were collected and processed with a BD Cytofix/Cytoperm Plus Fixation/Permeabilization Kit (BD Biosciences) according to the manufacturer’s instructions. Cells were stained with anti-mouse CD4 antibody conjugated with PerCP (BioLegend) and anti-mouse CD8 antibody conjugated with PE, respectively. Subsequently, cells were fixed, permeabilized and stained for granzyme B or IFN-γ with FITC-labeled anti-human/mouse granzyme B antibody or anti-mouse IFN-γ antibody conjugated with Alexa Fluor® 488 (BioLegend). Isotype-matched antibodies were used as a control. Data were collected using a Flow cytometer FC500 and analyzed by FlowJo software.
Statistical analysis
All data were analyzed by GraphPad Prism (version 5.03) software (GraphPad, USA). For statistical analysis, data were analyzed by one-way analysis of variance by Tukey’s multiple comparison post hoc test or two-tailed unpaired Student’s t-test. Probability (p) values lower than 0.05 were considered significant difference and were labeled with one asterisk, while two and three asterisks represented p-values of 0.01 and 0.001, respectively.
Results
Toxicity of OCNs in macrophages
OCNs were prepared as described by Arayachukeat et al7. The obtained OCNs were characterized by SEM and TEM and revealed an average size of 100–200 nm (data not shown). To investigate the possibility of cytotoxicity of OCNs in macrophages, different concentrations of OCNs were incubated with BMDMs for 24 hrs, and the cell viability was measured by MTT assay. The results showed that even at a relatively high dose of OCNs at 100 µg/mL, there is no detectable toxicity (Figure 1A). Similar results were obtained from the macrophage cell lines J774A.1 and RAW264.7 (data not shown). In addition, the toxicity of OCN was examined when OCNs were combined with OVA, a model antigen, and MPL or Poly(I:C), two potential adjuvants to be used in later experiments. As shown in Figure 1B, no acute toxicity against macrophages was detected in all conditions. Therefore, the concentrations that did not demonstrate acute cytotoxicity were used in the next experiments.
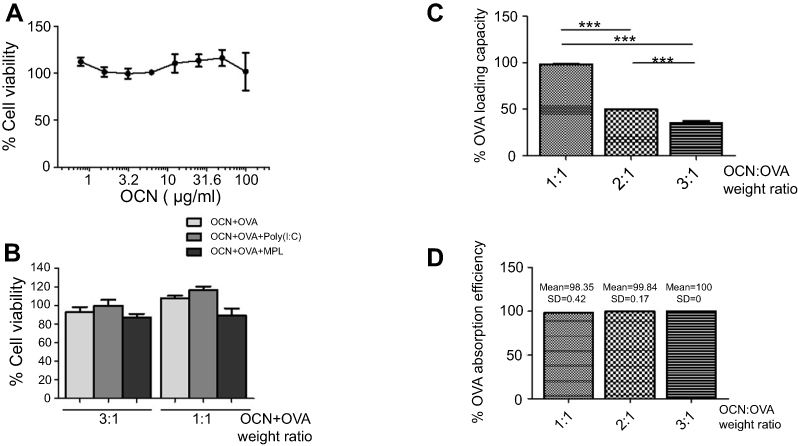 |
Figure 1 Cytotoxicity of OCNs on macrophages and the loading capacity of OCNs. (A) BMDMs were incubated with various concentrations of OCNs (0.8, 1.5, 3, 6.3, 12.5, 25, 50, and 100 µg/mL) for 24 hrs at 37°C. Cell viability was determined by the MTT assay. The results represent the mean ± SEM of the results from three independent experiments. (B) BMDMs were incubated with the mixtures of OCN+OVA at 3:1 and 1:1 (w/w) with or without indicated adjuvant for 24 hrs at 37°C. Cell viability was determined by MTT. The result represented the mean ± SEM from four independent experiments. (C, D) Three weight ratios of OCN+OVA (1:1, 2:1, and 3:1) were determined for the OVA-loading capacity (C) and OVA adsorption efficiency (D). Data represented the means ± SD from three independent experiments. The significance of differences between the groups was determined by one-way analysis of variance (ANOVA). ***p<0.001. Abbreviations: OCN, oxidized carbon nanosphere; Poly(I:C), polyinosinic polycytidylic acid; MPL, monophosphoryl lipid A; HEPES, (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; OVA, ovalbumin; MTT, thiazolyl blue tetrazolium bromide; PMA, phorbol myristate acetate; mβcd, methyl-ß-cyclodextrin; DMSO, dimethyl sulfoxide; w/w, weight-by-weight; EEA1, early endosomal antigen 1; LAMP1, lysosomal-associated membrane protein 1; LC3, microtubule-associated protein 1 light chain 3. |
Cellular uptake of OCN+OVA by macrophages
To apply OCNs for protein delivery, we first estimated the protein-loading capacity on OCNs and the protein adsorption efficacy on OCNs using OVA as a model antigen. The weight ratios of OCNs and OVA were examined at 1:1, 2:1, and 3:1. As shown in Figure 1C and D, as the weight of OCNs increased, the loading capacity decreased, suggesting that at 2:1 or 3:1 ratio, there are still spaces for OVA loading on OCNs. On the other hand, the OVA adsorption efficacy to OCNs showed comparable level in all ratios tested with the efficacy ranging between 98% and 100%.
