Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
Oxidative stress and free radicals in COPD – implications and relevance for treatment
Authors Domej W, Oetll K, Renner W
Received 8 June 2014
Accepted for publication 31 July 2014
Published 17 October 2014 Volume 2014:9(1) Pages 1207—1224
DOI https://doi.org/10.2147/COPD.S51226
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Wolfgang Domej,1 Karl Oettl,2 Wilfried Renner3
1Division of Pulmonology, Department of Internal Medicine, 2Institute of Physiological Chemistry, 3Clinical Institute of Medical and Chemical Diagnostics, Medical University of Graz, Graz, Austria
Abstract: Oxidative stress occurs when free radicals and other reactive species overwhelm the availability of antioxidants. Reactive oxygen species (ROS), reactive nitrogen species, and their counterpart antioxidant agents are essential for physiological signaling and host defense, as well as for the evolution and persistence of inflammation. When their normal steady state is disturbed, imbalances between oxidants and antioxidants may provoke pathological reactions causing a range of nonrespiratory and respiratory diseases, particularly chronic obstructive pulmonary disease (COPD). In the respiratory system, ROS may be either exogenous from more or less inhalative gaseous or particulate agents such as air pollutants, cigarette smoke, ambient high-altitude hypoxia, and some occupational dusts, or endogenously generated in the context of defense mechanisms against such infectious pathogens as bacteria, viruses, or fungi. ROS may also damage body tissues depending on the amount and duration of exposure and may further act as triggers for enzymatically generated ROS released from respiratory, immune, and inflammatory cells. This paper focuses on the general relevance of free radicals for the development and progression of both COPD and pulmonary emphysema as well as novel perspectives on therapeutic options. Unfortunately, current treatment options do not suffice to prevent chronic airway inflammation and are not yet able to substantially alter the course of COPD. Effective therapeutic antioxidant measures are urgently needed to control and mitigate local as well as systemic oxygen bursts in COPD and other respiratory diseases. In addition to current therapeutic prospects and aspects of genomic medicine, trending research topics in COPD are presented.
Keywords: reactive oxygen species, reactive nitrogen species, chronic obstructive pulmonary disease, antioxidants
Introduction
Oxygen was recognized almost 250 years ago by Carl Wilhelm Scheele (1771) and Joseph Priestley (1774), who were both studying combustion processes.1 Since the 1960s, when reactive oxygen species (ROS) were first described (Table 1), we have known that oxygen can both support and threaten life and is necessary for the generation of energy in aerobic life. Helmut Sies made an important contribution when he described oxidative stress as a potentially harmful disturbance in the oxidant–antioxidant balance in favor of the former.2 This disturbance is brought about by reactive molecules, including free radicals (FR) and non-radical reactive species, summarized as reactive species (RS) and comprising ROS and reactive nitrogen species (RNS). In addition, considerable research power was needed to understand the cross-linked connections between oxygen radical burst and related basic physiological and pathological mechanisms. In normal respiration, it is estimated that 2% of the inhaled oxygen appears in the form of ROS, of which half is suggested to damage proteins and one-quarter to damage DNA.3
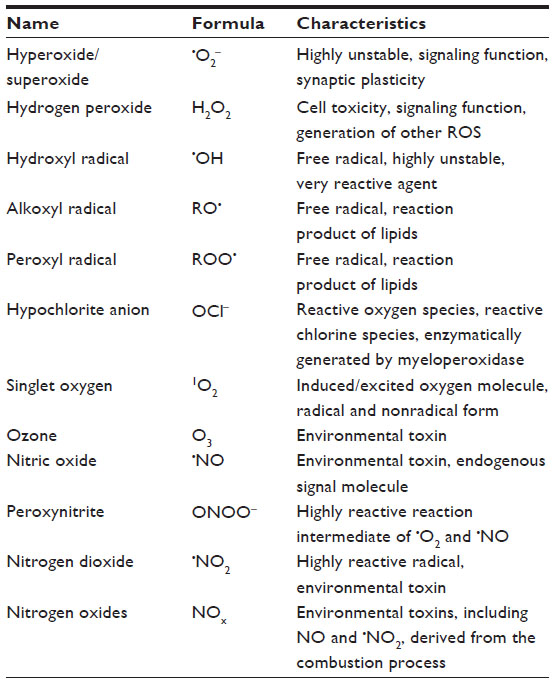 | Table 1 Free radicals (ROS, RNS) as contributors to oxidative stress |
From our present point of view, chronic obstructive pulmonary disease (COPD) is an incurable but preventable respiratory disease with high prevalence that is on the way to becoming the third most common cause of death worldwide by 2020.4 COPD is characterized by persistent progressive airflow limitation and hyperinflation, along with both systemic inflammation and chronic inflammation of the airways and lung parenchyma that is mainly caused by tobacco smoke and airborne particulate matter; both are responsible for the endogenous generation and release of oxidative stressors in the airways.5,6
Doubtless, there are still unanswered questions about the physiology and pathophysiology of the radical burden in the human body. In health, the respiratory system is balanced by both enzymatic and nonenzymatic mechanisms.7 The increased oxidative burden in COPD may contribute to a range of pathogenic processes starting with inactivation of antiproteases; enhancing bronchial inflammation by activating redox-sensitive transcription factors; mucus gland hyperplasia and hypersecretion; corticosteroid resistance; enhanced senescence; activation of neutrophils, macrophages, and fibroblasts; abnormal airway T-cell population; and small airway fibrosis culminating in direct damage to respiratory cells (apoptosis) with defective regeneration.8,9 In early stages of COPD, patients experience impairment of skeletal muscle function and physical performance due to a loss of oxidative type I muscle fibers and oxidative capacity (oxidative muscle phenotype/oxphen). In less advanced COPD, the muscle oxphen regulatory response to acute exercise is not blunted regardless of exercise-induced hypoxia.10 COPD has a special predilection for systemic involvement that may finally result in severe skeletal muscle dysfunction, muscle atrophy, sarcopenia, weight loss, and catabolic status.11,12 These frequent symptoms in advanced disease may also be explained by imbalances in the local redox system.13 Acute exacerbations of COPD triggered by viral and bacterial infections may be attributed to increased endogenous oxidants from macrophages and neutrophils sequestered from the circulating blood.14 Oxidant–antioxidant imbalances in both the lungs and the circulation, gene polymorphisms, and activation of transcription factors such as Nuclear factor kappa B NF-κB contribute as a bundle to the molecular pathogenesis of COPD.15–17 In smokers particularly, the distorted equilibrium between oxidants and antioxidants and associated pathogenic pathways may be of a high impact on the development of COPD or neoplastic lung disease and also on the search for novel therapeutic approaches.15,18
Characteristics of reactive species
Oxygen has the extraordinary ability to oxygenate other molecules; it can break chemical bonds and fragment molecules into smaller units. New radical agents are generated by electron transfer that in turn can oxidize other molecules in a self-limiting chain reaction. Molecular oxygen (O2) itself contains two unpaired electrons, making it a diradical. However, it has only weak oxidative potential compared to some of its powerful cell-damaging metabolites (Table 1).
FR are inorganic or organic chemical compounds with one or more unpaired electron(s), symbolized by an additional point (•); they tend to be highly reactive and short lived. They are generated both exogenously by a great variety of reactions and endogenously. FR may be formed by chemical cleavage of covalent bonds, when one electron each of a common electron pair then remains with the split-off piece. The hydroxyl radical (•OH) or hyperoxide/superoxide radical (•O2−) ROS are of particular biological importance. Singlet oxygen (1O2) is also a highly reactive agent generated in phagocytic processes as well as in various reactions of photosensitization; it has both a radical and nonradical form. Sulfur dioxide (SO2) itself is not a FR, but it may react to sulfite ions (SO32−) and upon reaction superoxide generates other radicals such as SO3•−, SO4•−, SO5•− that contribute to urban smog. Nitrogen dioxide (•NO2), a reactive FR, is a common indoor and outdoor gaseous pollutant (caused by combustion/traffic) that has been shown experimentally to cause epithelial damage and to induce inflammation in the airways. Chronic NO2 exposition may increase the incidence and severity of respiratory infections and worsen preexisting COPD. Similar inflammatory patterns and worsening of symptoms in patients with asthma and COPD have been demonstrated to be due to high concentrations of the oxidizing environmental pollutant ozone.19 A recent experimental study showed that mice exposed chronically to ozone developed emphysematous changes in their lungs and a loss of antioxidative stress response after 3–6 weeks of exposure.20 Nitrogen monoxide (•NO) and nitrogen dioxide (•NO2) are also highly reactive FR and main constituents of urban smog.
Hydroxyl radicals (•OH) are generated from hydrogen peroxide (H2O2), peroxyl radicals in enzymatic pathways (oxidoreductases, xanthine oxidase, nicotinamide adenine dinucleotide phosphate [NADPH] oxidase), and electron transmission from flavins and quinolones. Such reactions mainly occur in the context of inflammation processes and/or phagocytosis by macrophages. The •OH radical is one of the most reactive chemical agents. It may act as a physiological intracellular agent that, in excess, is considered to be a risk factor for several respiratory diseases (Table 1). The •OH radical may go into a reaction chain with organic molecules, where each reaction generates both a reaction product and a new free radical. Antioxidative agents can weaken and stop this chain reaction. The generation of •OH radicals is initiated by reactions of metal ions with hydrogen peroxide (Fenton reaction), ionizing radiation or by exposition to ozone. The most important sources of the superoxide radical (•O2−) are the mitochondrial respiratory chain and the endoplasmatic reticulum. In the mitochondria, the generation of •O2− is based on electron transport chains that function imprecisely and leak; that may cause a single electron transfer to oxygen. In the respiratory chain, this leaky location is mainly related to the NADH-coenzyme-Q-reductase complex. Superoxide in aqueous solution is less harmful. However, depending on its pH it may be protonated to the hydroperoxyl radical (HO•2) which is more reactive and highly soluble in a hydrophobic milieu. It can pass through membranes and has phospholipid damaging potential. In aqueous milieu, •O2− works as a reducing agent and produces H2O2 and O2 in a fast dismutation reaction. In addition, the generation of hydroxyl radicals by the reaction of H2O2 with •O2− catalyzed by transition metal ions, known as the Haber–Weiss reaction, provokes the harmful effect of superoxide. By the regeneration of bivalent iron, a precondition for the Fenton reaction, •O2− radicals also take part in the generation of alkoxyl radicals (RO•) from hydrogen peroxide. In the organism this is particularly important when lipid peroxidation causes a chain reaction that can lead to cell death.
A small amount of the iron in the body is not bound to proteins but occurs as a chelate complex with adenosine triphosphate, guanosine triphosphate, and citrate supporting the Fenton reaction as a potential source of FR. It is very likely that biochemical causes for the pathological effects are due to increased iron intake and generation of hydroxyl radicals.
