")
Back to Journals » OncoTargets and Therapy » Volume 9
Overexpression of Livin promotes migration and invasion of colorectal cancer cells by induction of epithelial–mesenchymal transition via NF-κB activation
Authors Ge Y, Cao X, Wang D, Sun W, Sun H, Han B, Cui J, Liu B
Received 4 August 2015
Accepted for publication 28 November 2015
Published 29 February 2016 Volume 2016:9 Pages 1011—1021
DOI https://doi.org/10.2147/OTT.S93738
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Yang Ge,1,2 Xiankui Cao,1 Dalu Wang,3 Wei Sun,1 Hongli Sun,1 Bing Han,1 Junpeng Cui,1 Baolin Liu1
1The Sixth Department of General Surgery, Shengjing Hospital of China Medical University, Shenyang, 2Department of General Surgery, General Hospital Under the Fushun Mining Affairs Bureau, Fushun, 3The Eighth Department of General Surgery, Shengjing Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China
Abstract: Livin is a novel member of the inhibitors of apoptosis protein family and has been implicated in the development and progression of colorectal cancer (CRC). However, the underlying mechanisms of Livin in CRC remain not fully understood. In this study, we investigated the effects of Livin expression on the proliferation and metastasis of CRC cells and also addressed its related molecular mechanism to metastasis. The expression of Livin in CRC cells (HCT116, SW480, and HT-29 cell lines) was determined by Western blot analysis. Our results show that the overexpression of Livin significantly promotes the proliferation, migration, and invasion of SW480 cells. Concurrently, the inhibition of Livin reduces the proliferation, migration, and invasion of HCT116 cells. In addition, Livin overexpression promotes the epithelial–mesenchymal transition, as evidenced by a decrease in epithelial E-cadherin expression and an increase in mesenchymal markers, including vimentin, Slug, and Snail. Furthermore, adding the NF-κB inhibitor, BAY 11-7028, or transfecting with small interfering RNA against p65 notably restores the expression level of E-cadherin and attenuates the invasive ability of Livin-overexpressing cells. Taken together, these results indicate that Livin potentiates migration and invasion of CRC cells partially through the induction of epithelial–mesenchymal transition via NF-κB activation. Livin may be a potential therapeutic target for CRC.
Keywords: Livin, colorectal cancer, migration, invasion, epithelial–mesenchymal transition, NF-κB
Introduction
Despite advances in cancer therapies, colorectal cancer (CRC) remains one of the most common and lethal tumors worldwide, with an estimated 1,200,000 new cases and 600,000 deaths every year.1,2 Moreover, like the other tumors, metastasis remains the major cause of CRC-related death.3 Metastasis renders primary tumors to the secondary metastatic sites, such as liver and lungs.4 Therefore, understanding the mechanisms involved in CRC metastasis is of great importance and may provide promising therapeutic targets for CRC.
Livin is a novel member of the inhibitors of apoptosis protein family, which selectively binds to the apoptotic regulators, including SMAC, caspase-3, caspase-7, and caspase-9, results in the inactivation and degradation of these enzymes, and finally inhibits cell apoptosis.5,6 A growing body of literature shows that Livin is abundantly expressed in tumor tissues but barely expressed in normal tissues,7–9 indicating an important role of Livin in cancer progression. More recently, Livin was shown to impact on multiple cellular behaviors, such as cell proliferation, invasiveness, and motility.10 Given the pleiotropic actions of Livin, it is now considered as a promising target for cancer treatment.5,11 In CRC, Livin expression is upregulated in colorectal carcinoma tissues and might correlate with CRC metastasis and prognosis.12–14 Myung et al recently demonstrated that Livin was associated with tumor stage and facilitated tumor progression via regulating cell motility and apoptosis in CRC.15 However, the molecular mechanisms of Livin’s involvement in the CRC metastasis remain to be elucidated.
In this study, we investigated the metastatic role of Livin and its underlying mechanism in CRC cells. Our results showed that Livin overexpression facilitates the migration, invasion, and epithelial–mesenchymal transition (EMT) of CRC cells. Furthermore, Livin-mediated EMT and metastasis was dependent on the activation of nuclear factor kappa B (NF-κB).
Methods
Cell culture
Three human CRC cell lines, namely HCT116, SW480, and HT-29 (Cell Bank of Chinese Academy of Sciences, Shanghai, People’s Republic of China), were cultured in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA). The cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 for subsequent experiments. The use of human CRC cell lines was approved by the ethics committee of China Medical University.
Plasmid construction and generation of Livin-overexpressing SW480 cell line
The coding sequence of Livin was obtained using reverse transcription-polymerase chain reaction (PCR) and was subsequently cloned into pcDNA3.1 vector. After sequencing confirmation, the SW480 cells were transfected with vector–Livin or vector empty using Lipofectamine 2000 Reagent (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instructions. Twenty-four hours after transfection, G418 (Life Technologies) solution was added to the culture to screen Livin- or vector-transfected cell clones.
RNA interference
The small interfering RNA (siRNA)-targeting NF-κB p65 (p65 siRNA) and scrambled control siRNA (GenePharma, Shanghai, People’s Republic of China) were transiently transfected into SW480 cells using Lipofectamine 2000 Reagent (Life Technologies). The short hairpin RNA (shRNA) construct-targeting Livin mRNA and scrambled control shRNA were transiently transfected into HCT116 cells using Lipofectamine 2000 Reagent (Life Technologies). The sequences of Livin shRNA are 5′-GATCCCCGGTGAGGTGCTTCTTCTGCTTCAAGAGAGCAGAAGAAGCACCTCACCTTTTT-3′, and the sequences of control shRNA are 5′-GATCCCCTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTT-3′.