Next, the weight ratio of OCNs to OVA was optimized for intracellular uptake by the two macrophage cell lines, J774A.1 and MH-S. As shown in Figure S1, the ratio of 1:1, 2:1, and 3:1 showed comparable % cellular uptake by macrophages. Therefore, the weight ratio of OCN:OVA at 1:1 and 3:1 was chosen further characterization.
To confirm the ability of OCNs to deliver protein into cells, we examined the kinetics of OVA delivery into BMDMs. As shown in Figure 2A and B, OVA alone was not detected inside the cells over 10 hrs of observation, while OVA delivered by OCNs was readily detected in the cytosol of macrophages as early as 30 mins, and the levels of OVA inside the cells increased as the incubation times became longer, up to 10 hrs. OVA delivered by OCNs was not detected inside the nuclei in all tested time points.
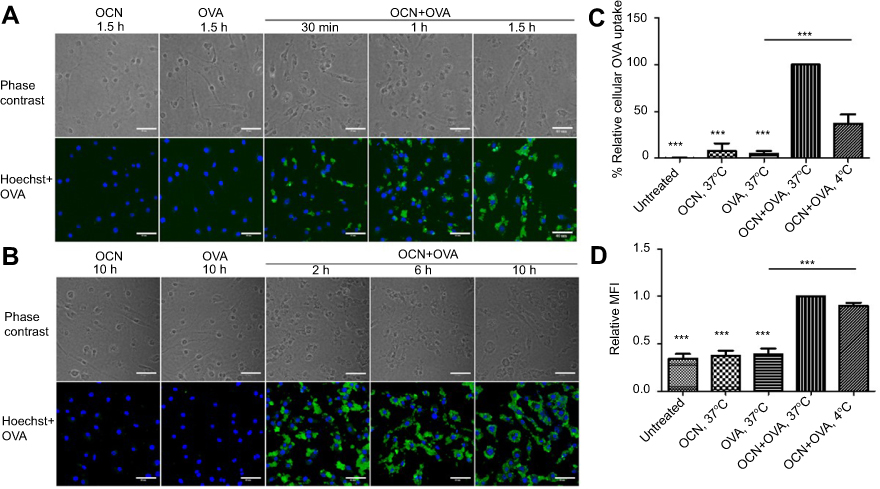 |
Figure 2 The kinetics of OVA uptake by BMDMs and the effect of temperature on the internalization of OVA delivered by OCNs. (A–B) BMDMs were incubated with OCN+OVA or OVA alone for the indicated durations. Cells were stained for detecting OVA or nuclei staining by Hoechst. (C–D) J774A.1, a macrophage cell line, was treated with OCNs, OVA, and OCN+OVA (1:1 w/w ratio) for 1 hr at 37°C or 4°C. The percentage of relative cellular OVA uptake (C) and the relative median fluorescent intensities (MFI) (D) were calculated relative to that of OCN+OVA at 37°C. Data were analyzed using one-way ANOVA followed by Tukey’s multiple comparison post hoc test, which represents the mean ± SEM from four independent experiments. ***p<0.001. |
To investigate how OCNs deliver OVA into phagocytes, uptake was performed at 4°C and 37°C in the J774A.1 macrophage cell line, and the relative cellular uptake (% relative percentages of cells that internalized OVA) and relative median fluorescent intensity (MFI) were compared. Representative gating strategy by flow cytometer to monitor the cellular uptake of OVA was shown in Figure S2. The results clearly demonstrated that delivery of OVA by OCNs, as determined by the relative cellular uptake, was significantly more efficient at 37°C than at 4°C (Figure 2C). In contrast, there was no significant difference in the MFI between the conditions at 4°C and 37°C (Figure 2D). This result suggested that the temperature independent transport of OVA by OCNs is partially responsible for the intracellular delivery of OVA by OCNs, but other mechanisms that are dependent on a temperature sensitive transport that relies on the active movement of cellular proteins may also be involved.