ROS are generated in aerobic cells and organisms by intracellular reactions. Though most oxygen and nitrogen radicals are structurally and thermodynamically stable, their intracellular behavior varies greatly. When they react with other compounds, their chemical lifetimes can vary from very short to very long. ROS are thermodynamically stable, but their reaction properties in the cellular compartment may be very different; the lifetime of the superoxide radical ranges from micro/milliseconds up to minutes and even hours in in vitro modes. There is a wide range of radicals, with life and observation times ranging from minutes up to years.21
Besides in COPD, RS as highly reactive molecules may have a role in the pathogenesis of various diseases (eg, idiopathic pulmonary fibrosis, Adult Respiratory Distress Syndrome (ARDS), diabetes, arterial hypertension, and cancer), in damage to cell structures (carbohydrates, nucleic acids, lipids, proteins), and in general in aging.22 In lung fibroses, it was shown that there is an abundance of oxidants and inflammatory cells that may produce a significantly greater amount of ROS than in healthy controls; inflammatory cells are also upregulated in asthmatic disease, increasing radical generation.
Since inflammatory cells and FR may also inactivate important antiproteases (α1-proteinase-inhibitor, secretory leukoprotease inhibitor), the development of lung emphysema may be greatly facilitated when protease activity is overwhelmed.23 ROS are also able to inactivate growth factors such as transforming growth factor (TGF)-beta, enhancing the fibrotic changes in the above-mentioned respiratory diseases and activating matrix metalloproteinases (MMP); simultaneously, antioxidative mechanisms such as dismutase or glutathione (GSH) peroxidase are activated.
However, oxidative stress is caused not only by an increased burden of oxidants but also by a decrease of the antioxidative potential. The protective antioxidant levels are significantly depleted in alveolar macrophages of COPD patients,24 and recent studies indicate that in inflammatory respiratory diseases such as COPD, antioxidative mechanisms are not sufficiently adapted as the increase of superoxide dismutase expression is lacking, so that oxidants subsequently may take over the leading role. Oxidative stress and COPD are closely related by several mechanisms. The most important of them induces increased expression of the proinflammatory genes that contribute to chronic inflammation in COPD (Table 2).25
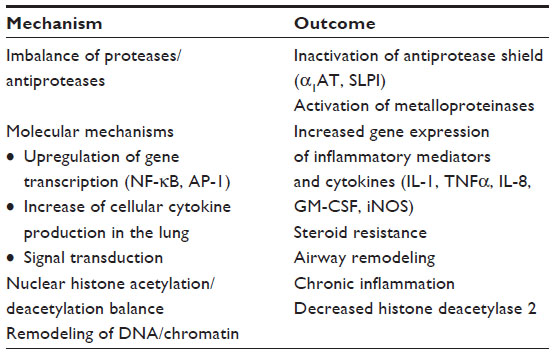 | Table 2 ROS-related hallmarks impacting COPD |
Within the endoplasmic reticulum, superoxide radicals are generated in the hydrolytic monooxygenase reactions, which transform major amounts of xenobiotics for detoxication. Hydroperoxides are stable under physiologic conditions, but may be rapidly decomposed under the catalytic impact of metals, eg, iron bound in hemoglobin, myoglobin, or cytochrome. Depending on the valence of the metal, alkoxyl radicals (R-O•) or peroxyl radicals may arise; the latter are not very reactive. Additionally cyclic peroxides may be formed from peroxide radicals. Sequential products are cyclic endoperoxides as found in prostaglandins. They are decomposed by O2, hydrolysis, or heat among other influences to aldehydes; of them, malondialdehyde (MDA) is very important as a marker for monitoring such processes. Superoxide generated by xanthine oxidase is involved in reperfusion injuries, as may occur in ischemic heart disease or cardiogenic shock. Oxygen supplementation for withdrawal from ischemic states increases the conversion of xanthine dehydrogenase to the oxidase, followed by increased generation of •O2−. Allopurinol, an inhibitor of xanthine oxidase, may be introduced when side effects of reperfusion are to be avoided.
Physiological tasks of radical species
All reactions of the different RS in the organism are closely connected. Continuous radical generation is a physiological task of cell metabolism (Figure 1).26 Besides, experimental studies have identified mitochondrial ROS as essential for O2 sensing. Inflammatory cells not only use but also release oxidizing agents (eg, myeloperoxidase [MPO] from neutrophils or eosinophilic peroxidase from eosinophilic leukocytes) to destroy microbes and to protect the organism. Thus, RS are necessary for efficient host defense against exogenous pathogens and infectious agents. In this context, RS can also react with molecular structures, rendering them dysfunctional. Since superoxide radicals participate in the pathogenesis of all kinds of membrane damage, lipid peroxidation is a central player. But superoxides may also help keep the organism functioning since they aid in maintaining defined redox potentials on the cellular level and so regulate development and differentiation processes. Synthesis of DNA requires tyrosine radicals, which are part of the subunit of ribonucleoside diphosphate reductase. RS also have the positive effect of contributing lipoxygenase to the synthesis of prostaglandins and leukotrienes. RS reactions are closely correlated to phagocytosis and RS are also vitally important for inducing apoptosis. Neutrophils and macrophages use much more oxygen to initiate phagocytic activity; at this point, a membrane-bound enzymatic complex, NADPH oxidase, is activated, which oxidizes NADPH. The released electrons may reduce oxygen to superoxide radicals, which can dismutate to hydrogen peroxide and O2. The latter may generate •OH radicals in the iron-catalyzed Haber–Weiss reaction. H2O2 and especially OH• damage RS in the bacteria walls. Phagocytic vacuoles contain the enzyme MPO, which in the presence of H2O2 and chloride ions generates hypochlorous acid (HOCl), also a highly reactive species. ROS and RNS play an important role in inflammatory processes but both function ambivalently and are not always harmful. Some of the RS are involved in important signaling pathways and have physiological tasks, eg, as signaling mediators, and the function of •NO as a potent vasodilator, neurotransmitter, and bactericide is well documented. There is increased generation of RS in inflammatory processes so that oxidative mechanisms cannot sufficiently protect •NO against oxidation; in turn, the strong oxidant and highly reactive intermediate peroxynitrite (ONOO−) is generated by diffusion-controlled reactions with •O2−. In contrast, ROS generated by macrophages in inflammatory processes may initiate intracellular signal transduction and reverse oxidation of intracellular enzymes. Then, this signaling initiates expression of cytokines and other mediators of inflammation. In this way, oxidative/antioxidative metabolism is integrated into the complex transduction of signaling pathways that are important for the understanding of the pathophysiology and progression of lung diseases.
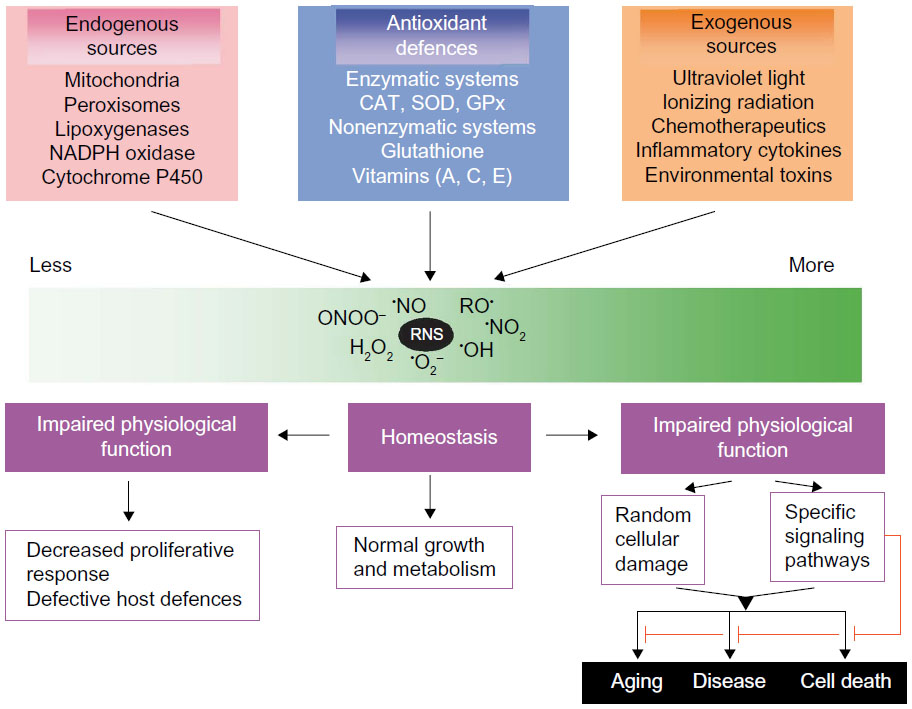 | Figure 1 Generating reactive oxygen and nitrogen species and defending against them. |
Tobacco smoke and exogenous radicals
With a large surface of 80–100 m2 and a daily breathing volume of 10,000 to 20,000 L, the lungs are very susceptible to injuries provoked by RS. The situation is drastic in smokers, since cigarette smoke, as the most important and preventable risk factor for COPD, contains about 5,000 different chemical compounds, as well as a high inhalative environmental burden of more than ten different oxidants and 15 FR.27,28 This environmental oxidant burden is augmented by additional release of endogenous oxidants from a larger number of inflammatory airway cells with high capacity for FR production, especially in COPD patients who smoke.8 Besides tobacco smoke with high contents of exogenous FR, urban (biomass fuel), industrial, and occupational air pollution provide various oxidants that probably are responsible for the high prevalence rates of COPD in nonsmokers in developing countries.29 Patients on long-term oxygen therapy are continuously exposed to large quantities of inhaled oxidants if they are supplemented with high inspiratory fractions of oxygen, but endogenously generated FR also contribute to the remarkable oxidant burden of airway structures in cigarette smokers (Table 3). One of the most important pathways to COPD is opened up by the high radical burden with a low antioxidant counterpart.30 In this context, cigarette smoke provokes a high oxidant burden and marked decrease in the antioxidant capacity even in the plasma. This is why oxidants can inactivate α1-proteinase inhibitor by reducing its ability to bind substrates like elastase from neutrophils. But the inflammatory process itself causes oxidant stress because there is great potential for excessive ROS to be generated. Thus, heavy smokers are at risk to develop COPD, suffer its impairments, and fail to respond to treatment of the disease. The initiation of the development of emphysema and COPD also points to the important role played by the strong exogenous oxidants that are inhaled in active and passive cigarette smoke (aldehydes, •NO2, SO2). Enzymes such as elastases released in excess by neutrophils are usually inactivated by the protective screen of antiproteases in the lung, so that they are not able to attack and destroy the invaluable elastic fibers in the lung. Oxidants damage the active elastase screen, and α1-proteinase inhibitor and neutrophil elastase facilitates the degradation of elastic fibers in a relentless process that ends in emphysema. An overload of exogenous and endogenous RS contributes to the imbalance in the redox status in smokers and patients with COPD. This distorted equilibrium between oxidants and antioxidants can damage the lung matrix and impair elastin synthesis as well as its repair. The potential of smoking or environmental smoke exposure to decrease antioxidants was demonstrated in a study of smokers, passive smokers, and nonsmokers. The study revealed significantly lower plasma ß-carotene and ascorbic acid but higher α-tocopherol concentrations in cigarette and passive smokers than in nonsmokers.31 The antioxidant levels found in the study population, however, were not related to differences in nutritional regimen, antioxidant intake, or other influencing factors such as age, sex, ethnicity, body mass index, alcohol consumption, or fruit and vegetable intake. Interestingly, changes in antioxidant values in plasma and erythrocytes may not be uniform in COPD patients. Radicals from cigarette smoke may also impact vitamin D metabolism in the lungs32 and so contribute to the increased prevalence of osteoporosis in smoking COPD patients.33 Recently, it has been shown that the vascular endothelial growth factor (VEGF) also seems to be involved in the pathogenesis of COPD; there is some evidence that VEGF and VEGF-2 receptor might be overexpressed in smokers with chronic bronchitis caused by upregulation of HIF-1α (hypoxia inducible factor-1α).34,35 Nrf2 (NF-E2-related factor 2), another redox-sensitive transcription factor inducing antioxidant expression, was negatively associated with the severity of COPD; novel therapeutic approaches to offset the effects of oxidative stress therefore should embrace substances that are directed toward enhancing Nrf2-regulated antioxidants36 while slowing upregulation of HIF-1α.
