")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Oxidative stress at high altitude: genotype–phenotype correlations
Received 27 December 2013
Accepted for publication 11 February 2014
Published 7 May 2014 Volume 2014:4 Pages 29—43
DOI https://doi.org/10.2147/AGG.S59775
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Priyanka Pandey,1,2 MA Qadar Pasha1,2
1CSIR-Institute of Genomics and Integrative Biology, Delhi, India; 2Department of Biotechnology, University of Pune, Ganeshkhind, Pune, India
Abstract: It has been well-documented that the hypobaric hypoxic environment at high altitude (HA) causes stress to both the permanent residents of HA and the sojourners. This oxidative stress primarily disturbs the oxygen-sensing and vascular homeostasis pathways, thereby upsetting normal human physiology, especially in sojourners. These environmental challenges have caused dynamic evolutionary changes within natives of HA, allowing them to develop adaptive plasticity. This review focuses on the genomic and biochemical features of the molecules involved in the oxygen-sensing and vascular homeostasis pathways with respect to HA pulmonary edema (HAPE) and adaptation. We review the role of genetic markers such as HIF-prolyl hydroxylase 2, endothelial PAS domain-containing protein 1, endothelial nitric oxide synthase, endothelin 1, cytochrome b-245 alpha polypeptide, and glutathione S-transferase pi 1, as well as three circulatory biomarkers (nitric oxide, endothelin 1, and 8-iso-prostaglandin F2α), by highlighting approaches such as candidate gene and genome-wide, adopted in deciphering the pathways. A disagreement between the two approaches has also been highlighted. In addition, we discuss that an overrepresentation of wild-type alleles in HA natives and mutant alleles of same polymorphisms in HAPE patients implies that the allelic variants at the same locus are involved in adaptation and HAPE, respectively. Moreover, healthy sojourners present a number of genomic features similar to HA natives, further strengthening the concept of genetic predisposition. A trend in correlation between protective and risk alleles and altered levels of circulatory markers clearly documents the phenomenon of genotype–phenotype correlations. We conclude that the genetic and biochemical markers discussed here both could prove to be potential targets for future diagnostic and therapeutic interventions.
Keywords: high altitude, oxidative stress, pulmonary edema, sojourners, natives
Introduction
Physiology at high altitude
The field of mountain medicine recognizes the high altitude (HA) region as comprising terrestrial elevations between 1,500 and 3,500 m.1 HA is characterized by extreme environmental conditions such as low atmospheric pressure, low partial pressure of oxygen, low temperature,1,2 and high ultraviolet radiation.3 The higher the altitude, the more likely the adverse effects. An altitude above 1,500 m affects the homeostasis of human physiology, and 2,500 m makes humans much more susceptible to disorders. Altitude above 5,500 m cannot be tolerated by the human body for a long period of time.1 The human body can adapt, as in HA natives, and acclimatize, as in sojourners (lowlanders), to the stressful environment of HA by having a higher breathing rate2,4,5 and higher heart rate1,2,4,5 and by making alterations in blood circulatory physiology.6,7 In this review, we discuss how permanent and temporary dwellings have shaped human physiology, differentially leading to a number of genetic and circulatory differences in HA natives and sojourners, respectively. It is these differences that make the two populations considerably distinct in their ability to adjust to the environmental stress at HA.
Hemoglobin in HA acclimatization and adaptation
Alterations in blood circulatory physiology are a key phenomenon observed in both the sojourners visiting HA and long-term residents of the place (ie, HA natives). Hemoglobin is one of the important factors involved in the discussion of HA acclimatization and adaptation. It has been noticed that modifications in hemoglobin functions play a key role in mediating an adaptive response to HA hypoxia.8,9 Hemoglobin is a specialized tetrameric protein found in the blood that is needed to transport oxygen molecules within the human body. The percentage saturation of hemoglobin with oxygen falls at HA, however, by increasing the concentration of hemoglobin, thus increasing the amount of oxygen that can be carried in the blood. Our body has devised a mechanism to overcome the effects of harsh environmental conditions at HA, but there is a negative aspect to this phenomenon as well, as too much hemoglobin makes blood viscous and difficult for the heart to pump effectively. This is a phenomenon commonly observed in a disease known as chronic mountain sickness. Population differences have been seen in hemoglobin levels, with lower hemoglobin levels found in Tibetans than in Aymaras and sojourners. This interpopulation difference has been accounted for by genetic factors for a very high proportion of the phenotypic variance found in hemoglobin concentration. The presence of significant genetic variance clearly indicates that natural selection leading to genetic adaptation, with respect to hemoglobin concentration, is going on in Tibetan and Aymara HA populations.10 Separate studies have reported haplotypes and variants of genes, such as endothelial PAS domain-containing protein 1 (EPAS1), HIF-prolyl hydroxylase 2 (EGLN1), and peroxisome proliferator-activated receptor alpha (PPARA) associating with hemoglobin concentrations. rs10803083 of EPAS1 is one of the most significant single nucleotide polymorphisms found at a significance level of a genome-wide study (GWAS) in the Aymara population.11,12
Hypobaric hypoxia and HA disorders
The inverse curvilinear correlation between barometric pressure, partial pressure of oxygen, and HA results in hypobaric hypoxia, which is the depletion of the oxygen supply to tissues, resulting from low atmospheric pressure. Hypobaric hypoxia has a number of deleterious effects on human physiology, and physical exertion in particular, in the unacclimatized sojourners, which creates respiratory and cerebral problems, eventually resulting in various mountain disorders.1,13 Among the various disorders, acute mountain sickness, HA pulmonary edema (HAPE), and HA cerebral edema1,2,13 afflict sojourners. In contrast, HA natives remain hale and hearty5 under such harsh conditions. In this review, we concentrate mainly on HAPE in sojourners and adaptation in HA natives.
HAPE and adaptation
HAPE is a life-threatening form of noncardiogenic pulmonary edema that occurs in unacclimatized sojourners on exertion.14–16 It is caused by acute exposure to low partial pressure of oxygen at HA. Speed of ascent and altitude attained are the key determinants in occurrence of HAPE,1 as on rapid ascent, the body does not have sufficient time for the acclimatization process, such as deeper breathing, increased respiratory rate, higher urine production, and increased red blood cell production.
In addition, at HA, because of the low pressure, water vapor is lost from the lungs at an increased rate, leading to dehydration, which also contributes to the symptoms of HAPE.17 Headache is the first symptom commonly experienced on ascent to HA and is often a result of dehydration. Exaggerated hypoxic pulmonary vasoconstriction is a hallmark of HAPE, characterized by abnormal build-up of fluid in the lungs, preventing air spaces from opening up and filling with fresh air after each breath.14–16,18 This abnormal fluid build-up leads to dyspnea, cough, congestion, tachypnea, tachycardia, central cyanosis, and wheezing while breathing. These symptoms constitute the diagnosis of HAPE.14–16,18,19 Chest radiographic infiltrates consistent with pulmonary edema confirm the disorder.14,16,18 The symptoms and signs disappear within 3 days of the start of treatment with supplemental oxygen and bed rest. The incidence rate of HAPE is variable but is more pronounced in unacclimatized sojourners than in HA natives.16,18 It seems that over the generations, HA natives have undergone positive natural selection for the beneficial traits (ie, an adaptive evolution has been undertaken). The adaptive plasticity inherited in subsequent generations has resulted in a large population of HA natives well-adapted to the strenuous HA settings.20–23 Visible adaptive changes observed in HA natives are higher resting ventilation,5 higher exhaled NO,24,25 blunted hypoxic vasoconstrictor response,5 and thin-walled pulmonary vasculature.26
While restricting ourselves to hypoxia-induced oxidative stress, we do recognize that adaptation in HA natives and HAPE in sojourners can be nongenetic, consisting of physiological, biochemical, or behavioral adjustments in response to environmental changes. Hence, it becomes difficult to differentiate the contribution of each marker to a particular phenotypic trait, which has convoluted the inference of genetic adaptation and HAPE. In this review, however, we have tried to look on and discuss a few of the important genes related to vascular homeostasis and oxygen-sensing pathways, respectively, as well as their associated circulatory biomarkers in HAPE and adaptation.