To identify the uptake mechanism, cytochalasin D, an inhibitor of actin polymerization that blocks phagocytosis, was used to pretreat cells before OVA delivery. As shown in Figure 3A and B, increasing doses of cytochalasin D had no impact on OVA uptake by macrophages in the presence of OCNs, indicating that phagocytosis is not the main mechanism of OCN delivery of proteins into cells. Next, the effect of mβcd, an agent that depletes membrane cholesterol and blocks lipid raft/caveolae-dependent endocytosis, was tested. The results showed that the percentage of OVA uptake in the presence of OCNs and the relative MFI of OVA did not change significantly by mβcd pretreatment, and unexpectedly, a slight increase in MFI was observed at the highest dose of mβcd (Figure 3C and D). Additionally, nystatin, an inhibitor of lipid raft/caveolae-mediated endocytosis, and chlorpromazine, an inhibitor of clathrin-mediated endocytosis was used to test for their effects on OCN-mediated OVA delivery. As shown in Figure S3, both inhibitors showed no impact on OCN-mediated OVA delivery into cells. Taken together, phagocytosis and lipid raft/caveolae- and clathrin-dependent endocytosis are not the main mechanism of OCN-mediated protein delivery into cells, while the temperature independent transport pathway plays partial role.
 |
Figure 3 The effect of inhibitors on the internalization of OVA delivered by OCNs. J774A.1 cell line was pretreated with various concentrations of cytochalasin D or mβcd for 30 mins before the addition of OCN+OVA (1:1). Cells were further incubated for another 1 hr. The percentage of relative cellular OVA uptake and the relative MFI were calculated relative to OCN+OVA without inhibitor. (A, B) The results obtained from cells treated with cytochalasin D. (C, D) The results obtained from cells treated with mβcd. The results represent the mean ± SEM from three experiments. The data were analyzed by one-way ANOVA followed by Tukey’s multiple comparison post hoc test. *p<0.05, **p<0.01, and ***p<0.001. |
Intracellular trafficking of OVA upon OCN-mediated delivery
To understand the fate of delivered proteins inside cells, immunofluorescence staining was used to track the trafficking of OVA delivered by OCNs in BMDMs. The potential cytosolic compartments that were examined were endosomes, lysosomes, and autophagy-associated autophagosomes using EEA1, LAMP1, and LC3 as markers. For endosomes, significant signals of OVA did not colocalize with EEA1, suggesting that OCNs did not deliver OVA to early endosome vesicles at least during the incubation periods of 30 mins and 1 hr (Figure 4A–P). Next, LAMP1, an abundant lysosomal membrane protein, was used to identify the lysosomal compartment, and as shown in Figure 5A–P, no colocalization between OVA and LAMP1 was observed at 3 and 8 hrs after incubation, suggesting that intracellular OVA did not reside in the lysosome. Cytosolic protein can be sequestered by the autophagic mechanism, and the contents are surrounded by autophagosomal membranes harboring LC3. To examine whether OCN-mediated delivery of OVA results in autophagosome formation surrounding OVA, an LC3 staining pattern was observed. As shown in Figure 6A–X, most LC3 and OVA did not colocalize, suggesting that OVA did not traffic to autophagosomes. Taken together, these results indicated that at least during the duration tested, OCNs did not deliver OVA into macrophages via endosomes, lysosomes, or autophagosomes.
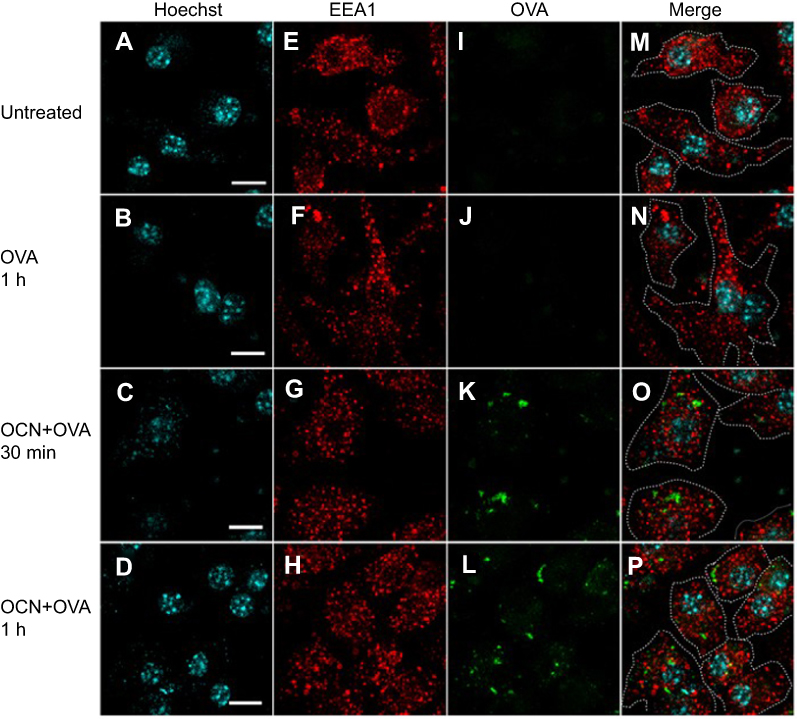 |
Figure 4 OVA delivered by OCNs did not colocalize with the endosomal compartment. OCN+OVA-fluorescein conjugate (ratio 3:1, green) were incubated with BMDMs. After 30 and 60 mins of incubation at 37°C, cells were subjected to immunofluorescence staining using rabbit monoclonal anti-EEA1 antibody and anti-rabbit IgG (H+L), F(ab’)2 fragment conjugated with Alexa Fluor® 555 for endosomes (red). Nuclei were stained with Hoechst (blue). Images were acquired by confocal microscopy. (A, E, I, M) Untreated cells, (B, F, J, N) cells incubated with OVA-fluorescein conjugate for 1 hr, (C, G, K, O) cells incubated with OCN+OVA-fluorescein conjugate for 30 mins, and (D, H, L, P) cells incubated with OCN+OVA-fluorescein conjugate for 1 hr. Scale bar =10 µm. |