 | Table 3 Environmental (exogenous) and endogenous causes of oxidative stress |
Oxidative stress biomarkers
It has continued to be difficult to evaluate and monitor oxidative stress with the aim of providing needed antioxidative support. Since RNS are highly reactive, quantifying them remains an unmet challenge. A wide range of different raw materials like exhaled air, exhaled breath condensate (EBC), sputum, serum/plasma, endobronchial biopsies, or lung sections have been used to determine and quantify oxidative burden in COPD.37,38 These materials differ considerably in availability and significance. A number of different markers have been used to determine oxidative stress. One of the most commonly determined biomarkers is plasmatic hydrogen peroxide, which is increased in COPD and in acute exacerbations of the disease.38,39 The concentration of hydrogen peroxide in EBC has been measured and was also found to be elevated in COPD patients.40–42 EBC analyses using EcoScreen technology demonstrated elevated hydrogen peroxide levels and lower pH values in both COPD and asthma patients compared to healthy controls; thus, neither H2O2 nor pH correlated significantly with pulmonary function or fractional exhaled nitric oxide. Nevertheless, scores on a COPD assessment test were significantly correlated to EBC-H2O2 levels.24 Unfortunately, this biomarker is not very specific and less indicative for COPD, coinciding with values for smokers without COPD. Samples of EBC may mirror biochemical changes in the airways and particularly in the epithelial lining fluid (ELF),37 whereby the limiting factor for obtaining adequate volumes of EBC is the ventilatory capacity over a given period of time. In this context, 8-isoprostane, representing an end product of lipid peroxidation, was determined in the EBC with controversial results on the relation to COPD stages as well as the variability of levels.43 The 8-isoprostane was also shown to be elevated in EBC of COPD patients compared to controls.40,44 Sputum is obtained noninvasively and as a raw material is also of increasing interest for determining oxidative stress markers. Among other eicosanoids, 8-isoprostane was measured in sputum but unfortunately the results are inconsistent. Tendencies or significant increases during acute exacerbations of COPD compared to stable subjects have been reported.45,46 Concentrations of 8-isoprostane are also increased in induced sputum of both asymptomatic cigarette smokers and COPD patients; thus, sputum 8-isoprostane concentrations do not clearly distinguish these two groups.47 Similarly, 4-hydroxy-2-nonenal, a highly reactive lipid peroxidation product, is increased in bronchial secretions of smokers with COPD, but 4-hydroxy-2-nonenal is also elevated in smokers without COPD compared to nonsmokers.48 Sputum MPO, a further biomarker mainly expressed by neutrophils, shows higher concentrations in COPD.49 In the case of MDA, it is not always clear whether thiobarbituric acid reactive substances (TBARS) or specifically MDA have been determined. In EBC, MDA/TBARS were found to be increased50 or in the control range.40 MDA may lead to cross-linkage and aggregation of membrane proteins by reacting with amino groups in proteins, and may be seen as the most important decomposition product of clinically relevant polyunsaturated fatty acids, since there is evidence that MDA allows discrimination between healthy smokers and COPD patients.51 Further biomarkers are available from plasma or serum. TBARS/MDA levels have been reported to be in the range of controls52,53 or elevated compared to healthy controls.54–56 The carbonyl content of plasma proteins and advanced oxidation protein products are frequently used biomarkers and have been found to be increased in COPD patients;52,54,56,57 on the other hand, the thiol content of plasma proteins was reported to be lower in COPD.53,54,56 Material from endobronchial biopsies has been investigated and while protein–MDA adducts and protein nitrotyrosine were in the control range, patients were found to have increased protein carbonyl content.57 Other biomarkers of lipid peroxidation such as gaseous flow-dependent ethane and pentane58 as well as increased levels of peroxynitrite have been determined in EBC of COPD patients.59 GSH levels have been proven to be increased in the broncho-alveolar lavage fluid (BALF) of smokers and stable COPD patients compared to nonsmokers, but are reduced in acute exacerbations.60 Diagnostic broncho-alveolar lavage (BAL) procedures would be inappropriately invasive for this purpose and less sensitive in the routine assessment of oxidative stress. ELF collected by EBC could be used to monitor both oxidative stress and inflammation.83 However, valid surrogate markers (biomarkers) for oxidative burden from the plasma, BALF, or ELF are not very reliable and do not reflect the overall oxidative burden. The search is ongoing for biomarkers of oxidative stress in general and in COPD in particular that would be reproducible and reflect changes due to pathology or therapeutic intervention. Despite the many biomarkers that have been investigated for their clinical usefulness to assess oxidative stress, none has turned out to be ideal, because none of the markers of oxidative damage can really discriminate between COPD and other inflammatory lung diseases such as asthma or pneumonia. Finally, there are as yet no relevant standardized clinical methods or treatment guidelines.
Physiological antioxidative screen
The human body has a range of endogenous enzymes as well as nonenzymatic antioxidants and radical scavengers that can help to prevent damage from FR. To prevent or at least mitigate damage by RS, the human body provides a range of antioxidative agents that may neutralize oxidants or change them into less harmful substances.61 In health, both the lungs and blood are sufficiently protected by a range of nonenzymatic (eg, ascorbic acid, α-tocopherol, ferritin, uric acid) and enzymatic antioxidants such as superoxide dismutases (SOD), catalases (CAT), and peroxidases (GPx) that can react with hydrogen peroxide. There is a sufficient physiological reserve of the constituents of the antioxidant screen in the airways such as intra- and extracellular SOD or GSH. Type II alveolar cells usually have more densely packed antioxidative enzymes than type I. CAT, a highly reactive intracytoplasmic enzyme against H2O2, is contained in both alveolar macrophage type II and inflamed respiratory cells; however, it is less inducible. Cigarette smoking may deplete this antioxidant screen and downregulate antioxidant pathways associated with COPD. While GPx reacts with H2O2, GSH reductase catalyzes from the reduction of GSH disulfide (GSSG) to GSH and thus is responsible for the stabilization of the body’s most important nonenzymatic antioxidant. The GSH system (GSH/GSSG) acts as the most important redox sensor on the cellular level (physiologically >90% in reduced form). Though GSH is considered to be one of the most important nonenzymatic antioxidants, even in the human lung (high levels in ELF), GSH homeostasis depends on gross interactions with other enzymes. SOD occurs to a certain extent in all cells of the respiratory system and may convert superoxide to H2O2 and O2. SODs are named according to their metal content (CuZnSOD, MnSOD, FeSOD, extracellular SOD [EcSOD]) and represent enzymes with activity against superoxide radicals.47 Increased sputum levels of EcSOD were typically found in smokers and COPD patients,62 and polymorphisms in the EcSOD gene were found to be associated with susceptibility to emphysema rather than with COPD.63 So MnSOD has confirmed antioxidant efficacy against damage to the lung alveolar epithelium and may be induced by both TNF-α and oxidants from tobacco smoke; conversely, it can even be inactivated by high oxidative burden. Since experimental EcSOD deficiencies led to emphysema,64 both EcSOD and MnSOD are strongly suggested to have an important protective role in COPD.65 Besides, there are a few endogenous and exogenous scavengers of RS, which are a disputed therapeutic option. Best known are the vitamins C (ascorbic acid) and E (tocopherol) and the vitamin A precursor ß-carotin, along with metal-binding proteins such as transferrin, ferritin, and ceruloplasmin that may also diminish RS generation.
In a second step, hydrogen peroxide is further degraded by enzymes like CAT or GPx, resulting in H2O and O2 or GSSG, respectively. GPx can only be activated in the presence of selenocysteine as well as GSH. The cellular redox system can only function when certain proteins, redoxins, are available. At the molecular level, ROS may stimulate inflammatory cytokines by increasing their gene transcription,66 while a protective counterpart works to maintain the steady state by increased activation of genes from the antioxidative screen. The ELF of the airways forms an important functional barrier for oxidants. This thin fluid film contains dismutases, GPx, and other enzymes, along with nonenzymatic radical scavengers like vitamin E, GSH, or ferritin. Antioxidants in the ELF may be responsible for individually different resistance of the airways to oxidative stress.
Therapeutic antioxidative approaches
Besides never smoking, stopping smoking is the best way to prevent onset and progression of COPD. Mortality rates for COPD vary depending on smoking habits, general air pollution, and dietary antioxidant intake of a population. The progress of airway inflammation, oxidant, and protease burden even months/years after stopping smoking, and nonresponsiveness to steroid inhalation have been recognized as hallmarks of a therapeutic challenge in COPD.67,68 Based on our present knowledge of the pathogenesis, the activity of COPD may be diminished or controlled either by downregulation of endogenous enzymatic ROS generation or by the introduction of novel powerful antioxidative treatments using single, or better, combined antioxidant/redox modulating regimens.69 Antioxidants may have preventative as well as therapeutic potential in COPD,38 can improve overall lung antioxidant capacity, and may find use in new therapeutic approaches (Table 4). A less-considered problem in prospective antioxidative substitution in COPD might be the heterogeneity of the disease based on several subphenotypes.70 Furthermore, an association of COPD with a variety of chronic comorbidities, especially in the elderly, may impede a uniform therapeutic approach. Many agents act as controllers of NF-kB; regulate antioxidant GSH biosynthesis genes but also remodel chromatin; and are involved in inflammatory gene expression. Though several antioxidant agents have been evaluated as potential treatment candidates, none could be shown convincingly to protect against COPD and oxidative burden. Most studies investigated the administration and efficacy of single antioxidative enzymes, but it is more likely that a combination of several antioxidants would have a better treatment outcome than a single substance.71 However, treatment options may either augment endogenous antioxidant enzymatic and nonenzymatic defenses or largely suppress the generation of ROS. This should be compatible with an increased thiol status, GSH/GSSG ratio, or enzymatic activity (SOD/GPx) indicative of a decrease in radical burden. There is already ample evidence for a genetic impact in the pathogenesis of COPD, and gene polymorphisms involving antioxidant genes encoding for SOD, CAT, glutathione-S-transferase, or cytochrome P450 are involved in FR neutralization (Table 5).5
 | Table 4 Aims of successful antioxidative treatment intervention in COPD |
 | Table 5 Agents with antioxidative potential |
An effective antioxidant regimen for therapeutic intervention should aim to target cardinal symptoms of COPD, such as mucus hypersecretion, mucostasis, chronic airway inflammation, and remodeling, and should aim to overcome steroid resistance.69 From among several radical scavengers with potential to slow down rapid aging, a number of possible candidate agents have emerged that could hold off the initiation and progression of COPD.72 Though there are a few supplemental antioxidants that might contribute to a breakthrough in COPD treatment, at present, there is no antioxidant regimen whose composition and duration of treatment can be recommended without reservation. Furthermore, the complex interactions combined with a still incomplete understanding of the subtle pathophysiology of COPD frequently stand in the way of better results.25 The clinical utility of antioxidants has to be proven in further studies and more antioxidative combination regimens need to be tested in clinical trials before any treatment guidelines can be formulated (Table 5).