Hypobaric hypoxia-induced oxidative stress
One of the most detrimental effects of hypobaric hypoxia is oxidative stress,27 which is the consequence of increased reactive oxygen species (ROS),28 reactive oxygen and nitrogen species (RONS),29 decreased antioxidants,30 and free radical-mediated reduction in pulmonary NO bioavailability.31,32 The ensuing oxidative stress primarily contributes to endothelial damage and vascular wall remodeling, which are potentially involved in the development of HA-related disorders in sojourners.1,13
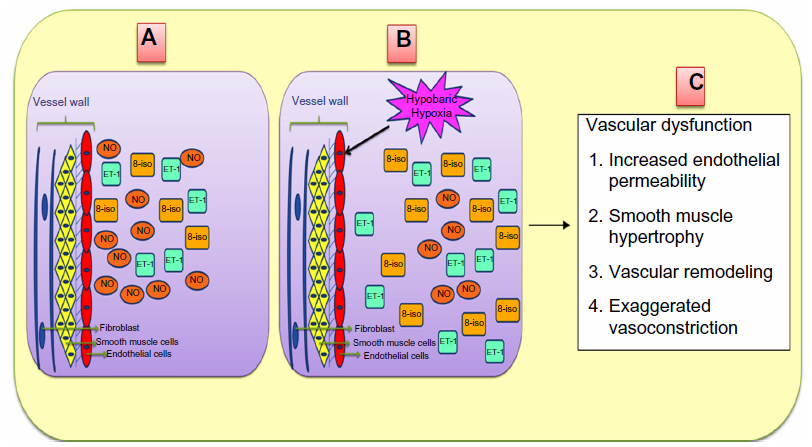
Thus, we can say that harsh climatic conditions at HA initiate a series of events in which there is an increased production of reactive intermediates,29,30,32,33 vasoconstrictors such as endothelin 1 (ET-1),32,34–36 5-hydroxytryptamine (5-HT), 8-iso-prostaglandin F2α (8-iso-PGF2α),34 and various other proinflammatory cytokines.29,35,37–39 In contrast, there is a decrease in the production of vasodilators such as NO,32,34,40,41 prostacyclins,38 antioxidants,29,30,38 and anti-inflammatory cytokines.37,38 This alteration in the production of these moieties is a fail-safe mechanism of the body to divert resources toward the energy-starved parts and to restore vascular homeostasis. However, this increase and decrease in the levels leads to an imbalance between the body’s ability to sustain the damage and detoxify the ROS/RONS and their continued manifestation in higher and higher amounts. An increase in the levels of vasoconstrictors and proinflammatory cytokines, in the absence of counteracting partners, leads to higher production of ROS/RONS and, hence, both to more damage to the vascular homeostasis and to vascular dysfunction.32,42 We hypothesize that this imbalance is a key mechanism implicated in different HA-related disorders. Thus, in effect, oxidative stress at HA is a vicious cycle that is reinforcing itself through a feedback loop, aggravating the damage in an effort to resolve the problem and giving rise to the initial set of conditions. Figure 1 explains HA-associated oxidative stress. Of note, there is a difference in levels of antioxidants expressed in sojourners and HA natives, which provides insight into the differential adaptation pattern and tolerance to HA by HA natives.28,43
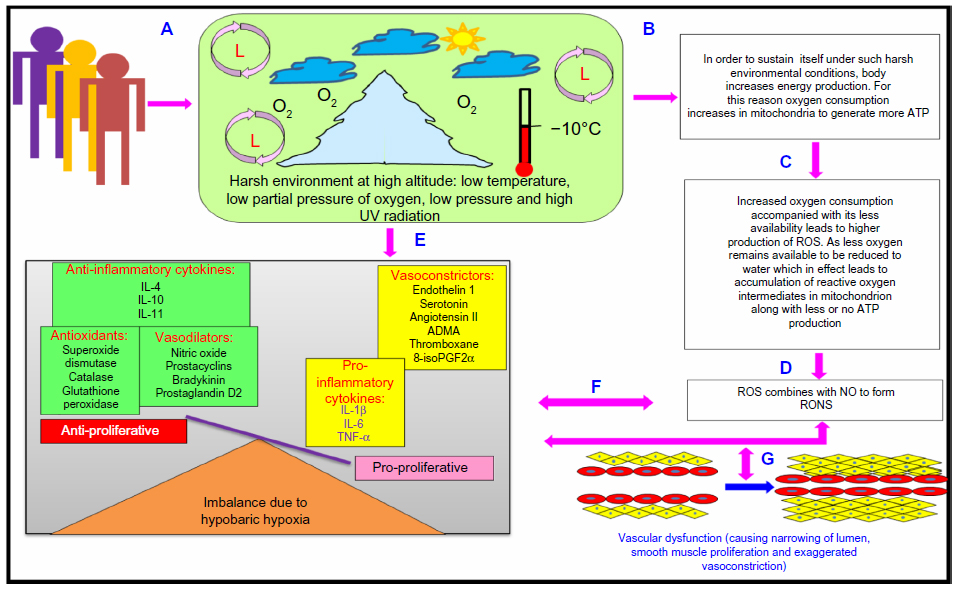 | Figure 1 High-altitude-associated oxidative stress. |
Gene–environment interaction
There is an interplay between gene and environment (ie, genes and the environment [G×E] interact to produce a phenotypic outcome, which is governed by both entities).44–47 One way to assess G×E interactions48 is to perform candidate gene studies. In such studies, genotyping is done in a case-control format, which tells about the risk or minor genotype (found more in cases) or protective or major genotype (found more in controls) of a gene. SPSS49,50 (IBM Corporation, Armonk, NY, USA) and MDR (Vanderbilt University Medical School, Nashville, TN, USA)51,52 are two well-known software programs that can be used to study such interactions. The input file format depends on the software being used. Accordingly, this tells how the environment interacts with genes or vice versa. Statistically significant interactions are indicated by P<0.05 or any other cut-off set by the user. Interaction can be synergistic or redundant, depending on the information gained or lost through the interaction, respectively.51
A common way of analyzing G×E interactions by multinomial logistic regression analysis follows:
- Problem: Understanding the effect of the mutant genotype of rs2238625 (T/C) on HAPE patients (cases).
- Solution: For this, a single nucleotide variant, rs2238625 (T/C), of GNA11 (guanine nucleotide binding protein [G protein], alpha 11 [Gq class]) will be genotyped in HAPE patients (cases) and related controls. For regression analysis, an input file needs to be prepared.
- Coding for an input file is such that:
- cases are coded as 0 and controls as 1 (reference),
- males are coded as 1 and females as 0, and
- a major genotype is coded as 1 and a minor genotype as 0.
- After genotyping, an initial counting of the common homozygotes, heterozygotes, and rare homozygotes is prepared, as shown in Table 1.
- ere, TT emerged as a major genotype (a genotype found majorly in both cases and controls) and CC as minor or concern genotype. While performing an analysis between major genotype TT and a heterozygote (ie, CT), we code TT as 1 and CT 0 (model 1), and we delete the CC genotype, as between TT and CC (TT =1, CC =0, CT=deleted; model 2).
- Input file formats are provided, as in Tables 2 and 3.
- Conclusion: A significant P-value suggests that there is an effect of mutant genotype (ie, CC) on cases. Since our cases are HAPE patients, it implies that the mutant genotype is causing a predisposition to HAPE in sojourners and there is a possible interaction between hypobaric, hypoxic environments at HA and GNA11.