 |
Figure 5 OVA delivered by OCNs did not colocalize with lysosomes. OCN+OVA-fluorescein conjugate (ratio 3:1, green) were incubated with BMDMs. After the indicated incubation times at 37°C, cells were subjected to immunofluorescence staining using rat anti-LAMP1 antibody and anti-rat IgG conjugated Alexa Fluor® 555 for lysosomes (red). Nuclei were stained with Hoechst (blue). Images were acquired by confocal microscopy. (A–D) Hoechst, (E–H) LAMP1 (I-L) OVA-fluorescein conjugate, (M–P) merged images. Scale bar =10 µm. |
 |
Figure 6 OVA delivered by OCNs did not colocalize with autophagosomes. OCN+OVA-fluorescein conjugate (ratio 3:1, green) were incubated with BMDMs. After the indicated incubation times at 37°C, cells were subjected to immunofluorescence staining using anti-LC3 antibody and anti-rabbit IgG (H+L), F(ab’)2 fragment conjugated with Alexa Fluor® 555 antibody for autophagosomes (red). Nuclei were stained with Hoechst (blue). Images were acquired by confocal microscopy. (A–F) Hoechst, (G–L) LC3, (M–R) OVA-fluorescein conjugate, (S–X) merged images. Scale bar=10 µm. |
OCNs enhance the OVA-specific immune response of cytotoxic T cells (CTLs) in combination with adjuvant MPL
To test the potential use of OCNs as a subunit protein antigen delivery system, the effect of OCNs on innate cytokine production by macrophages was first examined as the cytokine microenvironment can dictate the adaptive immune response. As shown in Figure S4, only basal level of IL-12p70 and TNFα was detected in OCN-treated BMDMs, compared to those activated by LPS.
In order to enhance an immune response raised by immunization, OCN+OVA were combined with two well-known adjuvants, Poly(I:C) or MPL, and used to immunize mice by the immunization schedule as depicted in Figure 7A. After three subcutaneous immunizations, blood samples and spleens were collected. The OVA-specific antibody response (IgG1 and IgG2a) between adjuvanted OVA and OCNs with adjuvanted OVA was not significantly different, except OCNs reduced the specific IgG1 antibody titer in the presence of Poly(I:C) (Figure S5A–C).
 |
Figure 7 OCNs in combination with MPL increase the frequency of OVA-specific granzyme B+ CD8+ T cells. (A) Immunization regimen. BALB/c mice were immunized with OVA+Poly(I:C), OVA+OCN+Poly(I:C), OVA+MPL or OVA+OCN+MPL via a subcutaneous route three times with 2-week interval. One week after the last immunization, splenocytes were harvested and stimulated with OVA (200 µg/mL) in vitro for 72 hrs. Cells were collected for CD8 staining and intracellular staining for granzyme B staining. (B) The gating strategy to identify the subset of CD8+ granzyme B+ T cells is shown. (C) Representative flow cytometry results are shown. The frequency of CD8+ granzyme B+ T cells in all CD8+ T cells is summarized in (D). Data represent the mean ± SEM. The significance of differences between two groups was determined by two-tailed unpaired Student’s t-test. **p<0.01. |
We, therefore, examined the frequency of OVA-specific CD8+ T cells in an in vitro re-stimulation assay in spleens harvested from the immunized mice. The gating strategy to identify CD8+ that expressed granzyme B, a key protease that CTL use to induce apoptosis of the target cells, is depicted in Figure 7B. As shown in Figure 7C and D, mice receiving OVA with Poly(I:C) with or without OCNs showed similar increasing percentages of CD8+ granzyme B+ T cells. In contrast, OVA with MPL induced lower percentages of CD8+ granzyme B+ T cells, while OCNs significantly increased the frequency of this cell subset to be as high as that in the Poly(I:C) group.