Further agents with antioxidative potential
GSH
Even though supplementation seems to be the logical therapeutic consequence in COPD, administration of GSH, whether by the oral, intravenous, or inhalatory route, has not proven to effectively increase its concentrations in either respiratory cells or ELF.28,73 Inhaled GSH produced some improvements in lung function, mainly in cystic fibrosis (CF) patients, but without any changes in the oxidative stress burden.74 Novel, possibly liposomal formulations to improve its bioavailability could have promise for the future.
N-acetyl-L-cysteine (NAC) and new thiols
Upon oral administration of this strong reducing agent and cellular precursor of GSH, NAC, a mucolytic, is deacetylated in the intestine to cysteine; as a thiol-containing α-amino acid and constituent of GSH, it improves both GSH redox balance and intracellular GSH synthesis in the lungs.75 However, NAC targets the replenishment of GSH in deficient cells and therefore may not be considered as an antioxidant agent in the proper sense. NAC is likely ineffective in GSH repleted cells.76 Though NAC is well tolerated, it has been found to impact COPD progression with varying success.77 In the BRONCUS (Bronchitis Randomized on NAC Cost-Utility Study) study, within 3 consecutive years, patients with COPD II and III could not significantly decrease their rate of exacerbation or slow the decline of forced expiratory volume (FEV1) versus placebo.78 In contrast, the intake of 600 mg NAC twice a day for 2 consecutive months led to a significant decrease in oxidative biomarkers in smoking COPD patients, but also to a reduction of bronchial hypersecretion and decline of FEV1.79 At the cellular level, NAC may also act as a prooxidant. NAC is ambivalent in function, which may explain the inconsistent efficacy results in several studies that were not due to methodological deficiencies alone. Nevertheless, there is strong and undebatable evidence of the clinical benefit of its mucolytic effect, and the reduction of the exacerbation rate is also convincing. In the PANTHEON study, which included 1,006 patients with moderate to severe stage II and III COPD (GOLD), the oral intake of 600 mg NAC twice a day was proven to be an efficient long-acting therapy, significantly decreasing both the exacerbation rate and duration of exacerbations independent of inhalative corticosteroids.80 But only animal studies have shown that NAC allows improvement of lung emphysema when elastase is sufficiently antagonized and α1-antitrypsin is protected against oxidation by cigarette smoke.81 Against all these conflicting results on NAC’s efficacy, its benefits include the increased concentration of the intracellular GSH, its remarkable mucolytic efficiency due to cysteine-mediated disruption of disulfide bonds in the glycoprotein matrix, and a general lack of side effects. Though some meta-analyses have shown a small but significant clinical benefit of NAC in COPD patients,82 the overall evidence for NAC in COPD remains inconclusive.83
Another thiol derivative with similar mucolytic and antioxidant properties is N-acystelyn (NAL), which was also found to fill up the intracellular GSH pool and to protect against ROS as H2O2. In healthy individuals, lung deposition of NAL was achieved via metered dose inhaler.84 NAL may be an effective therapeutic option in COPD, since it enhances intracellular GSH concentration of alveolar epithelial cells and strongly inhibits ROS formation,85 but its efficacy needs to be proven in more clinical trials. Procysteine may improve mitochondrial GSH in alveolar type II cells and serve as an adjunct therapeutic measure to improve macrophage function in COPD. The novel antioxidant and thiol derivate erdosteine is not only able to break disulfide bonds, but – when 300 mg is given twice/day for a minimum period of 8 months – is also a strong anti-inflammatory and antioxidant agent that considerably decreases the COPD exacerbation rate, significantly improving quality of life.86 The administration of this 600 mg/day significantly decreased blood ROS levels in smoking COPD patients. Erdosteine has already been widely introduced in clinical practice and is available in a number of countries worldwide due to its demonstrated benefits, particularly in patients with frequent, prolonged, and severe COPD exacerbations.87 Similar agents that are under clinical investigation are fudosteine and carbocysteine (carboxymethylcysteine [S-CMC]); in a large trial (PEACE study), the latter reduced the exacerbation rate of COPD by 25% when administered in a dosage of 1,500 mg daily for 1 year.88 A 6-month trial to demonstrate its antioxidant and anti-inflammatory potential showed that when S-CMC is given orally and activated in the gastrointestinal tract, it greatly decreases bronchial inflammation as well as the exhaled oxidant biomarkers 8-isoprostane and proinflammatory cytokines such as interleukin-6 (IL-6).89 Furthermore, ICAM-1 expression and adherence of microbes, eg, Moraxella catarrhalis and Streptococcus pneumoniae, are downregulated in vitro.90 Longer intake of S-CMC at higher dosage seems to be more effective in preventing acute exacerbations. Fudosteine, another mucolytic that strongly inhibits both mucus hypersecretion and the formation of peroxynitrite through radical scavenging, is available in several countries to treat COPD.59 Ergothioneine is an antioxidant derived from various vegetable and animal tissues. This compound was shown to inhibit ROS-mediated signaling in respiratory inflammation.91 It was recently demonstrated that the mucolytic expectorant ambroxol, commonly used to prevent acute exacerbations of COPD, enhances the plasmatic antioxidant potential as well as the levels of thioredoxin reductase at concentrations of 100–200 μM.92 Overall data on the efficacy of thiol antioxidants are inconsistent. These compounds aim rather at improving mucostasis and mucus secretion than acting on lung function itself. Nevertheless, there is evidence that carbocysteine particularly decreases COPD exacerbation rates.71
Nuclear factor 2 and Nrf2-activators
Nrf2 is a ubiquitous cytoplasmic transcription factor in normal cell populations and very important in protecting them against RS derived from tobacco smoke.71 Nrf2 concentrations in the lungs are typically decreased in COPD, and low Nrf2 levels are associated with redox modifications and degradation of histone deacetylase 2 (HDAC2), an enzyme that is involved in the regulation of steroid sensitivity.93 Compounds that activate Nrf2 may contribute to the cross-linked antioxidative screen of the lungs. Nrf2 as well as Nrf2 activators have great potential for defending against RNS in tobacco smoke, particularly in COPD patients, whose endogenous antioxidant defense has been overwhelmed and is less adaptable. Development of novel and efficient Nrf2 activators therefore should have high priority; they could then be combined with other therapeutic agents for a better outcome in COPD.94 Some synthetic and natural compounds, eg, sulforaphane (broccoli, cabbage), curcumin, or caffeic acid phenethyl ester, may induce Nrf2/ARE (antioxidative response element)-regulated gene expression; also, naturally occurring chalcones of the flavonoid family may activate the Nrf2 pathway inducing upregulation of intracellular GSH synthesis and hemoxygenase-1 activity.95
Lipid peroxidation inhibitors and protein carbonylation blockers
Neoplastic cells are remarkably resistant to lipid peroxidation, though their altered lipid composition decreases their enzymatic antioxidative screen. Their redox state, however, seems to be shifted far to the reduced side. Both edaravone and lazaroids are potent inhibitors of membrane lipid peroxidation.69 Edaravone operates as a strong FR scavenger that also may decrease carbonyl stress in COPD and so may protect against neutrophil lung infiltration and prevent pulmonary fibrosis.96 Edavarone as a novel substance may become increasingly important in lessening the severity of oxidative lung diseases and COPD. In animal studies, lazaroids were also able to protect against allergic bronchoconstriction, BAL eosinophilia in allergic sheep, and oxidative stress in hyperoxic states.97,98 Pharmacological approaches generally involve scavenging reactive aldehydes before they can carbonylate proteins. The carbonyl scavengers included some interesting compounds like the antihypertensive agents hydralazine (scavenger of acrolein) and captopril (scavenger of MDA), or carnosine. In a rat COPD model, overexpressed angiotensin converting enzyme was able to reduce oxidative stress and inflammation induced by cigarette smoke by inhibiting the NF-kB and p38 MAPK pathways.99 All these drugs have well known pharmacokinetically defined profiles, but neither short- nor long-term proven impact on COPD.