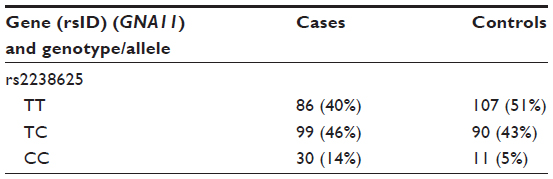 | Table 1 Initial counting of common homozygotes, heterozygotes, and rare homozygotes |
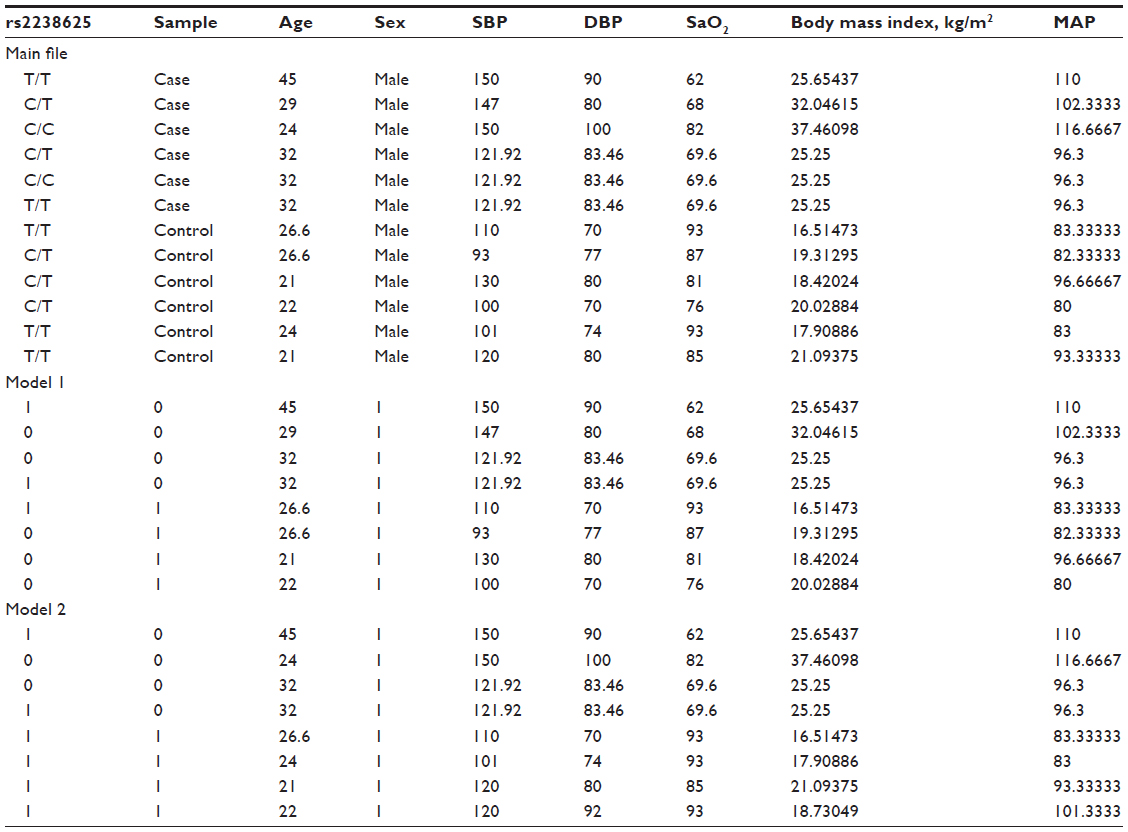 | Table 2 Input file formats |
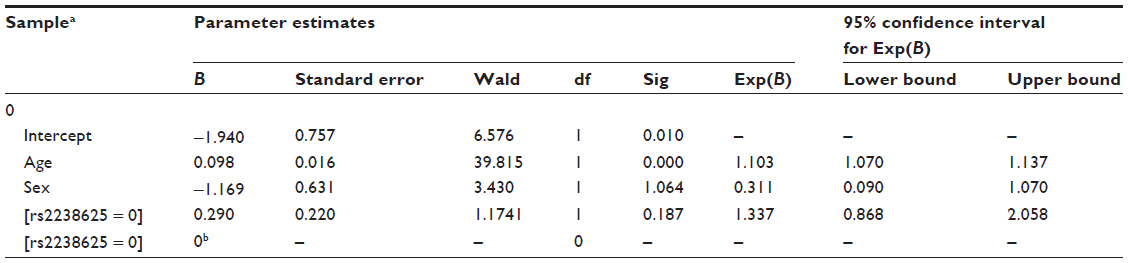 | Table 3 A typical result of a multinomial logistic regression analysis |
A snapshot of the SPSS software for performing the analysis is provided in Figure 2. Clinical characteristics influencing the results, such as age and sex, are entered in the covariate section of the multinomial regression analysis to nullify their effect.
 | Figure 2 Snapshot of SPSS software (IBM Corporation, Armonk, NY, USA). |
After performing the analysis, it is determined that what kind of G×E interaction is taking place may result in favorable as well as detrimental effects. In highland natives, this interaction has caused visible favorable changes, making them commodious enough to dwell in the HA settings, whereas the same interaction has remained neutral for a large number of sojourners but risky for a few. Differences in the sojourners’ response to the HA environment clearly demarcate the genetic predisposition, which has been well-documented in the HA natives11,46,53–55 and in one of the most common HA disorders (ie, HAPE).46,53,55 We attribute this genetic predisposition to evolutionary processes, suggesting that adaptation in HA natives stemmed from ancient ancestors favoring a “thrifty” genotype, which enabled survival and settlement under the harsh climatic conditions of HA. In contrast, lowlanders, who had never experienced such harsh environmental conditions, did not have the genotype. As a consequence, when unacclimatized sojourners (lowlanders) ascended to HA, they suffered with HA-related disorders such as HAPE. Thus, the thrifty genotype became protective in nature at HA.56 This may well be the case for the genes associated with the major physiological pathways that play crucial roles under the hypobaric hypoxic environment of HA, such as vascular homeostasis,34,46 oxygen sensing,57,58 and inflammatory pathways.40,42,59,60 The genes that majorly associate with these pathways can easily be recognized as ET-1,35,36,61 angiotensin-converting enzyme (ACE),62–65 endothelial nitric oxide synthase (NOS3),60,66,67 cytochrome b-245 alpha polypeptide (CYBA),68–70 glutathione S-transferase pi 1 (GSTP1),71–74 5-HT and the related 5-hydroxytryptamine transporter (5-HTT),75 bone morphogenetic receptor type isoforms (BMPRI and BMPRII),76–79 and rho-associated coiled coil kinase isoforms (ROCKI and ROCKII).75,80–83
The significance of these genes in the pulmonary vasculature has been well-established, which renders the lead for the ongoing studies. These genes can easily be identified as potential candidates in determining the phenomenon of adaptation and HAPE at HA. The selection of genetic variants from such genes that are facilitating adaptation at HA must be decisive to offset the detrimental consequences of hypoxia. Accordingly, the alleles favored at HA should be overrepresented in natives. Inversely, underrepresentation of the same genetic variants may lead to HA disorders such as HAPE. This can be established by association studies with three study groups: patients, related controls from lowland, and healthy highland natives. In view of these facts, we suggest that G×E toward acquiring the beneficial traits in HA natives is immense at the same time it is rendering sojourners susceptible to HAPE.
Candidate gene approach
A candidate gene approach is a hypothesis-driven approach that directly tests the effects of the genetic variants of a potentially contributing gene in an association study. These studies include either members of an affected family or unrelated cases and controls. These studies can be performed relatively quickly and inexpensively and may also allow identification of genes with small effects.66 The candidate gene approach has helped in identifying many candidates,6 but here we highlight only ET-1, NOS3, CYBA, and GSTP1, which are majorly implicated in vascular dysfunction resulting from hypoxia-induced oxidative stress at HA.
Criteria for marker inclusion
The reasons for studying these genetic and associated circulatory markers are as follows. First, ET-1,36,84 NOS3,85–87 CYBA,88 and GSTP174,88 are by far the four most well-characterized genes that differentially stimulate the physiological processes to bring in cellular changes at HA. Second, functionally counteracting partners have been chosen to study (eg, ET-1 counters NOS3 and GSTP1 counters CYBA) to understand in entirety the intriguing phenomenon of oxidative stress at HA89 (gene ID: 2950, gene ID: 1535). Third, these genes perform functions that are multifactorial in nature (gene ID: 4846, gene ID: 1906, gene ID: 2950, gene ID: 1535). Finally, the number of genes involved in physiological regulation, apart from these genes, is enormous and includes many genes with debatable functions. Circulatory biomarkers such as ET-1,90 NO,91 and 8-iso-PGF2α88,92 have been chosen because of their association with ET-1,90 NOS3,91 CYBA,88,92 and GSTP1,88 respectively, as well as because of their established multifactorial functions,93 such as vasoconstrictory and vasodilatory functions in oxidative stress at HA.
ET-1
ET-1 is a 21 amino acid long peptide hormone, the production of which is significantly regulated by hypoxia. It is a potent, long-lasting vasoconstrictor94–96 with mitogenic properties94,97–99 promoting smooth muscle cell proliferation,92,100 regulating vascular tone,94,95 and manifesting its effect through endothelin receptor type A.94,98 ET-1 has been studied at both the genetic101–104 and biochemical level.36,61,84,101,105 Three ET-1 polymorphisms (viz, CTn–CAn repeat, Lys198Asn, and G2288T) were studied in HA natives and HAPE patients, and the variants of these polymorphisms were found to be associated with adaptation or disease. The CTn-CAn repeats are the dinucleotide repeat polymorphism in ET-1. The (CT)n- (CA)n repeats were segregated and recognized as shorter (13–30 base pairs) and longer (31–45 base pairs).106,107 Among the variants, an overrepresentation of CTn-CAn shorter repeats, Asn198, and T alleles was reported in HAPE patients compared with in ethnically matched related healthy sojourners (HAPE-resistant, controls [HAPE-r]). Moreover, the HAPE patients had significantly higher ET-1 levels, which correlated positively with risk variants. In contrast, an overrepresentation of CTn-CAn longer repeats, G allele, and the combination of wild-type genotypes (ie, longer-repeats/GG, longer-repeats/Lys198Lys) were reported in both HA natives and healthy sojourners (HAPE-r). Consequent to this, plasma ET-1 levels were significantly lower in HA natives. The wild-type genotypes (ie, longer-repeats, GG, and Lys198Lys genotypes) associated with significantly lower ET-1 levels in the highlanders (HLs), as expected.106,107 It is obvious from these findings that elevated ET-1 levels cause exaggerated vasoconstriction and increased endothelial permeability (Figure 3). Moreover, the correlation of these variants (CTn-CAn shorter repeats, Asn198, and T alleles with elevated ET-1 levels in HAPE patients) accounts for the same function, and vice versa in HA natives and HAPE-r (Table 4).