Real-time (RT)-PCR
Total RNA from cells was extracted using a RNA simple Total RNA Kit (TIANGEN Co., Beijing, People’s Republic of China). The RNA was then converted into complementary DNA using Super M-MLV Reverse Transcriptase (BioTeke, Beijing, People’s Republic of China). The specific primers used for Livin and β-Actin were as follows: Livin, 5′-CAGTTCCTGCTCCGGTCAA-3′ (forward) and 5′-GGGCTCAAGAACCCACCAC-3′ (reverse); β-Actin, 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ (forward) and 5′-CTGTCACCTTCACCGTTCCAGTTT-3′ (reverse). The amplified products were quantitated with SYBR Green fluorescence (Solarbio, Beijing, People’s Republic of China) in an Exicycler™ 96 RT-PCR machine (Bioneer, Daejeon, Korea).
Western blot analysis
Cell lysis was prepared using the radioimmunoprecipitation analysis (RIPA) lysis buffer (Beyotime Institute of Biotechnology, Haimen, People’s Republic of China). The protein concentration was determined using the bicinchoninic acid kits (Beyotime Institute of Biotechnology). Equal amounts of protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (8% or 10% gel) and electrotransferred onto polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). After blocking with 5% nonfat milk at room temperature for 1 hour, the membranes were incubated with specific primary antibodies (anti-Livin, 1:1,000, Abcam, Cambridge, UK; anti-MMP-2, anti-E-cadherin, and anti-p65, 1:400, Boster, Wuhan, People’s Republic of China; anti-vimentin, anti-Slug, anti-Snail, and anti-Histone H3, 1:500, Bioss, Beijing, People’s Republic of China; and anti-β-Actin, 1:1,000, Santa Cruz Biotechnology Inc., Dallas, TX, USA) at 4°C overnight. The membranes were rinsed with Tween-Tris buffered saline (TTBS) three times and then incubated with appropriate secondary antirabbit or antimouse immunoglobulin G-horseradish peroxidase (IgG-HRP) antibody (1:5,000 diluted; Beyotime Institute of Biotechnology). The targeted protein bands were visualized using an enhanced chemiluminescence reagent (Qihai Biotec, Shanghai, People’s Republic of China) and analyzed using the Gel-Pro Analyzer software.
Colony formation assay
Briefly, the SW480, vector control, and Livin-overexpressing cells were seeded as single cells in 35 mm dishes (100 cells per dish) and cultured for 14 days until the cell colonies were visible. The colonies were fixed with 4% paraformaldehyde and stained with Giemsa solution. The colonies consisting of at least 50 cells were counted under a phase contrast microscope (Motic, Xiamen, People’s Republic of China). Colony formation efficiency was calculated as follows: (number of colonies/number of cells inoculated) ×100%.
MTT assay
The cells were seeded in 96-well plates at a density of 2×103 cells in each well and incubated for various time intervals (24, 48, 72, or 96 hours). Then the cells were treated with 0.2 mg/mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich Co., St Louis, MO, USA) for an additional 4 hours of incubation. Finally, the cell supernatant was removed, and the resulting formazan was dissolved in 200 μL of dimethylsulfoxide. The optical density was determined at 490 nm using a microplate reader.
Wound-healing assay
Cells were grown to 90% confluence in the plates. After aspirating the cell culture medium, a uniform wound was scratched on the cell monolayer using a 200 μL sterile pipette tip. The remaining cells were then washed twice with serum-free culture media and cultured in a medium containing 10% FBS. The wound closure photographs were captured at 0 hour and 24 hours after scratching using a phase contrast microscope. The percent of wound closure was calculated as the cell migration distance to the initial wound distance.
Transwell invasion assay
The invasive capacities of CRC cells were determined using Matrigel (BD Biosciences, San Jose, CA, USA)-coated Transwell chambers (8 μm pore size; Corning Incorporated, Corning, NY, USA). Briefly, the cells were resuspended with serum-free medium, 5×104 cells were seeded into the upper chamber, and the lower chamber was added with 800 μL of DMEM containing 20% FBS. After 24 hours of incubation, cells that remained on the upper chamber were scraped away with a cotton swab, and the cells that migrated to the bottom of the filter were fixed and stained with crystal violet dyes (AMRESCO, Solon, OH, USA). The number of invaded cells was photographed under an inverted phase contrast microscope.
Immunofluorescence staining
The distribution of E-cadherin and NF-κB p65 in SW480 cells was determined by immunofluorescence staining. Cells grown on the coverslips were fixed and permeabilized in 0.1% Triton X-100. After blocking with normal goat serum (Solarbio), the cell slides were incubated in anti-E-cadherin or anti-p65 antibody (dilution 1:200; Boster) at 4°C overnight. Subsequently, the cells were incubated with Cy3-labeled goat anti-rabbit secondary antibody (1:200; Beyotime Institute of Biotechnology) for 1 hour. After washing off the unbound secondary antibodies, all slides were counterstained with 4′,6-diamidino-2-phenylindole. The images of stained slides were visualized under a laser scanning confocal microscope (Olympus, Tokyo, Japan)
Electrophoretic mobility shift assay
Nuclear extracts from SW480, Livin-transfected, or vector-transfected cells were prepared using a nuclear protein extraction kit (Beyotime Institute of Biotechnology). The binding reaction was performed using a Nonradioactive NF-κB EMSA Kit (Viagene Biotech, Changzhou, People’s Republic of China). The core sequence of the probe-targeting NF-κB is 5′-AGTTGAGGGGACTTTCCCAGGC-3′. Five microliters of nuclear extracts (at a concentration of 5 μg/μL) were incubated in 15 μL of mixed binding buffer containing biotin-labeled NF-κB p65 probe for 20 minutes at room temperature. The DNA/protein complexes were separated from free DNA on 6.5% nondenaturing polyacrylamide gel in 0.25 mM tetrabromoethane (TBE) and electrotransferred onto a nylon membrane. After cross-linking using UV light for 30 minutes, the membranes were detected using streptavidin–HRP and an enhanced chemiluminescence solution (Qihai Biotec).
Statistical analysis
Data are presented as the mean ± standard deviation. One-way analysis of variance was used for the analyses between groups, and multiple comparisons were performed using the Bonferroni post hoc test. Data were processed using the GraphPad Prism 5.0 software (San Diego, CA, USA). A P-value <0.05 was considered statistically significant.