Dietary antioxidant polyphenols and vitamins
Plant polyphenols including the widespread group of flavonoids (flavone, flavonols, isoflavones) are well known anti-inflammatory and antioxidant agents with one or more hydroxyl groups, and if taken regularly may reduce risk of COPD.100 With increased natural dietary intake of polyphenols (catechin/green tea polyphenols, flavonol/quercetin, flavone/apigenin), COPD patients can improve their symptoms, lung function (FEV1),101 and arterial oxygen tension.102 Recently, the flavonol epicatechin contained in cocoa was found to reduce intracellular oxidative stress and to prevent glucocorticoid resistance.103 Other polyphenols like resveratrol, found in grapes and a proven strong inhibitor of inflammatory cytokine release from neutrophils and macrophages, are known to have a protective antioxidant effective in coronary heart disease. Since it has been shown that resveratrol improves GSH synthesis in respiratory epithelial cells by activating the Nrf2-pathway,104 its antioxidant benefit is also related to the scavenging of FR contained in cigarette smoke. Resveratrol was able to improve both corticosteroid efficacy and lung function in a general population.105 This suggests a positive association between moderate consumption of wine and high FEV1 and FEV1/FVC (forced vital capacity) ratio. In a similar way, curcumin, a phenolic yellowish pigment and ingredient of turmeric (rhizome of Curcuma longa), has been found to modulate NFκB, cyclooxygenase-2 expression, and neutrophil migration in the airways.106 Curcumin has long been known to be a potent scavenger of various endogenously generated RS (superoxide radicals, hydrogen peroxide, nitrogen centered radicals) and a modulator of antioxidant enzymes (SOD, CAT, GPx). Further, it has antibacterial, antiviral, antifungal, antineoplastic, and distinct anti-inflammatory properties. Still other useful characteristics are an ability to restore impaired steroid responsiveness in COPD by maintaining histone deacetylase activity in monocytes and to improve a reduced intracellular GSH level.106,107 Nanoparticle-encapsulated curcumin may provide novel therapeutic perspectives with modified absorption and improved bioavailability combined with greater efficacy in inhibiting NF-κB and greater suppression of NF-κB-regulated proteins that are, for instance, involved in angiogenesis or cell proliferation.108 The category of catechins (monomeric flavonols) has also proven to be effective in FR scavenging. Catechins, in particular vitamins C and E and further antioxidants (CAT or SOD), may contribute to the total antioxidant status of the body. Among the various green tea polyphenols, epigallocatechin gallate was identified as the most bioactive compound modulating inflammatory pathways and the expression of major inflammatory cytokines (IL-8).109 Like sulforaphane, epigallocatechin gallate might have future promise as a therapeutic option in COPD. The former occurs naturally in many vegetables including broccoli, kohlrabi, radish, cabbage, cauliflower, or mustard and is a potent antioxidant with additional antibiotic attributes that enhance bacterial elimination by macrophages.71 Sulforaphane can also improve corticosteroid sensitivity in patients with COPD.110 It is suggested that sulforaphane together with curcumin or phenyl-isothiocyanate provides a greater treatment benefit by downregulating inflammatory gene expression more effectively. Even if such therapeutic strategies are still in the developmental stage, they could have potential for treating COPD.111 There is also ample evidence that quercetin, a flavonol contained in apples, onions, green tea, and capers, has potent anti-inflammatory and antioxidant properties. In experimental studies in mice, quercetin prevented lipid peroxidation, expression of MMPs (MMP9 and 12), and development of emphysema,112 while also inhibiting influenza virus infection.113 Plant components such as flavonoids (fruits, vegetables) or lycopenes (tomato) also provide important dietary antioxidants. While flavonoids can protect cells from lipid peroxidation, lycopenes may downregulate inflammatory responses induced by cigarette smoke and also modulate signal transduction pathways of inflammation.114 Further studies on the apparently beneficial effect of lycopene in COPD would be desirable since tomato juice prevented lung damage in the offspring of female rats exposed to nicotine.115
Acai, from the acai berry indigenous to South America, was demonstrated to have high antioxidant capacity due to its high polyphenol, flavonol, and anthocyanin content. Ingestion of acai pulp has proven beneficial due to its high superoxide scavenging property with downregulating effects on inflammation through inhibition of cycloxygenases 1 and 2, NO generation, and inducible nitric oxide synthase isoenzymes (iNOS) activity.116
Unfortunately, the efficacy of many polyphenols both in vivo and in vitro is limited either by low bioavailability or by transformation in the gastrointestinal tract, where some may even become toxic or change their biological effects. A 12-week diet emphasizing fruits and vegetables failed to produce any significant effect on oxidative stress or airway inflammation in 75 patients with moderate to severe COPD.117 Long-term studies with dietary intake of polyphenols and their influence on COPD progression have yet to be made. There is some evidence that high intake of nutraceuticals is beneficial for cough, mucus production, or shortness of breath in COPD but there is no detailed information on the pharmacokinetics and bioavailability behind this. An interesting aspect is that dietary intake of minerals, mainly calcium, was found to be positively associated with a lower prevalence rate of COPD, while there was a negative relation for iron substitution.118
Vitamin D is likely linked to reversal of steroid resistance as well as airway remodeling. Since vitamin D regulates several genes, it is also involved in immune responses, inflammation, and cell proliferation, differentiation, and apoptosis. In this context, vitamin D acts as a ligand for nuclear hormone vitamin D receptor (VDR),119 controls a range of cellular functions, and is associated with accelerated impairment of lung function, particularly with the typically low VDR levels in the lungs of COPD patients. Animal studies using VDR knock-out mice saw increased influx of inflammatory cells, phosphoacetylation of NF-kB, increase of pro-inflammatory mediators, and upregulation of MMPs in the lung.120 So there are rational causes for vitamin D supplementation to prevent as well as to treat COPD. Vitamin E supplementation may decrease carbonyl and MDA levels in smokers and in mice with emphysema,121 and the risk for COPD by up to 10%.122 A positive association between intake of vitamin E and lung function was reported in a study population of 2,633 individuals.123 Overall, the intake of vitamin antioxidants may modulate oxidative stress in COPD, but general supplementation of vitamin C (ascorbic acid), vitamin E (tocopherols), provitamin A (ß-carotene), or selenium to augment the antioxidant screen and downregulate ROS-triggered inflammation seems to be only rational when pools are greatly depleted,11 yet such vitamin supplementation has so far failed to meet expectations. The majority of clinical trials aiming to improve the antioxidant status in COPD patients have been disappointing25 and it remains unclear whether dietary or pharmacological measures would improve endogenous antioxidant enzyme defense and enhance nonenzymatic defense. Since the studies were also controversial, it must be assumed that oral vitamin supplementation alone does not sufficiently target damaged lung regions (Table 3). Dietary antioxidant supplementation could potentially improve and support antioxidative defense mechanisms in the body but should only be instituted when lab work has substantiated low levels of antioxidants or their depletion.
Antioxidative pharmacological mimetics
Specialized small molecules may mimic a decreased or depleted intracellular antioxidant screen (SOD, CAT, GPx). Experimental agents with SOD-mimicking activity include salens, manganese-metalloporphyrins, and manganese-containing macrocyclic ligands;124 of them, the latter two seem to be very promising, with significant antioxidant and anti-inflammatory enzymatic properties that could be useful, even in COPD. Some SOD mimetics (manganese metalloproteins) are found to have additional catalase-like activity, neutralizing intracellular H2O2 and decomposing peroxynitrite (ONOO−).125 These antioxidative enzymatic properties have only been studied in models of airway inflammation, but there is an urgent need for novel recombinant SOD mimetics to prevent airway inflammation due to neutrophils and to decrease inflammatory cytokines and lipid peroxidation with inhaled tobacco smoke. Another challenge is to develop novel pharmacological mimetics with extracellular SOD activity, since EcSOD was found to attenuate lung inflammation and emphysema in mouse models exposed to cigarette smoke by decreasing the oxidative fragmentation of the extracellular matrix proteins and protecting against oxidative posttranslational modification of elastin.71,126 GSH peroxidase mimetics, eg, the selenium-based strong antioxidant ebselen, may decompose peroxynitrite and improve RS-induced inflammation by increasing GSH efficiency.127 Glutathione peroxidase-1 (GPx-1) may protect lungs from oxidative damage derived from cigarette smoke.28,128 In an experimental study, the GPx mimetic ebselen was able to inhibit smoke-induced airway inflammation, as shown with a decrease in the inflammatory cell content of BALF (macrophages, neutrophils, proteolytic burden and macrophages and neutrophil chemotactic factor gene expression) when administered prophylactically, but treatment with ebselen was also able to inhibit BALF inflammation.129
Another class of antioxidant enzyme mimetics with reported catalytic potential against peroxynitrite (peroxynitrite decomposition catalysts) in animal studies has to be investigated for its efficacy in alleviating the typically large peroxynitrite burden in COPD.130 However, except for peroxynitrite decomposition catalysts there is no evidence today that these compounds will ever be applicable to lung injuries induced by tobacco smoke.
Inhibition of MPO and nitric oxide synthase
MPO, a bactericidal enzyme from neutrophils and macrophages, is increased in mice exposed to cigarette smoke, and in the respiratory tract of patients with COPD as a consequence of ROS-mediated NF-κB activation.131 Synthetic 2-thioxanthines revealed strong potential to inhibit MPO.132 In response to cigarette smoke, 2-thioxanthines were shown to interrupt both progression of emphysema and small airways remodeling, but only in animal studies.133 MPO inhibitors including the caffeine metabolite 1,7-dimethylxanthine have therapeutic potential and may have a promising future as treatment for inflammation in COPD. In contrast to the decreased plasmatic NO level in smokers, iNOS are induced in lungs of COPD patients.134 Nitric oxide synthase (NOS) inhibitors reduced progression of emphysema experimentally and could be another future therapeutic option.135 These agents decrease NO availability and so reduce the S-nitrosylation of HDAC2 that occurs in COPD.136 Unfortunately, this mechanism seems not to affect HDAC2 carbonylation by cigarette smoke. None of the compounds of this class can yet be safely applied in ROS-mediated chronic lung diseases and steroid resistance in COPD, and require further study.
It is therefore suggested that selective iNOS-inhibitors with concomitant supplementation of antioxidant agents from other classes could constitute a future strategy for severe COPD aimed to slow progression of emphysema.135
Antioxidants of vegetable origin and antioxidant agents partly under investigation
The application of melatonin and dehydroepiandrosterone has been recommended, especially in USA. Both hormones were thought to be useful for FR scavenging and immune stimulation, and to slow aging.137,138 There could be future therapeutic use for the group of nitrogenous pterins, which also scavenge FR and alleviate oxidative stress on immune cells. Pterins are also used clinically as immunological biomarkers. Cigarette additives from the stone of the acai berry were able to reduce the harmful effects of cigarette smoke in mice (inflammation, emphysema) through the antioxidant enzymes MPO and GSH; in addition, neutrophil and macrophage elastase levels were significantly reduced.139 Apocynin, an organic compound of the small plant Picrorhiza kurroa and structural relative of vanillin was shown to have the anti-inflammatory property of inhibiting NADPH oxidase in an asthma model.140 Also, nebulized apocynin seems to be effective and safe as shown in asthmatics.141 Omega-3 polyunsaturated fatty acids are also known to be beneficial against oxidative stress and inflammatory mediators in smokers.142 In this context, an association between dietary intake of fatty acids, FEV1, and respiratory disease has been demonstrated.143 A novel group of proresolving mediators (metabolites of arachidonic acid and omega-3 fatty acids) inhibit inflammation, granulocyte invasion, and monocyte recruitment.
Spin traps and thioredoxin (Trx) mimetics
Spin traps have been developed to scavenge highly reactive FR and to form more stable radicals for accumulation and detection by electron spin resonance spectroscopy, and so may act as antioxidants. Most of the spin traps have a nitrone or nitroxide structure and are derivates of phenyl-based nitrones (eg, a-phenyl-N-tert-butyl nitrone), which have been shown to be beneficial in a variety of lung diseases, though only in animal studies.144 Besides the radical scavenging effects, spin trap compounds in vivo may impact on enzymes that probably play a more important therapeutic role than radical scavenging. Their efficacy in COPD, however, has not yet been established.