 | Figure 3 Significance of biomolecules in vascular homeostasis. |
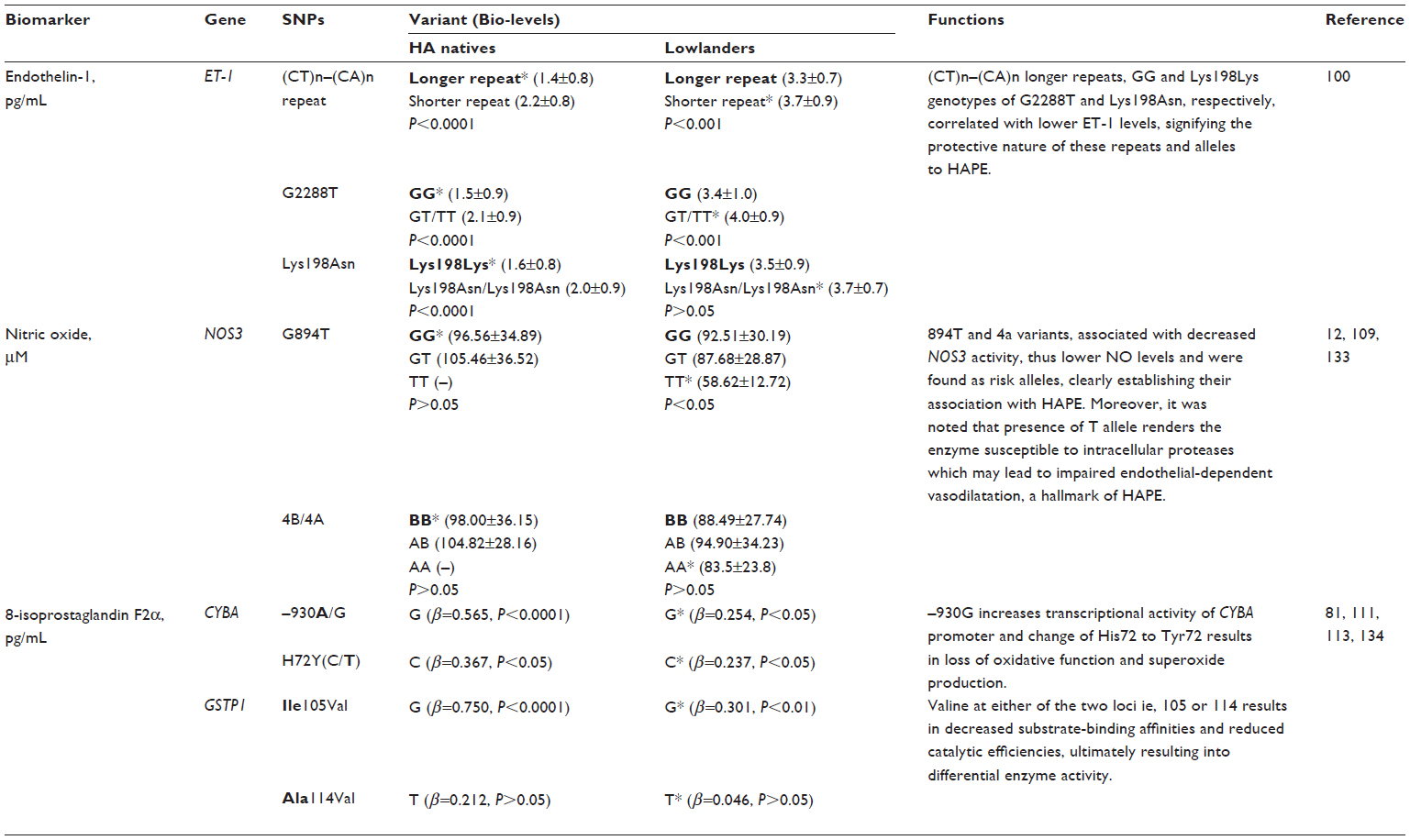 | Table 4 Single nucleotide polymorphisms (SNPs) and biomarkers under hypobaric hypoxia |
NOS3
NOS3 is an enzyme that generates NO, a potent vasodilator with multiple other functions in blood vessels, by converting L-arginine to L-citrulline, using nicotinamide adenine dinucleotide phosphate as an energy source.91,108 NO is involved in regulating vascular tone by inhibiting smooth muscle contraction,91,108–110 monocyte adhesion,108 and platelet aggregation.91,108,110 The gaseous molecule also counteracts many environmentally induced maladaptive changes such as smooth muscle cell proliferation91,108–110 and hypoxic pulmonary vasoconstriction.108,111 NOS3 gets activated by higher concentrations of divalent calcium ions,109,111 leading to vasodilation via NO and thereby countering exaggerated vasoconstriction, a hallmark of HAPE.18,112 It has been the most investigated candidate gene because of its relevance in the maintenance of vascular homeostasis.91,111
A number of polymorphisms are reported, but the favored polymorphisms in the gene were Glu298Asp (G894T)86,87,113,114 and 4b4a86,87,114 (a 27 bp variable number of tandem repeats). Glu298Asp has also been reported to have a functional effect on NOS3 protein by causing differential susceptibility of NOS3 protein to cleavage on allele change from G894T.115 It was reported that Asp298 and 4a alleles of Glu298Asp and 4b4a polymorphisms, respectively, were overrepresented in HAPE patients compared with in the healthy sojourners (HAPE-r). The interaction between these two polymorphisms was significant with an overrepresentation of the haplotype Glu2984b in the controls and Asp2984a in HAPE patients. In addition, it was also observed that NO levels were significantly reduced in HAPE patients; moreover, the HAPE patients who overrepresented the minor alleles had significantly lower levels of NO. Conversely, Glu298 and 4b alleles of Glu298Asp and 4b4a polymorphisms, respectively, were overrepresented in HA natives and healthy sojourners. The interaction between these two polymorphisms was significant, with an overrepresentation of the haplotype Glu2984b in the HA natives. A correlation analysis of these variants revealed that the subjects with wild-type alleles or their combinations 894GG+4bb 894G (298Glu) and 4b were associated with higher levels of circulating NO, whereas those with the minor alleles were associated with lower levels of NO.85,116,117 As elevated NO levels cause increased vasodilatation (Figure 3), it can be said that respective correlated variants (ie, Glu298and 4b in HA natives and HAPE-r; Table 4) also account for the same function, and vice versa in HAPE patients.
CYBA
CYBA encodes for CYBA, also known as p22phox subunit. This protein forms a critical component of the nicotinamide adenine dinucleotide phosphate oxidase system, which is involved in the production of superoxide anion, a reactive free radical.118 These superoxides are involved in causing oxidative stress, one of the debilitating effects of HA exposure, leading to HAPE.29 The gene has functional variants that have been associated with various oxidative stress-related diseases.69,70,118–121 The polymorphisms in focus are -930A/G and H72Y (C242T).
It has been found that elevated free radical formation (a hallmark of oxidative stress), along with antioxidant deficiency, can be reliably assessed by estimating the levels of 8-iso-PGF2α. 8-iso-PGF2α, a potent vasoconstrictor, is a prostaglandin-F2-like compound belonging to the F2 isoprostane class that is produced in vivo by the free radical-catalyzed peroxidation of arachidonic acid. It is a reliable biomarker of oxidative stress, and its concentration is increased in the bronchoalveolar lavage fluid and urine of patients with interstitial lung diseases and chronic obstructive pulmonary disease.122–124 8-iso-PGF2α levels were invariably elevated in HAPE patients; moreover, it has also been found to be associated with the polymorphisms of CYBA mentioned earlier. The genotype distributions of -930A/G and C242T differed significantly in HAPE patients. The alleles, G of -930A/G and C of C242T, were overrepresented in HAPE patients, as was the haplotype G-C. Furthermore, 8-iso-PGF2α has been found to be significantly increased in HAPE patients, and risk alleles G and C of -930A/G and C242T associated significantly with higher levels of 8-iso-PGF2α in HAPE patients. In contrast, allele A of -930A/G and T of C242T, respectively, and their respective haplotypes A-T were over-represented in HA natives and healthy sojourners. Furthermore, 8-iso-PGF2α has been found decreased significantly in HA natives and sojourners. Protective alleles A and T of -930A/G and C242T associated with significantly lower levels of 8-iso-PGF2α in HA natives and healthy sojourners.88 As apparent from the earlier text, elevated 8-iso-PGF2α levels cause exaggerated vasoconstriction and increased oxidative stress (Figure 3). For this reason, it can be said that respective correlated variants (ie, G of -930A/G and C of C242T in HAPE patients; Table 4) also account for the same function, and vice versa in HA natives and HAPE-r.