Because IFNγ is an important cytokine involved in the cell-mediated immune response and is produced mainly by helper T-cell type 1 and CD8+ T cells, the impact of using OCNs for immunization with protein was examined. A similar gating strategy, as depicted in Figure 8A, was used to identify CD8+ IFNγ+ T cells. As shown in Figure 8A and B, mice receiving OVA and Poly (I:C) with or without OCNs showed a significantly increased frequency of CD8+ IFNγ+ T cells upon restimulation with OVA. In contrast, for MPL, adding OCNs to the formulation significantly enhanced the effect of MPL on the frequency of CD8+ IFNγ+ T cells. Using ELISA to monitor overall cytokine production from an in vitro re-stimulation showed no significant difference in the presence or absence of OCNs (Figure S6).
 |
Figure 8 OCNs increased the frequency of IFNγ-producing CD8+ T cells when combined with adjuvant MPL. Splenocytes from mice treated as above were stimulated with OVA as shown in Figure 7. Cells were collected for intracellular IFN-γ staining and analyzed by flow cytometry. (A) The gating strategy to identify the subset of CD8+ FNγ+ T cells is shown. Representative flow cytometry results are shown. The frequency of CD8+ IFNγ+ T cells in all CD8+ T cells is summarized in (B). Data represent the mean ± SEM. The significance of differences between two groups was determined by two-tailed unpaired Student’s t-test. **p<0.01. |
Because we hypothesized that OCN-mediated delivery of OVA would facilitate antigen processing and presentation by MHC class I by endosomal leakage of OVA, we examined whether OVA colocalizes with MHC class I containing intracellular compartments, MHC class I and OVA were detected upon OCN-mediated delivery in BMDMs. As shown in Figure 9A–P, clear co-localization of both proteins was detected inside the cells. This result strongly supports our hypothesis on the potential enhancement of OVA being loaded onto MHC class I by OCNs.
 |
Figure 9 OVA delivered by OCNs colocalized with MHC-I containing compartments. OCN+OVA (ratio 3:1 ratio) were incubated with BMDMs. After the indicated incubation times at 37°C, cells were subjected to immunofluorescence staining using biotin conjugated anti-mouse H2Dd antibody and DyLight 488 conjugated streptavidin for MHC-I staining (green). OVA were detected using rabbit polyclonal anti-OVA antibody and anti-rabbit IgG antibody conjugated with Alexa Fluor® 555 (red). Nuclei were stained with Hoechst (blue). Images were acquired by confocal microscopy. (A–D) Hoechst, (E–H) MHC-I, (I–L) OVA and (M–P) the merged images. Arrow heads indicate colocalization between OVA and MHC-I (yellow). Scale bar =20 µm. |
Taken together, these results suggest that OCNs deliver OVA to MHC class I containing intracellular compartments and are highly potent in enhancing the impact of adjuvants such as MPL to increase the activation of CTL but do not have any additive effect with such adjuvants that already exhibit strong activity in inducing the CTL response.
Discussion
In this study, we confirmed the ability of OCNs to deliver proteins into APCs as macrophages and uncovered their potential use as protein subunit delivery for immunization. Previous reports by our group demonstrate that OCNs effectively deliver curcumin, PNAs, and antibodies into various human and murine cells.7–10 We have also reported the use of OCNs as a delivery system for M. tuberculosis recombinant protein antigens with enhancing effect on cell-mediated immune response.18 The ability of OCNs to cross lipid bilayer membrane themselves and also to bring with them cargoes across lipid bilayer membrane was previously confirmed through both cell-sized liposomes (possess lipid bilayer membrane similar to that of real cells) and the real cells.7–9 Previous characterizations of OCNs indicated that the particles could effectively adsorb some proteins and lipid molecules through their high surface energy.7–9 Here in this work, we also confirmed the OVA adsorption onto OCN by quantifying the supernatant of the OVA–OCN mixture after centrifugation. Only 1.65% of OVA at weight ratio of 1:1 was found left in the supernatant of the spin down OVA-loaded OCN suspension, comparing to 100% found in the supernatant of the control OVA alone.