Results
Construction of stably Livin-overexpressing SW480 cell line
To assess the functional role of Livin in CRC cells, we first examined the expression of Livin in three CRC cell lines and constructed a cell line that stably overexpressed Livin. As shown in Figure 1A, Livin expression was the lowest in SW480 cells and the highest in HCT116 cells among the cell lines tested. Thus, SW480 cells were chosen for Livin overexpression. After transfection, the mRNA level of Livin was significantly increased in Livin-transfected cells, which was about three times that of the vector control cells (Figure 1B, P<0.01). Similarly, Western blot results also revealed a marked upregulation of Livin protein level in Livin-transfected cells (Figure 1C, P<0.01). The results indicated that a stable Livin-overexpressing SW480 cell line was constructed.
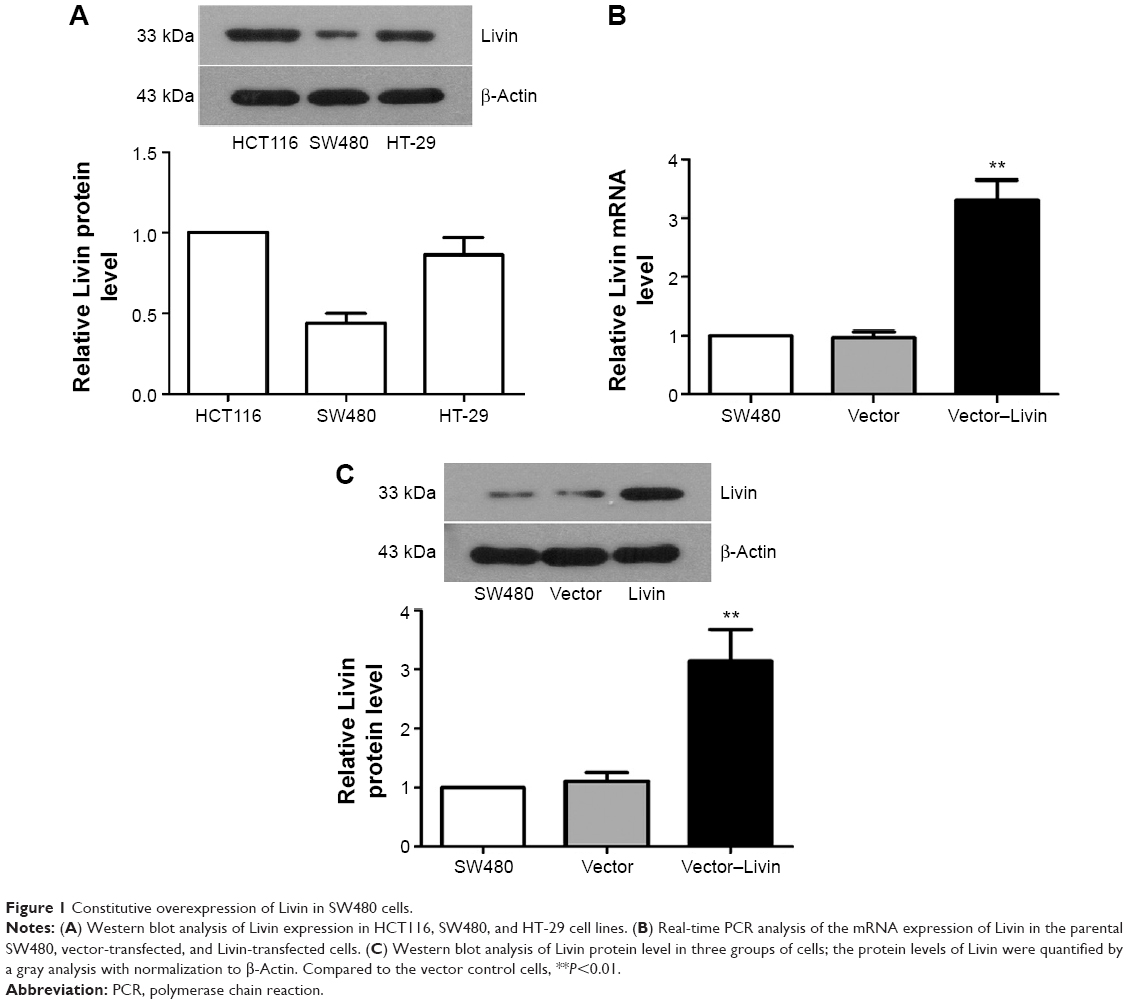 | Figure 1 Constitutive overexpression of Livin in SW480 cells. |
Livin promotes the proliferation of SW480 cells
The effect of Livin overexpression on cellular proliferation was determined by colony formation and MTT assays. The colony formation assay showed more colonies of the Livin-transfected cells in the plate (Figure 2A). MTT results indicated that fortified Livin expression increased the cell proliferation rate after 48 hours (Figure 2B, P<0.05), but there was no significant difference in proliferation between Livin-transfected and vector control cells at 24 hours.