GSTP1
GSTP1 encodes the enzyme glutathione S-transferase pi 1, which plays an important role in the detoxification of reactive intermediates by scavenging ROS with reduced glutathione, thereby maintaining homeostasis and preventing tissue damage125 (gene ID: 2950). GSTP1 is a polymorphic gene, and its exonic variants, I105V (A315G) and A114V (C342T), have been reported to be capable of altering catalytic activity.126,127 Moreover, these polymorphisms are associated with diseases involving hypoxia,73,74,128,129 and 8-iso-PGF2α, a reliable indicator of oxidative stress,122– 124 has been found to be associated with the two polymorphisms of GSTP1 mentioned earlier. In the case of HAPE, the G allele of I105V (A/G) and T allele of A114V (C/T) were prevalent in HAPE patients and associated with increased 8-iso-PGF2α levels. Haplotype G-T, formed of risk alleles of I105V (A/G) and the T allele of A114V (C/T), were overrepresented in HAPE patients. Contrary to these findings, the A allele of I105V (A/G) and C allele of A114V (C/T) were prevalent in HA natives and healthy sojourners and were associated with decreased 8-iso-PGF2α levels. Haplotypes A-C, formed of protective alleles of I105V (A/G) and A114V (C/T), were overrepresented in HA natives and healthy sojourners (lowlanders).88 In view of the fact that elevated 8-iso-PGF2α levels cause exaggerated vasoconstriction and increased oxidative stress (Figure 3), it can be said that respective correlated variants (ie, G allele of 105V [A/G] and T allele of A114V [C/T] in HAPE patients; Table 4) also account for the same function, and vice versa in HA natives and HAPE-r.
In view of the facts mentioned earlier, it may be inferred that the overrepresentation of wild-type alleles (protective variants) in HA natives and mutant alleles (risk variants) of the same polymorphism in HAPE patients are involved in adaptation and HAPE. The trend in correlation of alleles with biolevels further documents the phenomenon of genotype–phenotype correlations, as it was seen that protective variants correlated with lower ET-1 and 8-iso-PGF2α levels and higher NO levels, and vice versa with risk variants. Elevated NO, decreased ET-1, and 8-iso-PGF2α levels in HA natives suggest their crucial role in HA adaptation and acclimatization, and vice versa for decreased NO level, and increased ET-1 and 8-iso-PGF2α in HAPE patients, compared with in the healthy sojourners, signifies their role in hypobaric hypoxia-induced oxidative stress at HA. Figure 3 describes the consequences of increased and decreased levels of a few of these molecules under hypobaric hypoxia-induced oxidative stress at HA.
GWAS
The candidate gene approach is restricted in a large part by an understanding of the underlying and associated biology of the disease being investigated, GWAS are being performed. These are noncandidate-driven, unbiased studies. GWAS are one of the most exhaustive techniques, employing high-throughput technology to scan the whole genome for a large variety of variants such as copy number variations and single nucleotide polymorphisms.130–133 The application of genome-wide scans in the HA investigations has revealed two chromosomal regions associated with adaptive evolution in the two highland populations; that is, Andean and Tibetan. The identified genes are EGLN1 and EPAS1, of an oxygen-sensing pathway, and are particularly important in adapting to the hypobaric hypoxic environment at HA.21,53,134–138 In simultaneous investigations, it has been found that few variants in these genes compared with in the lowland population are associated with low hemoglobin concentration in blood, and thus with adaptation.134,139–141 Figure 4 mentions the oxygen-sensing pathway involving concerned genes such as erythropoietin, vascular endothelial growth factor, and platelet-derived growth factor B. Activation of these genes leads to the alteration in vascular homeostasis, leading to oxidative stress at HA.
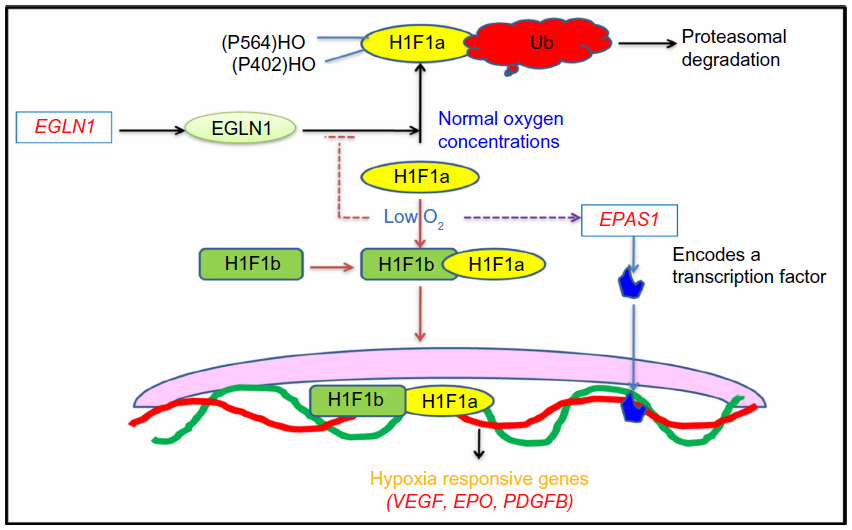 | Figure 4 Oxygen-sensing pathway. |
Candidate gene and genome-wide approach: the disagreement ensues
It is of consequence to add that a comparison between the candidate gene and genome-wide approaches has revealed astonishing differences. The candidate gene approach has identified several markers of significance; however, none could be validated through GWAS. Of note, we are not against GWAS, since this approach has identified novel genes and pathways in studies of other diseases and biologic phenotypes, but in the study of HA physiology and disease, the amount of knowledge gained has been negligible and conflicting. The basis for the discrepancy between candidate-gene and GWAS-identified genes is unknown. As adaptation and maladaptation of the complex diseases are the phenomena involving a number of genes, it raises a fundamental question and an argument that genome-wide approach is not yielding as many markers, despite the candidate gene approach representing the association of several loci. Can it be attributed to the method, the smaller sample size, or the statistical tools? We suggest that without compromising the stringency of the statistical measures, an appropriate approach needs to be found so that an enormous amount of capital being put up in such high-throughput experiments, such as GWAS, is more productive and that the information retrieved is helpful for the cause of human establishment. Table 5 represents a comparative view of candidate gene and genome-wide association studies, respectively.
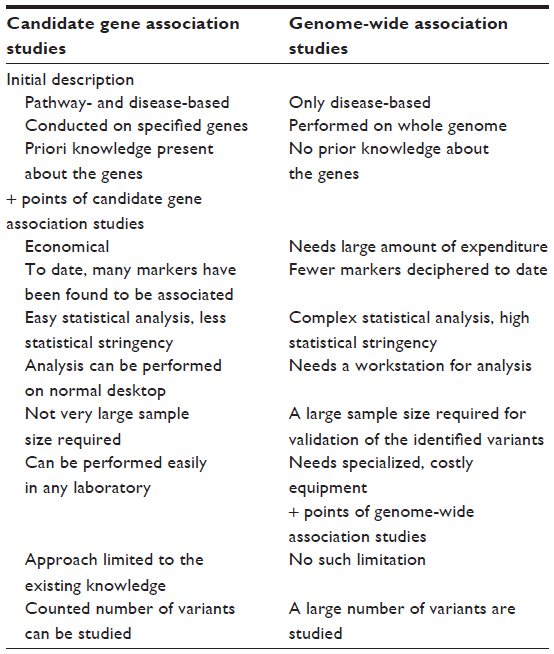 | Table 5 A comparative view of candidate gene association studies and genome-wide association studies |
Conclusion
To sum up, the last decade has experienced phenomenal progress in understanding the genetic association of variants with HA-related disorders and adaptation patterns at HA. Simultaneously, the molecular aspects and biochemical parameters of HA also received equal attention. Genetic and biochemical studies until now have highlighted the significance of few oxidative stress markers in relation to adaptation and disease; together, both could prove to be potential candidates for diagnostic and therapeutic intervention. The near absence of HA disorders such as HAPE in natives, the relative exaggerated susceptibility of few sojourners to HAPE, and the neutrality of a large number of sojourners clearly indicates the involvement of genetic variants in differentially regulating physiology, leading to disease in sojourners and adaptation in HA natives.
The overrepresentation of wild-type or protective alleles in HA natives and their association with desirable levels of circulatory biomarkers strengthens their involvement with HA adaptation. In contrast, overrepresentation of mutant or risk alleles and their association with disturbed levels of biomarkers in HAPE patients strengthens their involvement in hypoxia-induced oxidative stress, and thereby in HAPE. Moreover, healthy sojourners present a number of genomic features similar to HA natives, further strengthening the concept of genetic predisposition to HAPE. In this regard, we try to provide a succinct overview in Table 4, where a correlation between significantly associated variants of selected genes (ie, ET-1, NOS3, CYBA, and GSTP1) and associated biomarkers (NO, ET-1, and 8-iso-PGF2α) is provided for a quick understanding of HA adaptation and susceptibility to HA disorders. Genome-wide association studies have additionally facilitated deciphering newer pathways concerned with hypoxia-induced oxidative stress. Although human physiology is complex, and many genes are involved in vascular dysfunction resulting from hypoxia-induced oxidative stress, we could just provide a glimpse of a few established genes and biomarkers pertaining to HAPE, adaptation, and maladaptation. Studies would continue to identify, validate, and establish newer markers of global significance, and taken together, both genetic and biochemical markers discussed in the review could be potential targets for future diagnostic and therapeutic interventions for HA-related disorders.