Following the methods of Saraste et al and dos Santos et al, here we investigated the role of temperature independent and temperature sensitive mode of transport in OCN-mediated delivery of proteins.19,20 The results from incubating the cells at 4°C indicated that temperature independent transport was one of the mechanisms involved in the macrophage uptake of OVA in the presence of OCNs. The temperature sensitive transport is involved cellular movement which includes phagocytosis and endocytosis while temperature independent transport does not rely on cellular movement such as diffusion and pore formation on the plasma membrane.
Cytochalasin D was reported to inhibit phagocytosis and the macropinocytosis pathway, while mβcd was reported to block the clathrin- and caveolin-mediated pathway.21 Additional two endocytosis inhibitors, lipid raft/caveolae-mediated pathway inhibitor nystatin and clathrin-mediated pathway inhibitor chlorpromazine, were used. The results confirmed that OCN-mediated protein delivery was not involved lipid raft/caveolae-mediated pathway.22,23 These results imply that phagocytosis, macropinocytosis, clathrin- and lipid raft/caveolae-mediated pathways were not actively involved. Therefore, we proposed that the combination of temperature independent and clathrin/caveolae-independent endocytosis pathways are the major pathways in OCN-mediated protein delivery.24 We cannot exclude the possibility that other endocytic pathways may be involved.25
Other factors influence the recognition and uptake of nanoparticles by phagocytes, including the size, charge, and the interaction between nanoparticles and proteins. How do macrophages recognize the OVA complex with OCNs? Nanoparticles conjugated to proteins can be recognized by macrophages using surface receptors such as scavenger receptors, pattern recognition receptors, and Fc receptors.25 Because OCNs and OVA are not covalently conjugated, OVA can be absorbed onto the OCN surface and sensed by phagocytes.
The results from an intracellular trafficking study inside macrophages revealed that OVA only partially colocalized with the early endosome, lysosome, and autophagosome, while a large portion of OVA was found inside the cytosol. Based on previous findings using OCNs, we speculated that any lipid membrane surrounding OVA is penetrated by OCNs, allowing the protein to escape from membrane vesicles. This possibility led us to propose that OCN-mediated delivery of protein antigens would favor protein processing by the proteasome and that the resulting peptides are presented on MHC class I, as is the case for cytosolic proteins. Indeed, no OVA was found colocalized with endosomes, lysosomes or autophagosomes. We did not test the impact of OCN on acidification of lysosomal compartments, and it was likely that OCN leaked out from the compartment more efficiently when pH insides lysosome was decreased, and thus helped escaping of OVA from phagosome.8
The results from MHC class I and OVA co-localization experiments in BMDMs indicated that at least OVA delivered by OCN ends up in the same intracellular compartments as MHC class I. These compartments include ER and Golgi which are known as the intracellular trafficking of MHC class I.26 How OVA delivered by OCNs is located in the ER and/or Golgi needs further investigation but from our proposed model of OCN interaction with lipid on cellular membrane of intracellular vesicles, it is possible that OCNs may interrupt ER/Golgi membrane that allows entry of OVA into these MHC class I containing compartments.8
To use OCNs as a protein antigen delivery for immunization, they need to deliver protein to certain cytosolic compartments and should induce cytokines to direct T-cell responses.27 Our preliminary study found that OCNs alone induced only low to nondetectable levels of TNFα and IL-12 (Figure S4). This is not surprising given the nature of carbon materials that has as immunological inert property. OCNs are rather expected to deliver protein into cells and allow it to be processed and presented preferentially by cytosolic pathway. This observation led us to combine OCNs with two known adjuvants, Poly(I:C) and MPL, and investigate their impact on immune responses in in vivo. MPL is a potent TLR4 agonist, while Poly(I:C) is a TLR3 agonist. It is well known that Poly(I:C) as an adjuvant induces strong Th1 and cell-mediated immune responses, as observed with the antitumor response, compared with other TLR agonists, including MPL.28,29 When the immune response between the group of mice that received adjuvanted OVA with or without OCNs was compared, the combination of OCNs with MPL increased the activation of CTLs to a similar level as that observed with the Poly(I:C) group. OCNs did not further increase the impact of Poly(I:C).