Trxs are approximately 12-kDa polypeptides and ubiquitous in nature. They belong to the oxidoreductase family and act as antioxidant enzymes; they are important for the metabolism of peroxides because they facilitate the reduction of H2O2 and organic hydroperoxides, and regulate the cell redox state and growth. Recently, synthesized Trx mimetics have been shown to induce an upregulation of various redox-sensitive nuclear processes. Upregulation of Trx by some small synthetic molecules has been suggested to have therapeutic potential for airway diseases including COPD,145 though high levels of Trx expression have been associated with aggressive cancers and poor prognosis.
Improvement of steroid responsiveness
Corticosteroids provide a major anti-inflammatory treatment option in airway disease and are highly beneficial in COPD patients suffering from high exacerbation rates. Corticosteroids suppress inflammatory gene expression by inhibiting histone acetyltransferase and by recruiting HDAC2 to the NF-κB-activated gene complex. Oxidative stress may decrease the anti-inflammatory effects of corticosteroids as well as the steroid sensitivity itself. HDAC2, which usually suppresses inflammatory gene expression, is reduced in lung tissue of patients with COPD, and so prevents corticosteroids from suppressing inflammation in the lung;146 the high prevalence of steroid resistance in COPD hence is due to oxidation/S-nitrosylation of the HDAC2 at Cys-262 and Cys-274.136 The inability to control lung inflammatory response is dependent on the Nrf2–HDAC2 axis.93 However, a reversal of HDAC2 does not function if HDAC2 is carbonylated by cigarette smoke.
Therapeutic agents that could function as add-ons to improve steroid responsiveness in COPD might be a desirable therapeutic goal for the future. Nrf2-activators such as sulforaphane may improve steroid sensitivity in COPD, and NO-scavengers might also provide a future therapeutic option for reversing S-nitrosylation of HDAC2. Several other antioxidant compounds may enhance the efficacy of corticosteroids by mitigating endogenous oxidants and aldehydes.131 The flavonol epicatechin in cocoa was also found to ameliorate intracellular oxidative stress as well as the development of steroid resistance and may provide a strong rationale for dietary intake as an antioxidant.103 In this context, the polyphenol curcumin was shown to inhibit inflammation and to restore steroid sensitivity upon oxidative stress by maintaining HDAC2 activity in monocytes. Direct HDAC2-activating agents may be of high clinical impact as treatment for corticosteroid resistance in inflammatory lung disease including COPD. It is suggested that glucocorticosteroids together with polyphenols could become a strong therapeutic tool.
Pulmonary rehabilitation
Besides pharmacological treatment, the nonpharmacological treatment of COPD includes smoking cessation, pulmonary rehabilitation (PR), and nutritional support. PR is a multidisciplinary approach aiming to decrease dyspnea and improve physical exercise capacity and quality of life. However, patients with COPD show increased pulmonary and systemic oxidative burden both at rest and upon provocation by physical exercise, respectively.147 In this context the extraordinary value of pulmonary rehabilitation is beyond question and rehabilitation programs are an important issue in COPD, since PR may lead to better tolerance of physical exercise (peak workload) and concomitantly to a decrease in exercise-induced oxidative damage.148
Future (genomic) medicine in COPD
While cigarette smoking is clearly the number-one risk factor in the development of COPD, recent research suggests that genetic influences may also play an important role and could explain why some smokers develop COPD while others do not. The marked variability in lung function and risk of COPD in smokers together with studies of familial aggregation further support an important role for genetics in COPD.149,150
Although COPD is likely a genetically codetermined disease based on subtle differences in COPD phenotypes, α1-antitrypsin deficiency (eg, protease inhibitor Z) still remains the only proven genetic risk factor for COPD. Even among protease inhibitor Z individuals, there is substantial variability in lung function, suggesting that other genetic modifiers may influence the expression of lung disease in severe α1-antitrypsin deficiency.151 The variable development of COPD in smokers without α1-antitrypsin deficiency and the familial aggregation of lung-function measurements also suggest genetic influences on lung-function decline leading to COPD. Only a limited number of COPD pharmacogenetic studies have been published to date, with inconclusive results.152 Many candidate gene loci have been investigated as potential COPD genetic determinants by case-controlled genetic association studies, but again mostly with inconsistent results.153 Two potential reasons for the conflicting findings between association studies may be genetic heterogeneity and population stratification. Further clinical studies should focus on patients with specific COPD-related subphenotypes, such as predominant emphysema, bronchitis, or frequent exacerbation subtype, because potential novel antioxidative agents might have different effects on different types of COPD.120 Recently published linkage analysis studies have identified regions of the genome that contain COPD susceptibility genes. Future investigations of genetic impact in COPD should consider using family-based designs for association studies and studying positional candidate genes within regions of linkage.154 Genetic studies may also help to refine these COPD subtypes by identifying genetic variants associated with specific disease phenotypes. Furthermore, gene-expression profiling may also be an important method to distinguish molecular subtypes of COPD.
Disclosure
The authors report no conflicts of interest in this work.
References
Pilgrim E. Die Entdeckung der Elemente [The discovery of the elements]. Stuttgart: Mundus Verlag; 1950. German. | |
Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82(2):291–295. | |
Acworth IN, Bailey B. The Handbook of Oxidative Metabolism. Chelmsford (MA): ESA, Inc.; 1996. | |
Lopez AD, Murray CC. The global burden of disease, 1990–2020. Nat Med. 1998;4:1241–1243. | |
Lakhdar R, Denden S, Kassab A, et al. Update in chronic obstructive pulmonary disease: role of antioxidant and metabolizing gene polymorphisms. Exp Lung Res. 2011;37(6):364–375. | |
Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176(6):532–555. | |
Halliwell B. Antioxidants in human health and disease. Annu Rev Nutr. 1996;16:33–50. | |
MacNee W. Oxidants and COPD. Curr Drug Targets Inflamm Allergy. 2005;4(6):627–641. | |
Hansel TT, Barnes PJ. New drugs for exacerbations of chronic obstructive pulmonary disease. Lancet. 2009;374(9691):744–755. | |
Slot IG, van den Borst B, Hellwig VA, Barreiro E, Schols AM, Gosker HR. The muscle oxidative regulatory response to acute exercise is not impaired in less advanced COPD despite a decreased oxidative phenotype. PLoS One. 2014;9(2):e90150. | |
Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med. 1996;154(4 Pt 1):1055–1060. | |
Barreiro E. Protein carbonylation and muscle function in COPD and other conditions. Mass Spectrom Rev. 2014;33(3):219–236. | |
Langen RC, Korn SH, Wouters EF. ROS in the local and systemic pathogenesis of COPD. Free Radic Biol Med. 2003;35(3):226–235. | |
MacNee W, Wiggs B, Belzberg AS, Hogg JC. The effect of cigarette smoking on neutrophil kinetics in human lungs. N Engl J Med. 1989;321(14):924–928. | |
Repine JE, Bast A, Lankhorst I. Oxidative stress in chronic obstructive pulmonary disease. Oxidative Stress Study Group. Am J Respir Crit Care Med. 1997;156(2 Pt 1):341–357. | |
Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–280. | |
MacNee W, Rahman I. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S58–S65. | |
MacNee W. Oxidative stress and lung inflammation in airways disease. Eur J Pharmacol. 2001;429:195–207. | |
Dinh QT, Suhling H, Fischer A, Braun A, Welte T. [Innervation of the airways in asthma bronchiale and chronic obstructive pulmonary disease (COPD)]. Pneumologie. 2011;65(5):283–292. German. | |
Wiegman CH, Li F, Clarke CJ, et al. A comprehensive analysis of oxidative stress in the ozone-induced lung inflammation mouse model. Clin Sci (Lond). 2014;126(6):425–440. | |
Domej W, Földes-Papp Z, Flögel E, Haditsch B. Chronic obstructive pulmonary disease and oxidative stress. Curr Pharm Biotechnol. 2006;7(2):117–123. | |
Birben E, Sahiner UM, Sackesen C, Erzurum S, Katayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5(1):9–19. | |
MacNee W, Tuder RM. New paradigms in the pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6(6):527–531. | |
Täger M, Piecyk A, Köhnlein T, Thiel U, Ansorge S, Welte T. Evidence of a defective thiol status of alveolar macrophages from COPD patients and smokers. Chronic obstructive pulmonary disease. Free Radic Biol Med. 2000;29(11):1160–1165. | |
Loukides S, Bakakos P, Kostikas K. Oxidative stress in patients with COPD. Curr Drug Targets. 2011;12(4):469–477. | |
Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–247. | |
Churg DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126. | |
Vlahos R, Bozinovski S. Glutathione peroxidase-1 as a novel therapeutic target for COPD. Redox Rep. 2013;18(4):142–149. | |
Behrendt CE. Mild and moderate-to-severe COPD in nonsmokers: distinct demographic profiles. Chest. 2005;128(3):1239–1244. | |
Mak JC. Pathogenesis of COPD. Part II. Oxidative-antioxidative imbalance. Int J Tuberc Lung Dis. 2008;12(4):368–374. | |
Dietrich M, Block G, Norkus EP, et al. Smoking and exposure to environmental tobacco smoke decrease some plasma antioxidants and increase gamma-tocopherol in vivo after adjustment for dietary antioxidant intake. Am J Clin Nutr. 2003;77(1):160–166. | |
Hansdottir S, Monick MM, Lovan N, Powers LS, Hunninghake GW. Smoking disrupts vitamin D metabolism in the lungs. Am J Respir Crit Care Med. 2010;181(Meeting Abstracts):A1425. | |
Dimai HP, Domej W, Leb G, Lau KH. Bone loss in patients with untreated chronic obstructive pulmonary disease is mediated by an increase in bone resorption associated with hypercapnia. J Bone Miner Res. 2001;16(11):2132–2141. | |
Lee SH, Lee SH, Kim CH, et al. Increased expression of vascular endothelial growth factor and hypoxia inducible factor-1a in lung tissue of patients with chronic bronchitis. Clin Biochem. 2014;47(7–8):552–559. | |