Author contributions
Priyanka Pandey wrote the manuscript. MA Qadar Pasha conceptualized and edited the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Gallagher SA, Hackett PH. High-altitude illness. Emerg Med Clin North Am. 2004;22(2):329–355. | |
Peacock AJ. ABC of oxygen: oxygen at high altitude. BMJ. 1998;317(7165):1063–1066. | |
Blumthaler M, Ambach W, Ellinger R. Increase in solar UV radiation with altitude. J Photochem Photobiol B Biol. 1997;39(2):130–134. | |
Mason NP. The physiology of high altitude: an introduction to the cardio-respiratory changes occurring on ascent to altitude. Curr Anaesthesia Crit Care. 2000;11:34–41. | |
West JB. Respiratory and circulatory control at high altitudes. J Exp Biol. 1982;100:147–157. | |
Mortimer H, Patel S, Peacock AJ. The genetic basis of high-altitude pulmonary oedema. Pharmacol Ther. 2004;101(2):183–192. | |
Shivastava KK, Malhotra MS. Effect of adaptation to high altitude on components of blood and urine of lowlanders compared with high altitude natives. Int J Biometeorol. 1974;18(4):229–287. | |
Storz JF, Moriyama H. Mechanisms of hemoglobin adaptation to high altitude hypoxia. High Alt Med Biol. 2008;9(2):148–157. | |
Gonzales GF. Hemoglobina y testosterona: importancia en la aclimataciÓn y adaptaciÓn a la altura. [Hemoglobin and testosterone: importance on high altitude acclimatization and adaptation]. Rev Peru Med Exp Salud Publica. 2011;28(1):92–100. Spanish. | |
Beall CM, Brittenham GM, Strohl KP, et al. Hemoglobin concentration of high-altitude Tibetans and Bolivian Aymara. Am J Phys Anthropol. 1998;106(3):385–400. | |
Scheinfeldt LB, Tishkoff SA. Living the high life: high-altitude adaptation. Genome Biol. 20111(9):133. | |
Alkorta-Aranburu G, Beall CM, Witonsky DB, Gebremedhin A, Pritchard JK, Di Rienzo A. The genetic architecture of adaptations to high altitude in Ethiopia. PLoS Genet. 2012;8(12):e1003110. | |
Basnyat B, Murdoch DR. High-altitude illness. Lancet. 2003;361(9373):1967–1974. | |
BÄrtsch P, MairbÄurl H, Maggiorini M, Swenson ER. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol (1985). 2005;98(3):1101–1110. | |
Hultgren HN. High-altitude pulmonary edema: current concepts. Annu Rev Med. 1994;7:267–284. | |
Roach JM, Schoene RB. High-altitude pulmonary edema. In: Textbooks of Military Medicine: Medical Aspects of Harsh Environments. Volume 2. Lounsbury DE, Bellamy RF, Zajtchuk R, editors. Washington, DC: Office of The Surgeon General, Borden Institute; 2002:789–814. | |
Hackett PH, Roach RC. High-altitude illness. N Engl J Med. 2001;345(2):107–114. | |
Maggiorini M. High altitude-induced pulmonary oedema. Cardiovasc Res. 2006;72(1):41–50. | |
Jerome EH, Severinghaus JW. High-altitude pulmonary edema. N Engl J Med. 1996;334(10):662–663. | |
Beall CM. Detecting natural selection in high-altitude human populations. Respir Physiol Neurobiol. 2007;158(2–3):161–171. | |
Beall CM. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc Natl Acad Sci U S A. 2007;104 Suppl 1:8655–8660. | |
Simonson TS, McClain DA, Jorde LB, Prchal JT. Genetic determinants of Tibetan high-altitude adaptation. Hum Genet. 2012;131(4):527–533. | |
Stobdan T, Karar J, Pasha MA. High altitude adaptation: genetic perspectives. High Alt Med Biol. 2008;9(2):140–147. | |
Beall CM, Laskowski D, Strohl KP, et al. Pulmonary nitric oxide in mountain dwellers. Nature. 2001;414(6862):411–412. | |
Hoit BD, Dalton ND, Erzurum SC, Laskowski D, Strohl KP, Beall CM. Nitric oxide and cardiopulmonary hemodynamics in Tibetan highlanders. J Appl Physiol (1985). 2005;99(5):1796–1801. | |
Gupta ML, Rao KS, Anand IS, Banerjee AK, Boparai MS. Lack of smooth muscle in the small pulmonary arteries of the native Ladakhi. Is the Himalayan highlander adapted? Am Rev Respir Dis. 1992;145(5):1201–1204. | |
Jefferson JA, Simoni J, Escudero E, et al. Increased oxidative stress following acute and chronic high altitude exposure. High Alt Med Biol. 2004;5(1):61–69. | |
Sinha S, Ray US, Tomar OS, Singh SN. Different adaptation patterns of antioxidant system in natives and sojourners at high altitude. Respir Physiol Neurobiol. 2009;167(3):255–260. | |
Bakonyi T, Radak Z. High Altitude and Free Radicals. J Sports Sci Med. 2004;3(2):64–69. | |
Askew EW. Work at high altitude and oxidative stress: antioxidant nutrients. Toxicology. 2002;180(2):107–119. | |
Dweik RA. Nitric oxide, hypoxia, and superoxide: the good, the bad, and the ugly! Thorax. 2005;60(4):265–267. | |
Schiffrin EL. Oxidative stress, nitric oxide synthase, and superoxide dismutase: a matter of imbalance underlies endothelial dysfunction in the human coronary circulation. Hypertension. 2008;51(1):31–32. | |
Dosek A, Ohno H, Acs Z, Taylor AW, Radak Z. High altitude and oxidative stress. Respir Physiol Neurobiol. 2007;158(2–3):128–131. | |
Ali Z, Mishra A, Kumar R, et al. Interactions among vascular-tone modulators contribute to high altitude pulmonary edema and augmented vasoreactivity in highlanders. PLoS One. 2017(9):e44049. | |
Droma Y, Hayano T, Takabayashi Y, et al. Endothelin-1 and interleukin-8 in high altitude pulmonary oedema. Eur Respir J. 1996;9(9):1947–1949. | |
Sartori C, Vollenweider L, LÖffler BM, et al. Exaggerated endothelin release in high-altitude pulmonary edema. Circulation. 1999;99(20):2665–2668. | |
Behn C, Araneda OF, Llanos AJ, CeledÓn G, GonzÁlez G. Hypoxia-related lipid peroxidation: evidences, implications and approaches. Respir Physiol Neurobiol. 2007;158(2–3):143–150. | |
Sarada S, Himadri P, Mishra C, Geetali P, Ram MS, Ilavazhagan G. Role of oxidative stress and NFkB in hypoxia-induced pulmonary edema. Exp Biol Med (Maywood). 2008;233(9):1088–1098. | |
Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78(6):539–552. | |
Busch T, BÄrtsch P, Pappert D, et al. Hypoxia decreases exhaled nitric oxide in mountaineers susceptible to high-altitude pulmonary edema. Am J Respir Crit Care Med. 2001;163(2):368–373. | |
Duplain H, Sartori C, Lepori M, et al. Exhaled nitric oxide in high-altitude pulmonary edema: role in the regulation of pulmonary vascular tone and evidence for a role against inflammation. Am J Respir Crit Care Med. 2000;162(1):221–224. | |
Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99(7):675–691. | |
Padhy G, Sethy NK, Ganju L, Bhargava K. Abundance of plasma antioxidant proteins confers tolerance to acute hypobaric hypoxia exposure. High Alt Med Biol. 2013;14(3):289–297. | |
Gerke J, Lorenz K, Ramnarine S, Cohen B. Gene-environment interactions at nucleotide resolution. PLoS Genet. 2010;6(9):e1001144. | |
Hunter DJ. Gene-environment interactions in human diseases. Nat Rev Genet. 2005;6(4):287–298. | |
Jin G, Li S, Ge R, Albert M, Sun Y. High altitude disease: consequences of genetic and environmental interactions. N Am J Med Sci. 2009;2(3):74–80. | |
Rhen T, Schroeder A. Molecular mechanisms of sex determination in reptiles. Sex Dev. 2010;4(1–2):16–28. | |
Botto LD, Khoury MJ. Commentary: facing the challenge of gene-environment interaction: the two-by-four table and beyond. Am J Epidemiol. 2001;153(10):1016–1020. | |
Sam SS, Thomas V, Reddy KS, Surianarayanan G, Chandrasekaran A. Gene-environment interactions associated with CYP1A1 MspI and GST polymorphisms and the risk of upper aerodigestive tract cancers in an Indian population. J Cancer Res Clin Oncol. 2010;136(6):945–951. | |
Shi Y, Yuan Y, Xu Z, et al. Genetic variation in the calcium/calmodulin-dependent protein kinase (CaMK) pathway is associated with antidepressant response in females. J Affect Disord. 2012;136(3):558–566. | |
Hahn LW, Ritchie MD, Moore JH. Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics. 2003;19(3):376–382. | |