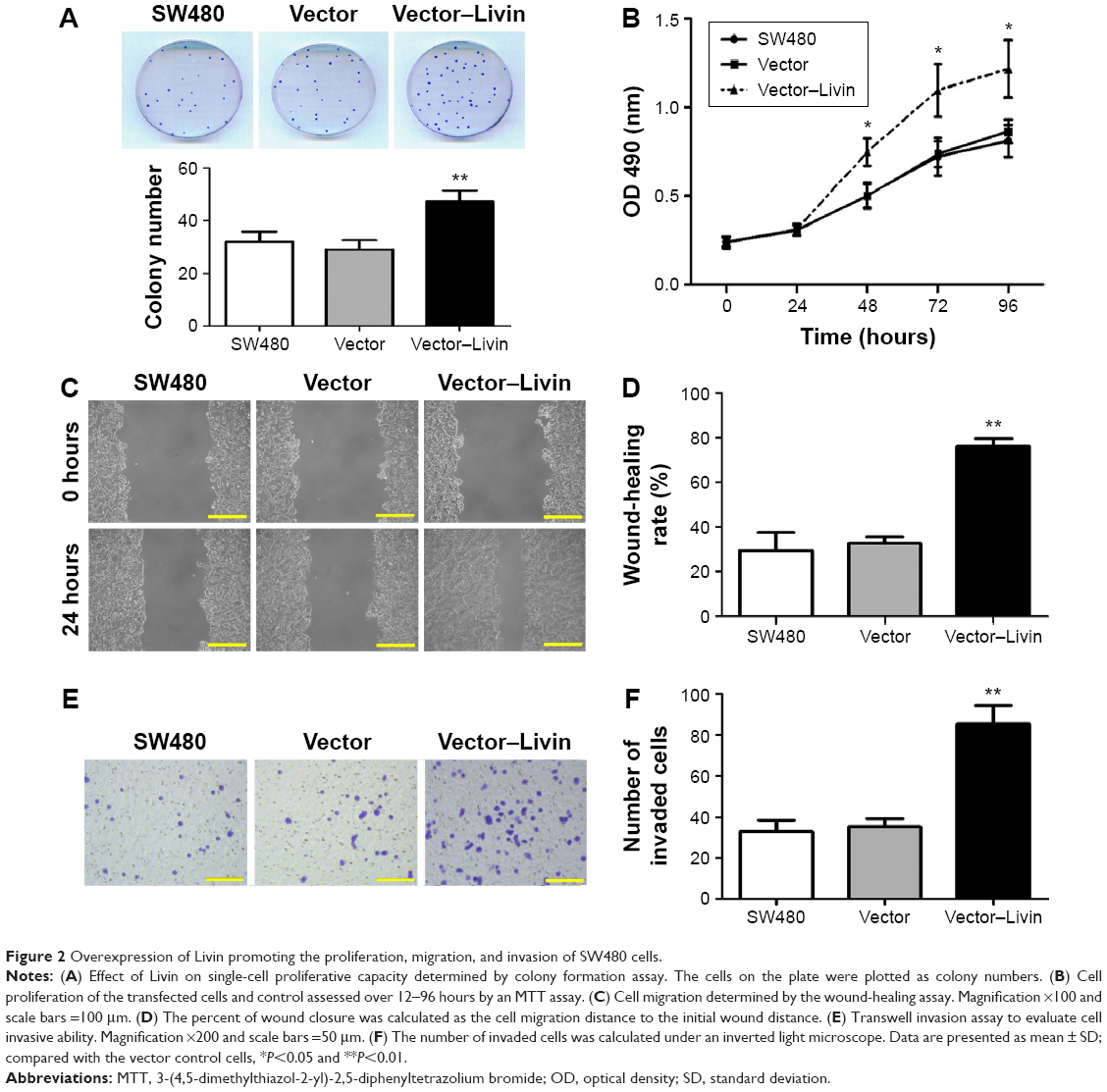 | Figure 2 Overexpression of Livin promoting the proliferation, migration, and invasion of SW480 cells. |
Overexpression of Livin promotes migration and invasion of SW480 cells
It has been demonstrated that Livin not only affects cell apoptosis but is also involved in tumor cell metastasis.16,17 To assess the functional role of Livin in CRC metastasis, we compared the wound-healing migratory and Matrigel invasive capacities of Livin-transfected cells with those of vector control or parental SW480 cells. Wound-healing assay results showed that Livin-transfected cells exhibited enhanced migratory capacity compared to the vector control cells (Figure 2C and D, P<0.01). Transwell invasion assay showed that more Livin-transfected cells penetrated through the Matrigel-coated chambers (Figure 2E and F, P<0.01), indicating that Livin overexpression increased the motility and invasive capacity of SW480 cells.
Downregulation of Livin inhibits migration and invasion of HCT116 cells
To confirm the metastatic role of Livin in CRC cells, we further used shRNA to knockdown Livin expression in HCT116 cells. As shown in Figure 3A and B, significant reduction in the mRNA and protein levels of Livin was found in Livin shRNA-transfected HCT116 cells as determined by RT-PCR and Western blot analyses (P<0.01 vs control cells). Consistently, knockdown of Livin caused a notable reduction in cell proliferation after 48 hours (Figure 3C). Furthermore, silencing of Livin expression dramatically inhibited the cell motility in wound-healing assay (Figure 3D) and the invasiveness in Transwell invasion assay (Figure 3E). Taken together, these results demonstrate that the knockdown of Livin inhibits CRC cell metastasis, highlighting the metastatic role of Livin in CRC cells.