The likely explanation for why OCNs did not enhance the effect of poly(I:C) may be because Poly(I:C) alone is sufficient to trigger a CTL response at the maximum level. Another possibility is the differential interaction between OCNs and these adjuvants. OCNs show preference to interact with lipid while its negative charge precludes it from making a direct contact with DNA or RNA.8 As MPL has lipid components, it is possible that OCNs can also adsorb MPL together with OVA in this setting while OCNs cannot do so with Poly(I:C). Furthermore, the receptors for MPL and Poly(I:C) are differentially located in the cells. TLR4 is expressed on the cell surface and intracellular vesicles where TLR3 can be found mainly in the endocytic compartments. This difference may result in little impact of OCNs on Poly(I:C)-triggered immune response. As our results suggested, OVA delivered by OCNs did not colocalize with any endocytic compartments.30 Lastly, Poly(I:C) and MPL activate different downstream signaling cascades that drives distinct outcomes. MPL/TLR4 interaction leads to activation of Myd88/TRAF6-dependent signaling pathways and the MAPK/NF-kB pathways, resulting in pro-inflammatory cytokine production. On the other hand, Poly(I:C)/TLR3 interaction activates TRIF-dependent IRF3 pathway and type I IFN.31 This difference in the signaling pathway between these two TLR agonists may be responsible for the different outcomes observed in our study.
When OCNs were used in combination with Poly(I:C), the serum titer of OVA-specific IgG1 was lower than that without OCNs. Consistent with this decreasing in IgG1 antibody titer (a Th2-driven isotype switching), decreasing Th2 cytokine, IL-5, was observed in an in vitro T-cell re-stimulation assay (Figure S6A). Thus, it is possible that the use of OCNs decreases Th2 cytokine response to antigen, leading to decreased Th2-driven antibody production.
In this study, we performed ex vivo analysis of CTL response in a re-stimulation assay. The results clearly demonstrated that OCN-mediated delivery of OVA enhanced memory CTL. Further study in vivo recall responses may be needed to confirm this ex vivo observation.
We proposed that OCN-mediated delivery of OVA feeds the protein antigen to the cytosolic antigen processing pathway and loads peptides onto MHC class I. As a result, more CD8+ T cells are activated. In fact, we observed colocalization of OVA with intracellular MHC class I in macrophages. In this study, we used macrophages as a model for antigen-presenting cells and presumed that dendritic cells interact and respond similarly to OCNs. Therefore, OCNs when combined with MPL are effective in triggering cell-mediated responses. Moreover, soluble antigens, in general, provide poor cross-presentation, while particulate antigens effectively induce cross-presentation.5,32 Nanoparticles and antigens can generate particulates that increase the frequency of cross-presentation.32 These findings indicate that OCNs are a good candidate for subunit-protein vaccines targeting cell-mediated immune responses, such as vaccines against intracellular pathogens and cancer.
Conclusion
This study shows that OCNs are effective in protein delivery into the cytosol of innate immune cells using both temperature independent and sensitive transport pathways. Endocytosis and clathrin- and caveolae-mediated uptake are not the mechanism of protein delivery by OCNs. Intracellular trafficking revealed that OCN-mediated delivery of OCNs did not colocalize with endosomes, lysosomes, and autophagosomes but found in MHC class I containing compartments. When combined with adjuvant MPL, OCNs enhanced the cell-mediated immune response by increasing antigen-specific CD8+ T cells. Taken together, the OCN has potential as a protein subunit delivery system that can be applied in vaccines to induce an effective cell-mediated immune response.
Acknowledgments
This work was supported by the Thai Government Annual Budget (GRB-APS-18-59-23-01) and the Newton Fund-Institutional Links (UK), and PS is supported by the 100th Anniversary Chulalongkorn University Fund for Doctoral Scholarship.
Disclosure
The authors report that there are no conflicts of interest in this work.
References
1. Kim BYS, Rutka JT, Chan WCW. Nanomedicine. New Engl J Med. 2010;363(25):2434–2443. doi:10.1056/NEJMoa1011205
2. Clark TG, Cassidy-Hanley D. Recombinant subunit vaccines: potentials and constraints. Dev Biol (Basel). 2005;121:153–163.
3. Zhao L, Seth A, Wibowo N, et al. Nanoparticle vaccines. Vaccine. 2014;32(3):327–337. doi:10.1016/j.vaccine.2013.11.069
4. Black M, Trent A, Tirrell M, Olive C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Rev Vaccines. 2010;9(2):157–173. doi:10.1586/erv.09.160
5. Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787. doi:10.1038/nri2868
6. Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic nanoparticles for vaccines and immunotherapy. Chem Rev. 2015;115(19):11109–11146. doi:10.1021/cr5004419
7. Arayachukeat S, Palaga T, Wanichwecharungruang SP. Clusters of carbon nanospheres derived from graphene oxide. ACS Appl Mater Interfaces. 2012;4(12):6808–6815. doi:10.1021/am3012406
8. Arayachukiat S, Seemork J, Pan-In P, et al. Bringing macromolecules into cells and evading endosomes by oxidized carbon nanoparticles. Nano Lett. 2015;15(5):3370–3376. doi:10.1021/nl504066f
9. Amornwachirabodee K, Tantimekin N, Pan-In P, et al. Oxidized carbon black: preparation, characterization and application in antibody delivery across cell membrane. Sci Rep. 2018;8(1):2489. doi:10.1038/s41598-018-20650-4
10. Amornwachirabodee K, Khramchantuk S, Pienpinijtham P, Israsena N, Palaga T, Wanichwecharungruang S. Enhancing passive transport of micro/nano particles into cells by oxidized carbon black. ACS Omega. 2018;3(6):6833–6840. doi:10.1021/acsomega.8b00487
11. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958. doi:10.1038/nri2448
12. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762. doi:10.1038/nri3070
13. Itano AA, Jenkins MK. Antigen presentation to naive CD4 T cells in the lymph node. Nat Immunol. 2003;4:733. doi:10.1038/ni957
14. König R, Huang L-Y, Germain RN. MHC class II interaction with CD4 mediated by a region analogous to the MHC class I binding site for CD8. Nature. 1992;356:796. doi:10.1038/356796a0