Su MW, Tung KY, Liang PH, Tsai CH, Kuo NW, Lee YL. Gene-gene and gene-environmental interactions of childhood asthma: a multifactor dimension reduction approach. PLoS One. 2012;7(2):e30694. | |
Aggarwal S, Negi S, Jha P, et al; Indian Genome Variation Consortium, Prasher B, Mukerji M. EGLN1 involvement in high-altitude adaptation revealed through genetic analysis of extreme constitution types defined in Ayurveda. Proc Natl Acad Sci U S A. 2010;107(44):18961–18966. | |
Li Q, Liu SY, Lin KQ, et al. [Association between six single nucleotide polymorphisms of EGLN1 gene and adaptation to high-altitude hypoxia]. Yi Chuan. 2013;35(8):992–998. Chinese. | |
Mishra A, Mohammad G, Thinlas T, Pasha MA. EGLN1 variants influence expression and SaO2 levels to associate with high-altitude pulmonary oedema and adaptation. Clin Sci (Lond). 2013;124(7):479–489. | |
Qi L, Cho YA. Gene-environment interaction and obesity. Nutr Rev. 2008;66(12):684–694. | |
HÖpfl G, Ogunshola O, Gassmann M. Hypoxia and high altitude. The molecular response. Adv Exp Med Biol. 2003;543:89–115. | |
Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–354. | |
Kubo K, Hanaoka M, Hayano T, et al. Inflammatory cytokines in BAL fluid and pulmonary hemodynamics in high-altitude pulmonary edema. Respir Physiol. 1998;111(3):301–310. | |
Ziesche R, Petkov V, Williams J, et al. Lipopolysaccharide and interleukin 1 augment the effects of hypoxia and inflammation in human pulmonary arterial tissue. Proc Natl Acad Sci U S A. 1996;93(22):12478–12483. | |
Modesti PA, Vanni S, Morabito M, et al. Role of endothelin-1 in exposure to high altitude: Acute Mountain Sickness and Endothelin-1 (ACME-1) study. Circulation. 2006;114(13):1410–1416. | |
Droma Y, Hanaoka M, Basnyat B, et al. Adaptation to high altitude in Sherpas: association with the insertion/deletion polymorphism in the Angiotensin-converting enzyme gene. Wilderness Environ Med. 2008;19(1):22–29. | |
King SJ, Booyse FM, Lin PH, Traylor M, Narkates AJ, Oparil S. Hypoxia stimulates endothelial cell angiotensin-converting enzyme antigen synthesis. Am J Physiol. 1989;256(6 Pt 1):C1231–C1238. | |
Morrell NW, Atochina EN, Morris KG, Danilov SM, Stenmark KR. Angiotensin converting enzyme expression is increased in small pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. J Clin Invest. 1995;96(4):1823–1833. | |
Oparil S, Narkates AJ, Jackson RM, Ann HS. Altered angiotensin-converting enzyme in lung and extrapulmonary tissues of hypoxia-adapted rats. J Appl Physiol (1985). 1988;65(1):218–227. | |
Liu R, Evgenov OV, Ichinose F. NOS3 deficiency augments hypoxic pulmonary vasoconstriction and enhances systemic oxygenation during one-lung ventilation in mice. J Appl Physiol (1985). 2005;98(2):748–752. | |
Steudel W, Scherrer-Crosbie M, Bloch KD, et al. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest. 1998;101(11):2468–2477. | |
San JosÉ G, FortuÑo A, Beloqui O, DÍez J, Zalba G. NADPH oxidase CYBA polymorphisms, oxidative stress and cardiovascular diseases. Clin Sci (Lond). 2008;114(3):173–182. | |
Vibhuti A, Arif E, Mishra A, et al. CYP1A1, CYP1A2 and CYBA gene polymorphisms associated with oxidative stress in COPD. Clin Chim Acta. 2010;411(7–8):474–480. | |
Wyche KE, Wang SS, Griendling KK, et al. C242T CYBA polymorphism of the NADPH oxidase is associated with reduced respiratory burst in human neutrophils. Hypertension. 2004;43(6):1246–1251. | |
Holley SL, Fryer AA, Haycock JW, Grubb SE, Strange RC, Hoban PR. Differential effects of glutathione S-transferase pi (GSTP1) haplotypes on cell proliferation and apoptosis. Carcinogenesis. 2007;28(11):2268–2273. | |
Nagai F, Kato E, Tamura HO. Oxidative stress induces GSTP1 and CYP3A4 expression in the human erythroleukemia cell line, K562. Biol Pharm Bull. 2004;27(4):492–495. | |
Vibhuti A, Arif E, Deepak D, Singh B, Qadar Pasha MA. Genetic polymorphisms of GSTP1 and mEPHX correlate with oxidative stress markers and lung function in COPD. Biochem Biophys Res Commun. 2007;359(1):136–142. | |
Yan H, Sun X, Den W, Fan X, Liu T. GSTP1, GSTM1, and GSTT1 polymorphisms in tibetan mountaineers. Biol Sport. 2006;23(3):303–311. | |
Guilluy C, Eddahibi S, Agard C, et al. RhoA and Rho kinase activation in human pulmonary hypertension: role of 5-HT signaling. Am J Respir Crit Care Med. 2009;179(12):1151–1158. | |
Frank DB, Lowery J, Anderson L, Brink M, Reese J, de Caestecker M. Increased susceptibility to hypoxic pulmonary hypertension in Bmpr2 mutant mice is associated with endothelial dysfunction in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol. 2008;294(1):L98–L109. | |
Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19(1):116–127. | |
Long L, Crosby A, Yang X, et al. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation. 2009;119(4):566–576. | |
Upton PD, Morrell NW. The transforming growth factor-β-bone morphogenetic protein type signalling pathway in pulmonary vascular homeostasis and disease. Exp Physiol. 2013;98(8):1262–1266. | |
McMurtry IF, Abe K, Ota H, Fagan KA, Oka M. Rho kinase-mediated vasoconstriction in pulmonary hypertension. Adv Exp Med Biol. 2010;661:299–308. | |
McMurtry IF, Bauer NR, Fagan KA, Nagaoka T, Gebb SA, Oka M. Hypoxia and Rho/Rho-kinase signaling. Lung development versus hypoxic pulmonary hypertension. Adv Exp Med Biol. 2003;543:127–137. | |
Vanderpool RR, Kim AR, Molthen R, Chesler NC. Effects of acute Rho kinase inhibition on chronic hypoxia-induced changes in proximal and distal pulmonary arterial structure and function. J Appl Physiol (1985). 2011;110(1):188–198. | |
Wang Z, Jin N, Ganguli S, Swartz DR, Li L, Rhoades RA. Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 2001;25(5):628–635. | |
Goerre S, Wenk M, BÄrtsch P, et al. Endothelin-1 in pulmonary hypertension associated with high-altitude exposure. Circulation. 1995;91(2):359–364. | |
Ahsan A, Mohd G, Norboo T, Baig MA, Pasha MA. Heterozygotes of NOS3 polymorphisms contribute to reduced nitrogen oxides in high-altitude pulmonary edema. Chest. 2006;130(5):1511–1519. | |
Droma Y, Hanaoka M, Basnyat B, et al. Genetic contribution of the endothelial nitric oxide synthase gene to high altitude adaptation in sherpas. High Alt Med Biol. 2006;7(3):209–220. | |
Droma Y, Hanaoka M, Ota M, et al. Positive association of the endothelial nitric oxide synthase gene polymorphisms with high-altitude pulmonary edema. Circulation. 2002;106(7):826–830. | |
Mishra A, Ali Z, Vibhuti A, et al. CYBA and GSTP1 variants associate with oxidative stress under hypobaric hypoxia as observed in high-altitude pulmonary oedema. Clin Sci (Lond). 2012;122(6):299–309. | |
Alonso D, Radomski MW. The nitric oxide-endothelin-1 connection. Heart Fail Rev. 2003;8(1):107–115. | |
Stow LR, Jacobs ME, Wingo CS, Cain BD. Endothelin-1 gene regulation. FASEB J. 2011;25(1):16–28. | |
FÖrstermann U, MÜnzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113(13):1708–1714. | |
PiÉrola J, Alemany A, YaÑez A, et al. NADPH oxidase p22phox polymorphisms and oxidative stress in patients with obstructive sleep apnoea. Respir Med. 2011;105(11):1748–1754. | |
Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31 Suppl 2:S170–S180. | |
Agapitov AV, Haynes WG. Role of endothelin in cardiovascular disease. J Renin Angiotensin Aldosterone Syst. 2002;3(1):1–15. | |
Kourembanas S, Marsden PA, McQuillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest. 1991;88(3):1054–1057. | |
Remuzzi G, Benigni A. Endothelins in the control of cardiovascular and renal function. Lancet. 1993;342(8871):589–593. | |
Iwasaki H, Eguchi S, Ueno H, Marumo F, Hirata Y. Endothelin-mediated vascular growth requires p42/p44 mitogen-activated protein kinase and p70 S6 kinase cascades via transactivation of epidermal growth factor receptor. Endocrinology. 1999;140(10):4659–4668. | |