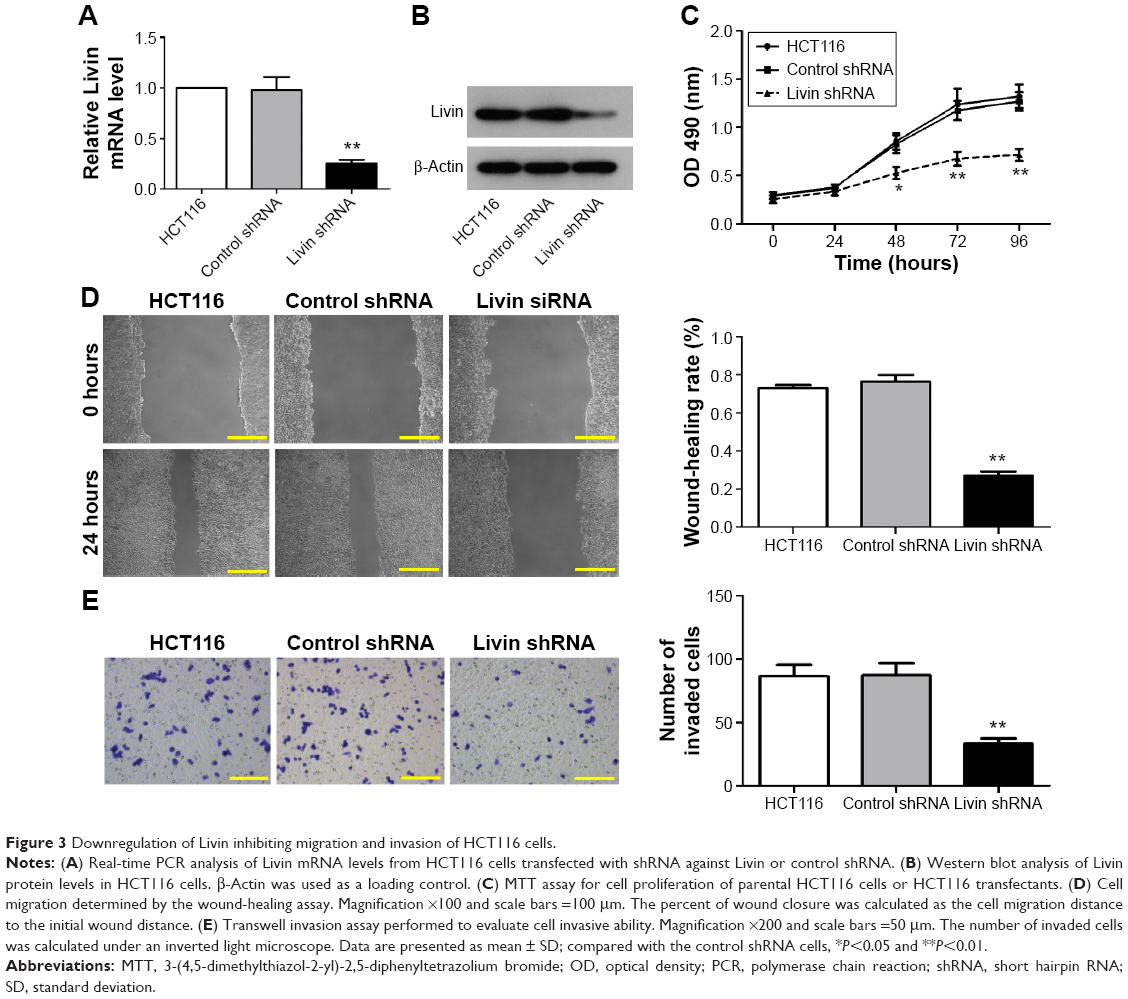 | Figure 3 Downregulation of Livin inhibiting migration and invasion of HCT116 cells. |
Livin promotes the expression of EMT-related markers in SW480 cells
Considering that EMT is a crucial step for mediating cancer metastasis, we further determined whether Livin overexpression affects the expression of EMT-related markers in SW480 cells. Our results showed that the overexpression of Livin significantly decreased the epithelial E-cadherin protein level but upregulated the expression levels of mesenchymal marker vimentin and the related transcription factors, including Snail and Slug in Livin-transfected cells (Figure 4A and B, P<0.01 vs vector group). Consistently, immunofluorescence staining also revealed a reduction of E-cadherin expression in Livin-transfected cells (Figure 4C). Together, these data suggest that Livin overexpression promotes the EMT in CRC cells, which might further mediate cell migration and invasion.
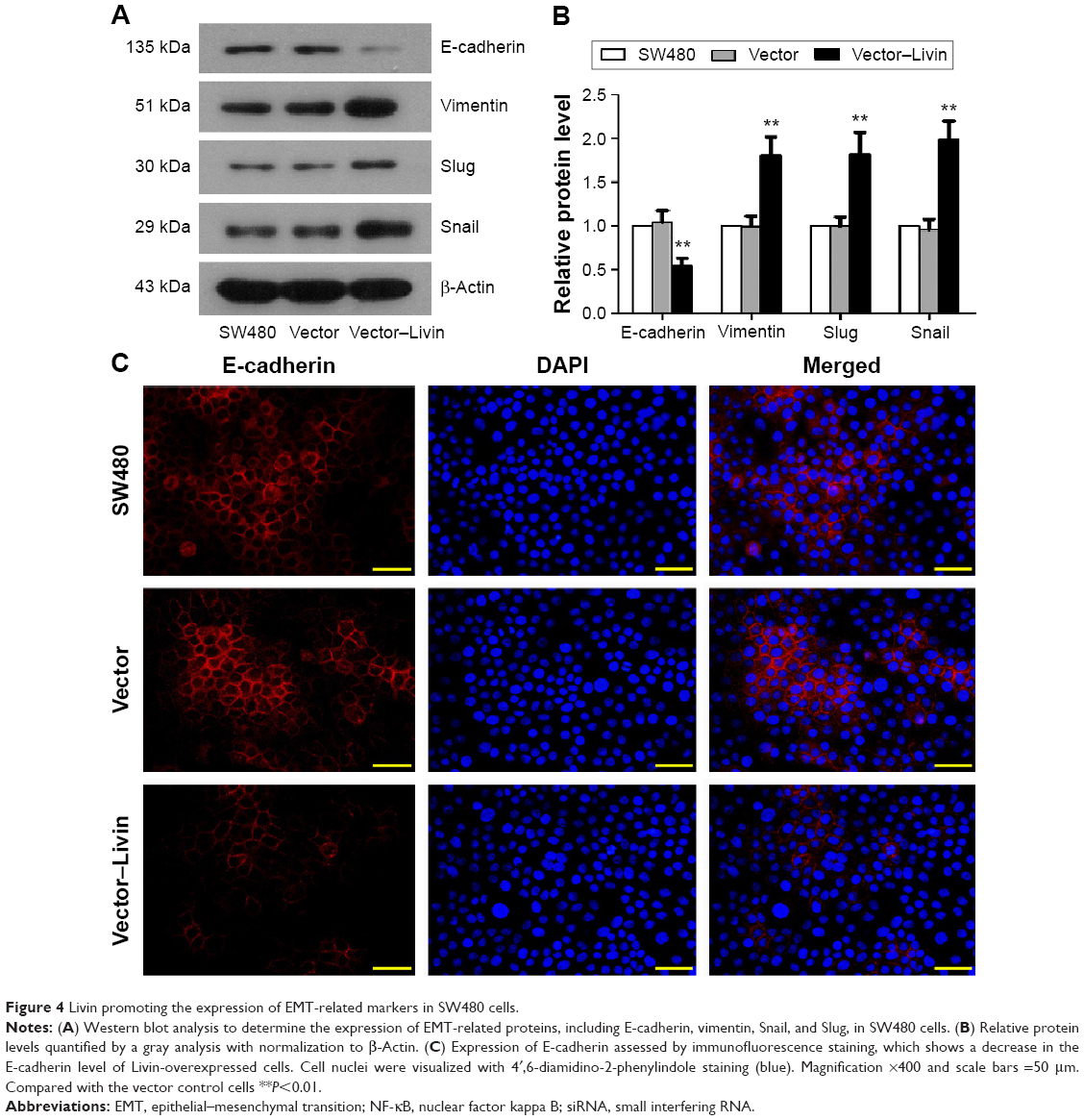 | Figure 4 Livin promoting the expression of EMT-related markers in SW480 cells. |
Livin activates the NF-κB pathway in SW480 cells
The NF-κB pathway has been shown to play a pivotal role in cancer metastasis.18 To assess whether Livin overexpression is related to NF-κB activation, we further determined the expression and transcriptional activity of NF-κB. Our data showed that the overexpression of Livin significantly increased the nuclear p65 levels as compared with that of the vector control cells (Figure 5A and B, P<0.01). Additionally, electrophoretic mobility shift assay (EMSA) results showed the low basal levels