15. Kasturi SP, Pulendran B. Cross-presentation: avoiding trafficking chaos? Nat Immunol. 2008;9:461.
16. Fehres CM, Unger WWJ, Garcia-Vallejo JJ, van Kooyk Y. Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front Immunol. 2014;5(149). doi:10.3389/fimmu.2014.00149
17. Boonyatecha N, Sangphech N, Wongchana W, Kueanjinda P, Palaga T. Involvement of notch signaling pathway in regulating IL-12 expression via c-Rel in activated macrophages. Mol Immunol. 2012;51(3–4):255–262. doi:10.1016/j.molimm.2012.03.017
18. Sawutdeechaikul P, Cia F, Bancroft G, Wanichwecharungruang S, Sittplangkoon C, Palaga T. Oxidized carbon nanosphere-based subunit vaccine delivery system elicited robust Th1 and cytotoxic T cell responses. J Microbiol Biotechnol. 2019;29(3):489–499. doi:10.4014/jmb.1810.10053
19. Saraste J, Palade GE, Farquhar MG. Temperature-sensitive steps in the transport of secretory proteins through the Golgi complex in exocrine pancreatic cells. Proc Natl Acad Sci U S A. 1986;83(17):6425–6429. doi:10.1073/pnas.83.17.6425
20. dos Santos T, Varela J, Lynch I, Salvati A, Dawson KA. Effects of transport inhibitors on the cellular uptake of carboxylated polystyrene nanoparticles in different cell lines. PLoS One. 2011;6(9):e24438. doi:10.1371/journal.pone.0024438
21. Kuhn DA, Vanhecke D, Michen B, et al. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J Nanotechnol. 2014;5:1625–1636. doi:10.3762/bjnano.5.174
22. Dutta D, Donaldson JG. Search for inhibitors of endocytosis: intended specificity and unintended consequences. Cell Logist. 2012;2(4):203–208. doi:10.4161/cl.23967
23. Kanlaya R, Sintiprungrat K, Chaiyarit S, Thongboonkerd V. Macropinocytosis is the major mechanism for endocytosis of calcium oxalate crystals into renal tubular cells. Cell Biochem Biophys. 2013;67(3):1171–1179. doi:10.1007/s12013-013-9630-8
24. Sandvig K, Kavaliauskiene S, Skotland T. Clathrin-independent endocytosis: an increasing degree of complexity. Histochem Cell Biol. 2018;150(2):107–118. doi:10.1007/s00418-018-1678-5
25. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle uptake: the phagocyte problem. Nano Today. 2015;10(4):487–510. doi:10.1016/j.nantod.2015.06.006
26. Adiko AC, Babdor J, Gutierrez-Martinez E, Guermonprez P, Saveanu L. Intracellular transport routes for MHC I and their relevance for antigen cross-presentation. Front Immunol. 2015;6:335. doi:10.3389/fimmu.2015.00335
27. Silva JM, Vandermeulen G, Oliveira VG, et al. Development of functionalized nanoparticles for vaccine delivery to dendritic cells: a mechanistic approach. Nanomedicine (Lond). 2014;9(17):2639–2656. doi:10.2217/nnm.14.135
28. Longhi MP, Trumpfheller C, Idoyaga J, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med. 2009;206(7):1589–1602. doi:10.1084/jem.20090247
29. Gnjatic S, Sawhney NB, Bhardwaj N. Toll-like receptor agonists: are they good adjuvants? Cancer J. 2010;16(4):382–391. doi:10.1097/PPO.0b013e3181eaca65
30. Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. 2019;19(5):291–304. doi:10.1038/s41577-019-0124-9
31. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
32. Nembrini C, Stano A, Dane KY, et al. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proc Natl Acad Sci USA. 2011;108(44):E989–E997. doi:10.1073/pnas.1104264108
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.