Zepeda AB, Pessoa A Jr, Castillo RL, Figueroa CA, Pulgar VM, Farías JG. Cellular and molecular mechanisms in the hypoxic tissue: role of HIF-1 and ROS. Cell Biochem Funct. 2013;31(6):451–459. | |
Malhotra D, Thimmulappa RK, Mercado N, et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocortiosteroid sensitivity in macrophages from COPD patients. J Clin Invest. 2011;121(11):4289–4302. | |
Louhelainen N, Myllärniemi M, Rahman I, Kinnula VL. Airway biomarkers of the oxidant burden in asthma and chronic obstructive pulmonary disease: current and future perspectives. Int J Chron Obstruct Pulmon Dis. 2008;3(4):585–603. | |
Lin JL, Thomas PS. Current perspectives of oxidative stress and its measurement in chronic obstructive pulmonary disease. COPD. 2010;7(4):291–306. | |
Gerritsen WB, Asin J, Zanen P, van den Bosch JM, Haas FJ. Markers of inflammation and oxidative stress in exacerbated chronic obstructive pulmonary disease patients. Respir Med. 2005;99(1):84–90. | |
Inonu H, Doruk S, Sahin S, et al. Oxidative stress levels in exhaled breath condensate associated with COPD and smoking. Resp Care. 2012;57(3):413–419. | |
Antczak A, Ciebiada M, Pietras T, Piotrowski WJ, Kurmanowska Z, Górski P. Exhaled eicosanoids and biomarkers of oxidative stress in exacerbation of chronic obstructive pulmonary disease. Arch Med Sci. 2012;8(2):277–285. | |
Murata K, Fujimoto K, Kitaguchi Y, Horiuchi T, Kubo K, Honda T. Hydrogen peroxide content and pH of expired breath condensate from patients with asthma and COPD. COPD. 2014;11(1):81–87. | |
Kostikas K, Papatheodorou G, Psathakis K, Panagou P, Loukides S. Oxidative stress in expired breath condensate of patients with COPD. Chest. 2003;124(4):1373–1380. | |
Ko FW, Lau CY, Leung TF, Wong GW, Lam CW, Hui DS. Exhaled breath condensate levels of 8-isoprostane, growth related oncogene alpha and monocyte chemoattractant protein-1 in patients with chronic obstructive pulmonary disease. Resp Med. 2006;100:630–638. | |
Tufvesson E, Ekberg M, Bjermer L. Inflammatory biomarkers in sputum predict COPD exacerbations. Lung. 2013;191:413–416. | |
Drozdovsky O, Barta I, Antus B. Sputum eicosanoid profiling in exacerbations of chronic obstructive pulmonary disease. Respiration. 2014;87:408–415. | |
Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167(12):1600–1619. | |
Rahman I, van Schadewijk AA, Crowther AJ, et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in the lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(4):490–494. | |
Metso T, Rytilä P, Peterson C, Haahtela T. Granulocyte markers in induced sputum in patients with respiratory disorders and healthy persons obtained by two sputum-processing methods. Respir Med. 2001;95(1):48–55. | |
Bartoli ML, Novelli F, Costa F, et al. Malondialdehyde in exhaled breath condensate as a marker of oxidative stress in different pulmonary diseases. Mediators Inflamm. 2011;2011:891752. | |
Corradi M, Rubinstein I, Andreoli R, et al. Aldehydes in exhaled breath condensate of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167(10):1380–1386. | |
Stanojkovic I, Kotur-Stevuljevic J, Milenkovic B, et al. Pulmonary function, oxidative stress and inflammatory markers in severe COPD exacerbation. Resp Med. 2011;105 Suppl 1:S31–S37. | |
Ben Moussa B, Sfaxi I, Tabka Z, Ben Saad H, Rouatbi S. Oxidative stress and lung function profiles of male smokers free from COPD compared to those with COPD: a case-control study. Libyan J Med. 2014;9:23873. | |
Ahmad A, Shameem M, Husain Q. Altered oxidant-antioxidant levels in the disease prognosis of chronic obstructive pulmonary disease. Int J Tuberc Lung Dis. 2013;17(8):1104–1109. | |
Woźniak A, Górecki D, Szpinda M, Mila-Kierzenkowska C, Woźniak B. Oxidant-antioxidant balance in the blood of patients with chronic obstructive pulmonary disease after smoking cessation. Oxid Med Cell Longev. 2013;2013:897075. | |
Nadeem A, Raj HG, Chhabra SK. Increased oxidative stress and altered levels of antioxidants in chronic obstructive pulmonary disease. Inflammation. 2005;29(1):23–32. | |
Barreiro E, Fermoselle C, Mateu-Jimenez M, et al. Oxidative stress and inflammation in the normal airways and blood of patients with lung cancer and COPD. Free Radic Biol Med. 2013;65:859–871. | |
Paredi P, Kharitonov SA, Leak D, Ward S, Cramer D, Barnes PJ. Exhaled ethane, a marker of lipid peroxidation, is elevated in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162(2 Pt 1):369–373. | |
Osoata GO, Hanazawa T, Brindicci I, et al. Peroxynitrite elevation in exhaled breath condensate of COPD and its inhibition by fudosteine. Chest. 2009;135(6):1513–1520. | |
Drost EM, Swarski KM, Sauleda J, et al. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax. 2005;60(4):293–300. | |
Biswas SK, Rahman I. Environmental toxicity, redox signaling and lung inflammation: the role of glutathione. Mol Aspects Med. 2009;30(1–2):60–76. | |
Regan EA, Mazur W, Meoni E, et al. Smoking and COPD increase sputum levels of extracellular superoxide dismutase. Free Radic Biol Med. 2011;51(3):726–732. | |
Sorheim IC, DeMeo DL, Washo G, et al. Polymorphisms in the superoxide dismutase-3 gene are associated with emphysema in COPD. COPD. 2010;7(4):262–268. | |
Yao H, Arunachalam G, Hwang JW, et al. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci U S A. 2010;107(35):15571–15576. | |
Gongora MC, Lob HE, Landmesser U, et al. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. Am J Pathol. 2008;173(4):915–926. | |
Rahman I, MacNee W. Role of transcription factors in inflammatory lung disease. Thorax. 1998;53(7):601–612. | |
Louhelainen N, Rytilä P, Haahtela T, Kinnula VL, Djukanovic R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulm Med. 2009;9:25. | |
Louhelainen N, Stark H, Mazur W, Rytilä P, Djukanovic R, Kinnula VL. Elevation of sputum matrix metalloproteinease-9 persists up to 6 months after smoking cessation: a research study. BMC Pulm Med. 2010;10:13. | |
Rahman I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim Biophys Acta. 2012;1822(5):714–728. | |
Makita H, Nasuhara Y, Nagai K, et al. Characterisation of phenotypes based on severity of emphysema in chronic obstructive pulmonary disease. Thorax. 2007;62(11):932–937. | |
Biswas S, Hwang JW, Kirkham PA, Rahman I. Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr Med Chem. 2013;20(12):1496–1530. | |
Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135(1):173–180. | |
Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000;16(3):534–554. | |
Griese M, Ramakers J, Krasselt A, et al. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am J Respir Crit Care Med. 2004;169:822–828. | |
Black PN, Morgan-Day A, McMillan TE, Poole PJ, Young RP. Randomized, contolled trial of N-acetylcysteine for treatment of acute exacerbations of chronic obstructive pulmonary disease [ISRCTN21676344]. BMC Pulm Med. 2004;4:13. | |
Rushworth GF, Megson IL. Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmakol Ther. 2014;141(2):150–159. | |
Dekhuijzen PN, van Beurden WJ. The role for N-acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):99–106. | |
Decramer M, Rutten-van Mölken M, Dekhuijzen PN, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trial. Lancet. 2005;365(9470):1552–1560. | |
Stey C, Steurer J, Bachmann S, Medici TC, Tramèr MR. The effect of oral N-acetylcysteine in chronic bronchitis: a quantitative systematic review. Eur Respir J. 2000;16(2):253–262. | |
Zheng JP, Wen FQ, Bai CX, et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON); a randomised, double-blind placebo-controlled trial. Lancet Respir Med. 2014;2(3):187–194. | |
Rubio ML, Martin-Mosquero MC, Ortega M, Peces-Barba G, González-Mangado N. Oral N-acetylcysteine attenuates elastase-induced pulmonary emphysema in rats. Chest. 2004;125(4):1500–1506. | |
Grandjean EM, Berthet P, Ruffmann R, Leuenberger P. Efficacy of oral long-term N-acetylcysteine in chronic bronchopulmonary disease: a meta-analysis of published double-blind, placebo-controlled clinical trials. Clin Ther. 2000;22(2):209–221. | |
Turner RD, Bothamley GH. N-acetylcysteine for COPD: the evidence remains inconclusive. Lancet Respir Med. 2014;2(4):e3. | |
Hardy JG, Newman SP, Knoch M. Lung deposition from four nebulizers. Respir Med. 1993;87(6):461–465. | |
Gillissen A, Jaworska M, Orth M, et al. Nacystelyn, a novel lysine salt of N-acetylcysteine, to augment cellular antioxidant defence in vitro. Respir Med. 1997;91(3):159–168. | |
Moretti M, Bottrighi P, Dallari R, et al. The effect of long-term treatment with erdosteine on chronic obstructive pulmonary disease: the EQUALIFE Study. Drugs Exp Clin Res. 2004;30(4):143–152. | |
Cazzola M, Floriani I, Page CP. The therapeutic efficacy of erdosteine in the treatment of chronic obstructive bronchitis: a meta-analysis of individual patient data. Pulm Pharmacol Ther. 2010;23(2):135–144. | |
Zheng JP, Kang J, Huang SG, et al. Effect of carbocysteine on acute exacerbations of chronic obstructive pulmonary disease (PEACE-Study): a randomised placebo-controlled study. Lancet. 2008;371(9629):2013–2018. | |
Carpagnano GE, Resta O, Foschino-Barbaro MP, et al. Exhaled interleukin-6 and 8-isoprostane in chronic obstructive pulmonary disease; effect of carbocysteine lysine salt monohydrate (SCMC-Lys). Eur J Pharmacol. 2004;505(1–3):169–175. | |
Tatsumi K, Fukuchi Y; PEACE Study Group. Carbocisteine improves quality of life in patients with chronic obstructive pulmonary disease. J Am Geriatr Soc. 2007;55(11):1884–1886. | |
Aruoma OI, Spencer JP, Mahmood N. Protection against oxidative damage and cell death by the natural antioxidant ergothioneine. Food Chem Toxicol. 1999;37(11):1043–1053. | |
Huang J, Xu J, Tian L, Zhong L. A thioredoxin reductase and/or thioredoxin system-based mechanism for antioxidant effects of ambroxol. Biochimie. 2014;97:92–103. | |
Adenuga D, Caito S, Yao H, et al. Nrf2 deficiency influences susceptibility to steroid resistance via HDAC2 reduction. Biochem Biophys Res Commun. 2010;403(3–4):452–456. | |
Milara J, Navarro A, Almudéver P, Lluch J, Morcillo EJ, Cortijo J. Oxidative stress-induced glucocorticoid resistance is prevented by dual PDE3/PDE4 inhibition in human alveolar macrophages. Clin Exp Allergy. 2011;41(4):535–546. | |
Yang YC, Lii CK, Lin AH, et al. Induction of glutathione synthesis and heme oxygenase 1 by the flavonoids butein and phloretin is mediated through the ERK/Nrf2 pathway and protects against oxidative stress. Free Radic Biol Med. 2011;51(11):2073–2081. | |
Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. 2011;254(2):72–85. | |
Fujimoto K, Kubo K, Okada K, Kobayashi T, Sekiguchi M, Sakai A. Effects of the 21-aminosteroid U-74006F on antigen induced bronchoconstriction and bronchoalveolar eosinophilia in allergic sheep. Eur Respir J. 1996;9(10):2044–2049. | |
van Klaveren RJ, Roelant C, Boogaerts M, Pype JL, Demedts M, Nemery B. Protective effects of the lazaroid U-74389G against hyperoxia in rat type II pneumocytes. Pulm Pharmacol Ther. 1998;11(1):23–30. | |
Xue T, Wie N, Xin Z, Qingyu X. Angiotensin-converting enzyme-2 overexpression attenuates inflamation in rat model of chronic obstructive pulmonary disease. Inhal Toxicol. 2014;26(1):14–22. | |
Arts IC, Hollmann PC. Polyphenols and disease risk in epidemiologic studies. Am J Clin Nutr. 2005;81(Suppl 1):317S–325S. | |
Tabak C, Arts IC, Smit HA, Heederik D, Kromhout D. Chronic obstructive pulmonary disease and intake of catechin, flavonols, and flavones: the MORGEN Study. Am J Respir Crit Care Med. 2001;164(1):61–64. | |
Santus P, Sola A, Carlucci P, et al. Lipid peroxidation and 5-lipoxygenase activity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171(8):838–843. | |
Ruijters EJ, Haenen GR, Weseler AR, Bast A. The cocoa flavanol (−)-epicatechin protects the cortisol response. Pharmacol Res. 2014;79:28–33. | |
Kode A, Rajendrasozhan S, Caito S, Yang SR, Megson IL, Rahman I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294(3):L478–L488. | |
Siedlinski M, Boer JM, Smit HA, Postma DS, Boezen HM. Dietary factors in lung function in the general population: wine and resveratrol intake. Eur Respir J. 2012;39(2):385–391. | |
Biswas SK, McClure D, Jimenez LA, Megson IL, Rahman I. Curcumin induces glutathione biosynthesis and inhibits NF-kappaB activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radical scavenging activity. Antioxid Redox Signal. 2005;7(1–2):32–41. | |
Meja KK, Rajendrasozhan S, Adenuga D, et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintainig HDAC2. Am J Respir Cell Mol Biol. 2008;39(3):312–323. | |
Anand P, Nair HB, Sung B, et al. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol. 2010;79(3):330–338. | |
Quin S, Alcorn JF, Craigo JK, et al. Epigallocatechin-3-gallate reduces airway inflammation in mice through binding to proinflammatory chemokines and inhibiting inflammatory cell recruitment. J Immunol 2011;186(6):3693–3700. | |
Malhotra D, Thimmulappa RK, Navas-Acien A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 2008;178(6):592–604. | |
Cheung LK, Khor TO, Kong AN. Synergistic effect of combinations of phenethyl isothiocyanate and sulforaphane or curcumin and sulforaphane in the inhibition of inflammation. Pharm Res. 2009;26(1):224–231. | |
Ganesan S, Faris AN, Comstock AT, et al. Quercetin prevents progression of disease in elastase/LPS-exposed mice by negatively regulating MMP expression. Respir Res. 2010;11:131. | |
Kumar P, Khanna M, Srivastava V, Tyagi YK, Raj HG, Ravi K. Effect of quercetin supplementation on lung antioxidants after experimental influenca virus infection. Exp Lung Res. 2005;31(5):449–459. | |
Palozza P, Simone R, Catalano A, et al. Lycopene prevention of oxysterol-induced proinflammatory cytokine cascade in human macrophages: inhibition of NF- κB nuclear binding and increase in PPARγ expression. J Nutr Biochem. 2011;22(3):259–268. | |
Maritz GS, Mutemwa M, Kayigire AX. Tomato juice protects the lungs of the offspring of female rats exposed to nicotine during gestation and lactation. Pediatr Pulmonol. 2011;46(10):976–986. | |
Kang J, Xie C, Nagarajan S, Schauss AG, Wu T, Wu X. Flavonoids from acai (Euterpeoleracea Mart.) pulp and their antioxidant and anti-inflammatory activities. Food Chem. 2011;128(1):152–157. | |
Baldrick FR, Elborn JS, Woodside JV, et al. Effect of fruit and vegetable intake on oxidative stress and inflammation in COPD: a randomised controlled trial. Eur Respir J. 2012;39(6):1377–1384. | |
Hirayama F, Lee AH, Oura A, Mori M, Hiramatsu N, Taniguchi H. Dietary intake of six minerals in relation to risk of chronic pulmonary disease. Asia Pac J Clin Nutr. 2010;19(4):572–577. | |
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266–281. | |
Sundar IK, Hwang JW, Wu S, Sun J, Rahman I. Deletion of vitamin D receptor leads to a premature emphysema/COPD by increased matrix metalloproteinases and lymphoid aggregates formation. Biochem Biophys Res Commun. 2011;406(1):127–133. | |
Nyunoya T, March TH, Tesfaigzi Y, Seagrave J. Antioxidant diet protects against emphysema, but increases mortality in cigarette smoke-exposed mice. COPD. 2011;8(5):362–368. | |
Agler AH, Kurth T, Gaziano JM, Buring JE, Cassano PA. Randomized vitamin E supplementation and risk of chronic lung disease in the Women’s Health Study. Thorax. 2011;66(4):320–325. | |
Britton JR, Pavord LD, Richards KA, et al. Dietary antioxidant vitamin intake and lung function in the general population. Am J Respir Crit Care Med. 1995;151(5):1383–1387. | |
Rahman I, Kinnula VL. Strategies to decrease ongoing oxidant burden in chronic obstructive pulmonary disease. Expert Rev Clin Pharmakol. 2012;5(3):293–309. | |
Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys. 1997;347(2):256–262. | |
Yao H, Arunachalam G, Hwang JW, et al. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci U S A. 2010;107(35):15571–15576. | |
Sharpe MA, Ollosson R, Stewart VC, Clark JB. Oxidation of nitric oxide by oxomanganese-salen complexes: a new mechanism for cellular protection by superoxide-dismutase/catalase mimentic. Biochem J. 2002;366(Pt 1):97–107. | |
Hillas G, Nikolakopoulou S, Hussain S, Vassilakopoulos T. Antioxidants and mucolytics in COPD management: when (if ever) and in whom? Curr Drug Targets. 2013;14(2):225–234. | |
Duong C, Seow HJ, Bozinovski S, Crack PJ, Anderson GP, Vlahos R. Glutathione peroxidase-1 protects against cigarette smoke-induced lung inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2010;299(3):L425–L433. | |
Stefanutti G, Pierro A, Smith VV, Klein NJ, Eaton S. Peroxynitrite decomposition catalyst FeTMPyP provides partial protection against intestinal ischemia and reperfusion injury in infant rats. Pediatr Res. 2007;62(1):43–48. | |
Rahman I, MacNee W. Antioxidant pharmacological therapies for COPD. Curr Opin Pharmacol. 2012;12(3):256–265. | |
Tiden AK, Sjögren T, Svensson M, et al. thioxanthines are mechanism-based inactivators of myeloperoxidase that block oxidative stress during inflammation. J Biol Chem. 2011;286(43):37578–37589. | |
Churg A, Sin DD, Wright JL. Everything prevents emphysema: are animal models of cigarette smoke-induced chronic obstructive pulmonary disease any use? Am J Respir Cell Mol Biol. 2011;45(6):1111–1115. | |
Brindicci C, Kharitonov SA, Ito M, et al. Nitric oxide synthase isoenzyme expression and activity in peripheral lung tissue of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(1):21–30. | |
Seimetz M, Parajuli M, Pichl A, et al. Inducable NOS inhibition reverses tobacco smoke induced emphysema and pulmonary hypertension in mice. Cell. 2011;147(2):293–305. | |
Malhotra D, Thimmulappa RK, Mercado N, et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest. 2011;121(11):4289–4302. | |
Rózanowska M, Sarna T, Land EJ, Truscott TG. Free radical scavenging properties of melanin interaction of eu- and pheo-melanin models with reducing and oxidising radicals. Free Radic Biol Med. 1999;26(5–6):518–525. | |
Singh AK, Ghosh S, Basu P, Haldar C. Daily variation in melatonin level, antioxidant activity and general immune response of peripheral blood mononuclear cells and lymphoid tissues of Indian goat Capra hircus during summer and winter. J Exp Biol. 2014;52(5):467–477. | |
de Moura RS, Pires KM, Santo Ferreira T, et al. Addition of açaí (Euterpe oleracea) to cigarettes has a protective effect against emphysema in mice. Food Chem Toxicol. 2011;49(4):855–863. | |
Kim SY, Moon KA, Jo HY, et al. Anti-inflammatory effects of apocynin, an inhibitor of NADPH oxidase, in airway inflammation. Immunol Cell Biol. 2012;90(4):441–448. | |
Stefanska J, Sarniak A, Wlodarczyk A, et al. Apocynin reduces reactive oxygen species concentrations in exhaled breath condensate in asthmatics. Exp Lung Res. 2012:38(2):90–99. | |
de Batile J, Sauleda J, Balcells E, et al. Association between Ω3 and Ω6 fatty acid intakes and serum inflammatory markers in COPD. J Nutr Biochem. 2012;23(7):817–821. | |
MacKeever TM, Lewis SA, Cassano PA, et al. The relation between dietary intake of individual fatty acids, FEV1 and respiratory disease in Dutch adults. Thorax. 2008;63(3):208–214. | |
Shi H, Timmins G, Monske M, et al. Evaluation of spin trapping agents and trapping conditions for detection of cell-generated reactive oxygen species. Arch Biochem Biophys. 2005;437(1):59–68. | |
Bachnoff N, Trus M, Atlas D. Alleviation of oxidative stress by potent and selective thioredoxin-mimetic peptides. Free Radic Biol Med. 2011;50(10):1355–1367. | |
Barnes PJ. Histone deacetylase-2 and airway disease. Ther Adv Respir Dis. 2009;3(5):235–243. | |
Pinho RA, Chiesa D, Mezzomo KM, et al. Oxidative stress in chronic obstructive pulmonary disease patients submitted to a rehabilitation program. Respir Med. 2007;101(8):1830–1835. | |
Mercken EM, Hageman GJ, Schols AM, Akkemans MA, Bast A, Wouters EF. Rehabilitation decreases exercise-induced oxidative stress in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(8):994–1001. | |
Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144(1):266–273. | |
Zhou JJ, Cho MH, Castaldi PJ, Hersh CP, Silverman EK, Laird NM. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am J Respir Crit Care Med. 2013;188(8):941–947. | |
Brebner JA, Stockley RA. Recent advances in α-1-antitrypsin deficiency-related lung disease. Expert Rev Respir Med. 2013;7(3):213–229. | |
Hersh CP. Pharmacogenetics of chronic obstructive pulmonary disease: challenges and opportunities. Pharmacogenomics. 2010;11(2):237–247. | |
Berndt A, Leme AS, Shapiro SD. Emerging genetics of COPD. EMBO Mol Med. 2012;4(11):1144–1155. | |
Agusti A, Sobradillo P, Celli B. Addressing the complexity of chronic obstructive pulmonary disease: from phenotypes and biomarkers to scale-free networks, systems biology, and P4 medicine. Am J Respir Crit Care Med. 2011;183(9):1129–1137. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.