of NF-κB-binding activity in parental SW480 and vector control cells, whereas the binding activity was significantly increased in Livin-transfected cells (Figure 5C and D, P<0.01). Immunofluorescence staining showed that Livin overexpression decreased the cytoplasmic p65 levels and increased the nuclear p65 levels as compared to the parental SW480 and vector control cells (Figure 5E), which were consistent with Western blot and EMSA results. Taken together, these results indicate that Livin overexpression induces NF-κB activation in SW480 cells.
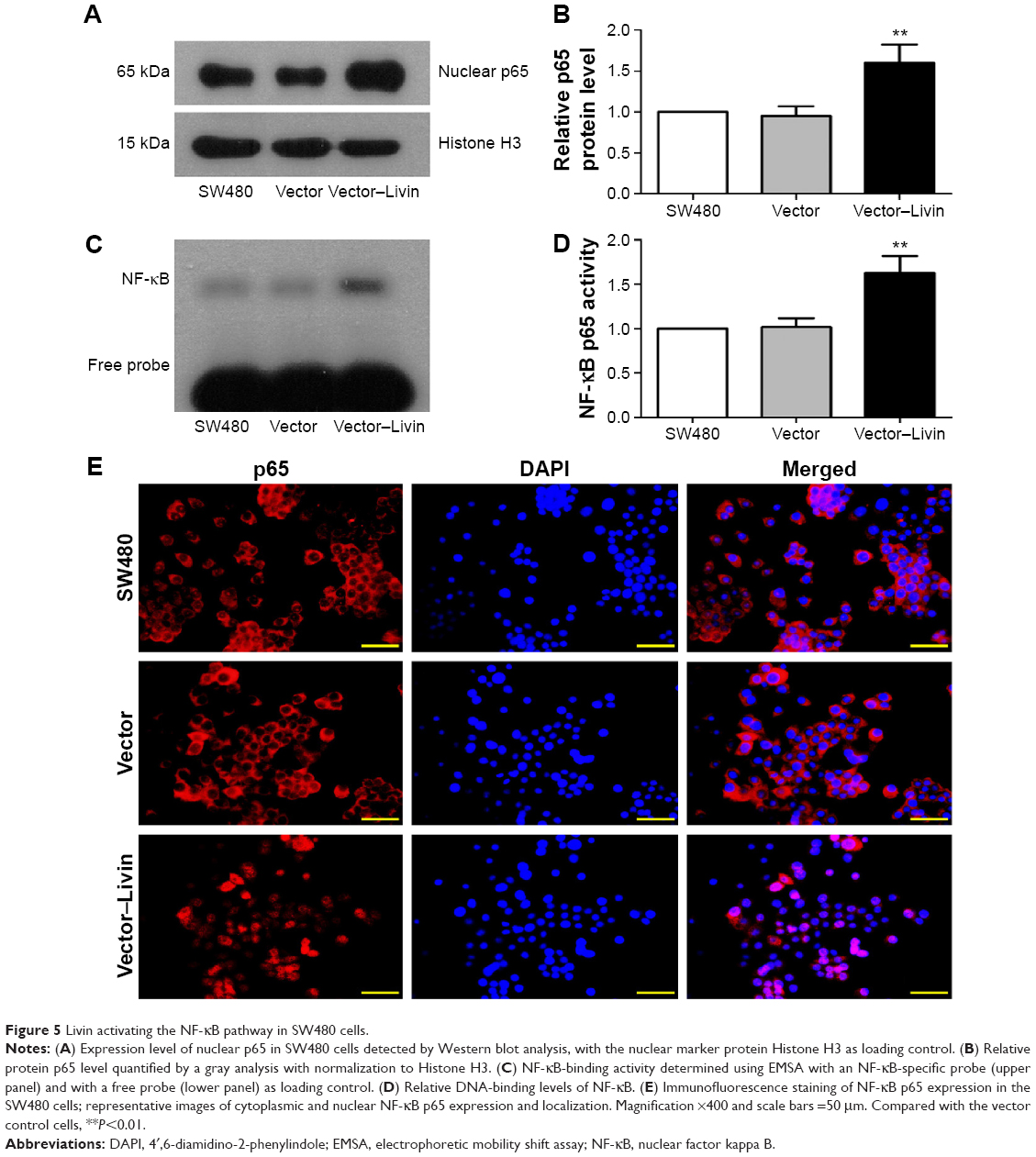 | Figure 5 Livin activating the NF-κB pathway in SW480 cells. |
NF-κB activation contributes to Livin-induced metastasis and EMT in CRC cells
To assess the involvement of NF-κB in Livin-induced CRC cell metastasis and EMT, we performed the in vitro inhibition assays using an NF-κB inhibitor and siRNA-targeting NF-κB p65. Western blot analysis showed that there was an obvious increase in NF-κB p65 with a concomitant decrease in E-cadherin levels in the Livin-transfected cells when compared to the vector or vector control siRNA cells (Figure 6A and B). Inhibition of NF-κB p65 by BAY 11-7028 or siRNA p65 significantly reduced the level of NF-κB p65 and increased the level of E-cadherin as compared to the Livin-overexpressing cells (Figure 6A and B, P<0.05). Moreover, Transwell invasion assays revealed that the inhibition of NF-κB reversed the invasive ability induced by Livin overexpression (Figure 6C and D, P<0.01). Overall, these data indicate that Livin-mediated cellular metastasis and EMT are partially dependent on NF-κB activation.