Simonson MS, Herman WH. Protein kinase C and protein tyrosine kinase activity contribute to mitogenic signaling by endothelin-1. Cross-talk between G protein-coupled receptors and pp60c-src. J Biol Chem. 1993;268(13):9347–9357. | |
Sugawara F, Ninomiya H, Okamoto Y, et al. Endothelin-1-induced mitogenic responses of Chinese hamster ovary cells expressing human endothelinA: the role of a wortmannin-sensitive signaling pathway. Mol Pharmacol. 1996;49(3):447–457. | |
Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem. 2001;276(16):12645–12653. | |
Barden AE, Herbison CE, Beilin LJ, Michael CA, Walters BN, Van Bockxmeer FM. Association between the endothelin-1 gene Lys198Asn polymorphism blood pressure and plasma endothelin-1 levels in normal and pre-eclamptic pregnancy. J Hypertens. 2001;19(10):1775–1782. | |
Sampsonas E, Lykouras D, Drakatos P, Moschopoulou A, Spiropoulos K, Karkoulias K. Endothelin-1 polymorphisms involved in impaired exercise tolerance in COPD patients. A pilot study. Eur Rev Med Pharmacol Sci. 2011;15(2):123–128. | |
Tiret L, Poirier O, Hallet V, et al. The Lys198Asn polymorphism in the endothelin-1 gene is associated with blood pressure in overweight people. Hypertension. 1999;33(5):1169–1174. | |
Zhu G, Carlsen K, Carlsen KH, et al. Polymorphisms in the endothelin-1 (EDN1) are associated with asthma in two populations. Genes Immun. 2008;9(1):23–29. | |
Morganti A, Giussani M, Sala C, et al. Effects of exposure to high altitude on plasma endothelin-1 levels in normal subjects. J Hypertens. 1995;13(8):859–865. | |
Charu R, Stobdan T, Ram RB, et al. Susceptibility to high altitude pulmonary oedema: role of ACE and ET-1 polymorphisms. Thorax. 2006;61(11):1011–1012. | |
Rajput C, Najib S, Norboo T, Afrin F, Qadar Pasha MA. Endothelin-1 gene variants and levels associate with adaptation to hypobaric hypoxia in high-altitude natives. Biochem Biophys Res Commun. 2006;341(4):1218–1224. | |
Ricciardolo FL, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev. 2004;84(3):731–765. | |
Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci U S A. 1997;94(12):6529–6534. | |
Podesser BK, HallstrÖm S. Nitric oxide homeostasis as a target for drug additives to cardioplegia. Br J Pharmacol. 2007;151(7):930–940. | |
Bird IM. Endothelial nitric oxide synthase activation and nitric oxide function: new light through old windows. J Endocrinol. 2011;210(3):239–241. | |
Scherrer U, Sartori C, Lepori M, et al. High-altitude pulmonary edema: from exaggerated pulmonary hypertension to a defect in transepithelial sodium transport. Adv Exp Med Biol. 1999;474:93–107. | |
Wang P, Ha AY, Kidd KK, Koehle MS, Rupert JL. A variant of the endothelial nitric oxide synthase gene (NOS3) associated with AMS susceptibility is less common in the Quechua, a high altitude Native population. High Alt Med Biol. 2010;11(1):27–30. | |
Yu-jing S, Ming-wu F, Wen-quan N, et al. Endothelial nitric oxide synthase gene polymorphisms associated with susceptibility to high altitude pulmonary edema in Chinese railway construction workers at Qinghai-Tibet over 4,500 meters above sea level. Chinese Med Sci J. 2010;25(4):215–222. | |
Tesauro M, Thompson WC, Rogliani P, Qi L, Chaudhary PP, Moss J. Intracellular processing of endothelial nitric oxide synthase isoforms associated with differences in severity of cardiopulmonary diseases: cleavage of proteins with aspartate vs glutamate at position 298. Proc Natl Acad Sci U S A. 2000;97(6):2832–2835. | |
Ahsan A, Charu R, Pasha MA, et al. eNOS allelic variants at the same locus associate with HAPE and adaptation. Thorax. 2004;59(11):1000–1002. | |
Ahsan A, Norboo T, Baig MA, Qadar Pasha MA. Simultaneous selection of the wild-type genotypes of the G894T and 4B/4A polymorphisms of NOS3 associate with high-altitude adaptation. Ann Hum Genet. 2005;69(Pt 3):260–267. | |
Goliasch G, Wiesbauer F, Grafl A, et al. The effect of p22-PHOX (CYBA) polymorphisms on premature coronary artery disease (≤40 years of age). Thromb Haemost. 2011;105(3):529–534. | |
San JosÉ G, Moreno MU, OlivÁn S, et al. Functional effect of the p22phox -930A/G polymorphism on p22phox expression and NADPH oxidase activity in hypertension. Hypertension. 2004;44(2):163–169. | |
He MA, Cheng LX, Jiang CZ, et al. Associations of polymorphism of P22(phox) C242T, plasma levels of vitamin E, and smoking with coronary heart disease in China. Am Heart J. 2007;153(4):640. e1–e6. | |
Moreno MU, San JosÉ G, FortuÑo A, Beloqui O, DÍez J, Zalba G. The C242T CYBA polymorphism of NADPH oxidase is associated with essential hypertension. J Hypertens. 2006;24(7):1299–1306. | |
Montuschi P, Barnes PJ, Roberts LJ 2nd. Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18(15):1791–1800. | |
Morrow JD, Roberts LJ. The isoprostanes: their role as an index of oxidant stress status in human pulmonary disease. Am J Respir Crit Care Med. 2002;166(12 Pt 2):S25–S30. | |
Roberts LJ, Morrow JD. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med. 2000;28(4):505–513. | |
Lo HW, Stephenson L, Cao X, Milas M, Pollock R, Ali-Osman F. Identification and functional characterization of the human glutathione S-transferase P1 gene as a novel transcriptional target of the p53 tumor suppressor gene. Mol Cancer Res. 2008;6(5):843–850. | |
Van Emburgh BO, Hu JJ, Levine EA, et al. Polymorphisms in CYP1B1, GSTM1, GSTT1 and GSTP1, and susceptibility to breast cancer. Oncol Rep. 2008;19(5):1311–1321. | |
Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase Pi gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem. 1997;272(15):10004–10012. | |
CustÓdio AC, Almeida LO, Pinto GR, et al. GSTP1 Ile105Val polymorphism in astrocytomas and glioblastomas. Genet Mol Res. 2010;9(4):2328–2334. | |
Ishii T, Matsuse T, Teramoto S, et al. Glutathione S-transferase P1 (GSTP1) polymorphism in patients with chronic obstructive pulmonary disease. Thorax. 1999;54(8):693–696. | |
Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. | |
Mansiaux Y, Carrat F. Contribution of genome-wide association studies to scientific research: a bibliometric survey of the citation impacts of GWAS and candidate gene studies published during the same period and in the same journals. PLoS One. 2012;7(12):e51408. | |
Kwon JM, Goate AM. The candidate gene approach. Alcohol Res Health. 2000;24(3):164–168. | |
Zhu M, Zhao S. Candidate gene identification approach: progress and challenges. Int J Biol Sci. 2007;3(7):420–427. | |
Beall CM, Cavalleri GL, Deng L, et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci U S A. 2010;107(25):11459–11464. | |
Bigham A, Bauchet M, Pinto D, et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010;6(9):e1001116. | |
Peng Y, Yang Z, Zhang H, et al. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol Biol Evol. 2011;28(2):1075–1081. | |
Xu S, Li S, Yang Y, et al. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol Biol Evol. 2011;28(2):1003–1011. | |
Yi X, Liang Y, Huerta-Sanchez E, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329(5987):75–78. | |
Ge RL, Simonson TS, Cooksey RC, et al. Metabolic insight into mechanisms of high-altitude adaptation in Tibetans. Mol Genet Metab. 2012;106(2):244–247. | |
Simonson TS, Yang Y, Huff CD, et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329(5987):72–75. | |
Xiang K, Ouzhuluobu, Peng Y, et al. Identification of a Tibetan-specific mutation in the hypoxic gene EGLN1 and its contribution to high-altitude adaptation. Mol Biol Evol. 2013;30(8):1889–1898. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.