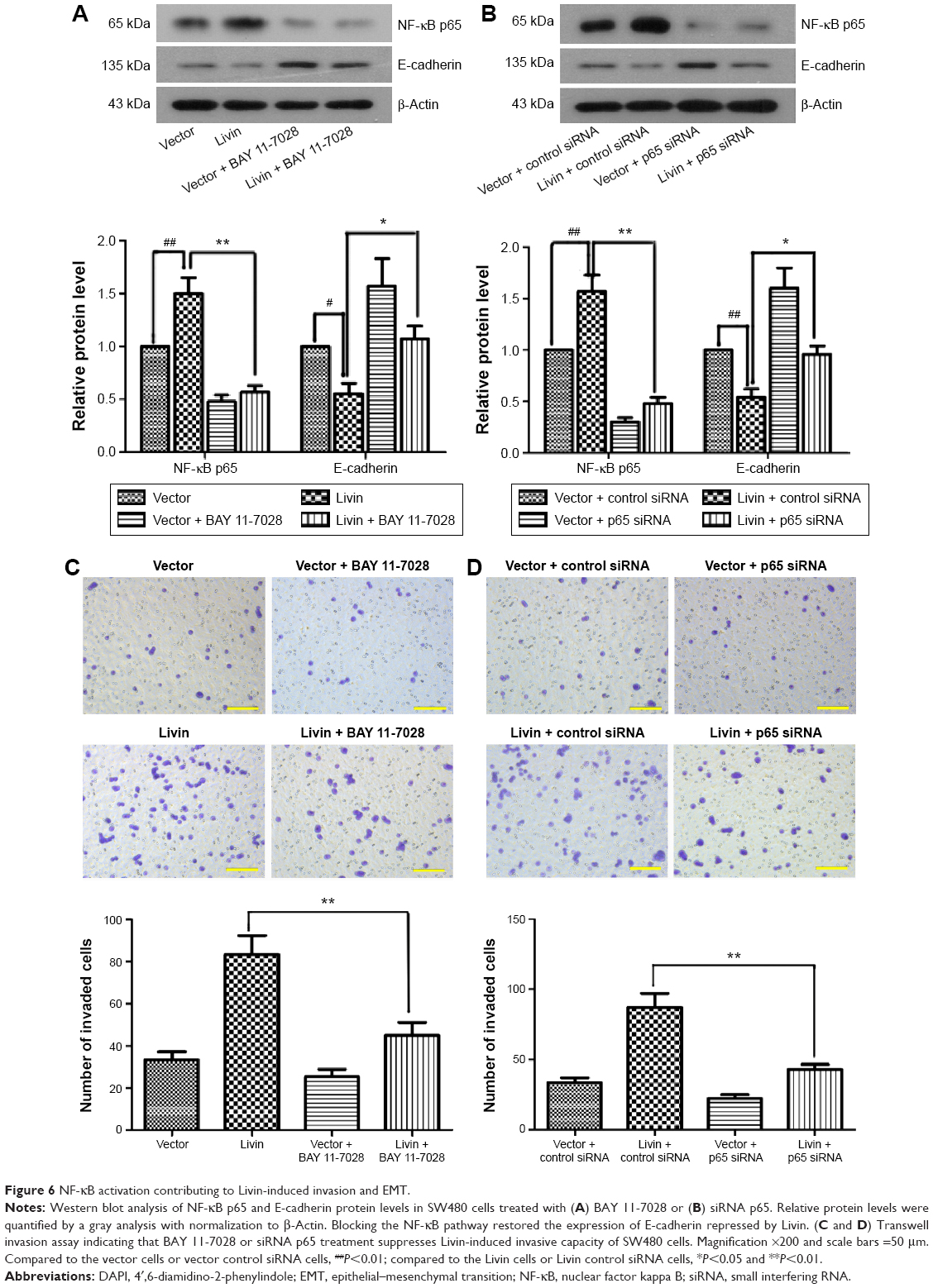 | Figure 6 NF-κB activation contributing to Livin-induced invasion and EMT. |
Discussion
Livin, a novel member of the inhibitors of apoptosis protein family, has been implicated in the development and progression of various human cancers. In this study, we demonstrated the potential metastatic role of Livin in CRC cells. Livin significantly facilitated the proliferation, motility, and invasiveness of CRC cells. Furthermore, Livin overexpression induced EMT in SW480 cells, which was partially dependent on NF-κB activation. These results indicate an important mediator of Livin in CRC cell migration and invasion.
Previous studies have linked Livin to metastasis in multiple solid tumors, such as lung cancer,19 breast cancer,20,21 gastric cancer,17 and laryngohypopharyngeal cancer.16 In CRC, ectopic overexpression of Livin correlates with tumor metastasis and implies poor prognosis.12–14 Recently, Myung et al further demonstrated that Livin expression is associated with the tumor stage and facilitated tumor progression via regulating CRC cell motility and apoptosis.15 However, the metastatic mechanism of Livin in CRC cells still needs investigation. Here, we showed that Livin overexpression significantly increased the proliferation, migration, and invasion of SW480 cells. Concurrently, downregulation of Livin inhibited the cell proliferation rate and suppressed the motility and invasiveness in HCT116 cells. These results demonstrate that Livin promotes cell proliferation and metastasis in CRC cells, highlighting the metastatic role of Livin in CRC and establishing that our results are in line with those of a previous report.20
EMT occurs in the early stage of tumor metastasis and plays a key role in cancer progression.4,22 The hallmark features of EMT include reduction in cell adhesion and acquirement of cell motility.23 E-cadherin is the most vital epithelial marker, which can be repressed by the transcriptional repressors, such as Snail and Slug.24,25 Vimentin is one of the mesenchymal markers during EMT. In this study, ectopic overexpression of Livin resulted in a significant downregulation of E-cadherin and upregulation of vimentin, Snail, and Slug at the protein levels in Livin-overexpressed SW480 cells. Immunofluorescence analysis also confirmed the decreased E-cadherin expression in Livin-overexpressed SW480 cells. Our results are in line with those of a recent study showing Livin-induced EMT in breast cancer cells21 and highlight a functional link between Livin and EMT in CRC cell metastasis.
Multiple pathways and molecules contribute to the EMT process,26,27 among which the NF-κB pathway has been shown to play an essential role for EMT process. NF-κB can regulate the expression of EMT-related proteins, such as Snail, Slug, Twist, and Zeb1.18,28 NF-κB closely correlates with cancer development and progression.29,30 Recently, Chen et al found that Livin regulated invasion of prostate cancer cells via the NF-κB signaling pathway.31 In this study, we found that the overexpression of Livin resulted in cytoplasmic NF-κB p65 translocation to the nucleus of CRC cells. In addition, Livin increased the NF-κB-binding activity, suggesting the activation of NF-κB in the Livin-overexpressing cells. Furthermore, by using the NF-κB inhibitor BAY 11-7028, we found that NF-κB inhibition restored the expression level of E-cadherin and inhibited the invasiveness of Livin-overexpressed cells. Similar results were observed in the siRNA-mediated NF-κB knockdown experiment. Our results suggest that Livin-induced EMT is at least partially dependent on NF-κB activation. Yet, it is also noteworthy that Livin regulated EMT in breast cancer cells through AKT (the serine/threonine kinase), also konwn as protein kinase B (PKB) signaling;21 further characterizations are required to illustrate the mechanisms related to Livin-induced EMT.
Conclusion
In summary, we demonstrated that Livin contributes to CRC cells’ migration, invasion, and EMT. The activation of NF-κB is at least partially responsible for Livin-induced EMT. Our results indicate a critical role of Livin in the regulation of CRC metastasis, which may provide a potential target for CRC therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Labianca R, Merelli B. Screening and diagnosis for colorectal cancer: present and future. Tumori. 2010;96(6):889–901. | ||
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. | ||
Gavert N, Ben-Ze’ev A. Epithelial-mesenchymal transition and the invasive potential of tumors. Trends Mol Med. 2008;14(5):199–209. | ||
Chang H, Schimmer AD. Livin/melanoma inhibitor of apoptosis protein as a potential therapeutic target for the treatment of malignancy. Mol Cancer Ther. 2007;6(1):24–30. | ||
Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3(6):401–410. | ||
Xi RC, Sheng YR, Chen WH, et al. Expression of survivin and Livin predicts early recurrence in non-muscle invasive bladder cancer. J Surg Oncol. 2013;107(5):550–554. | ||
Gazzaniga P, Gradilone A, Giuliani L, et al. Expression and prognostic significance of LIVIN, SURVIVIN and other apoptosis-related genes in the progression of superficial bladder cancer. Ann Oncol. 2003;14(1):85–90. | ||
Tanabe H, Yagihashi A, Tsuji N, Shijubo Y, Abe S, Watanabe N. Expression of survivin mRNA and Livin mRNA in non-small-cell lung cancer. Lung Cancer. 2004;46(3):299–304. | ||
Liu H, Wang S, Sun H, et al. Inhibition of tumorigenesis and invasion of hepatocellular carcinoma by siRNA-mediated silencing of the livin gene. Mol Med Rep. 2010;3(6):903–907. | ||
Nachmias B, Ashhab Y, Ben-Yehuda D. The inhibitor of apoptosis protein family (IAPs): an emerging therapeutic target in cancer. Semin Cancer Biol. 2004;14(4):231–243. | ||
Wang Y, Li Y, Zhou B, et al. Expression of the apoptosis inhibitor Livin in colorectal adenoma-carcinoma sequence: correlations with pathology and outcome. Tumour Biol. 2014;35(12):11791–11798. | ||
Xi RC, Biao WS, Gang ZZ. Significant elevation of survivin and Livin expression in human colorectal cancer: inverse correlation between expression and overall survival. Onkologie. 2011;34(8–9):428–432. | ||
Li JH, He WJ, He YJ. [Expression and clinical significance of Survivin and Livin in DukesoB colorectal cancer]. Ai Zheng/Chin J Cancer. 2007;26(5):547–551. Chinese. | ||
Myung DS, Park YL, Chung CY, et al. Expression of Livin in colorectal cancer and its relationship to tumor cell behavior and prognosis. PLoS One. 2013;8(9):e73262. | ||
Yoon TM, Kim SA, Lee DH, et al. Expression of Livin and the inhibition of tumor progression by Livin silencing in laryngohypopharyngeal cancer. In Vivo. 2014;28(5):751–759. | ||
Ou JM, Ye B, Qiu MK, et al. Knockdown of Livin inhibits growth and invasion of gastric cancer cells through blockade of the MAPK pathway in vitro and in vivo. Int J Oncol. 2014;44(1):276–284. | ||
Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104(3):733–744. | ||
Lin X, Li HR, Lin XF, et al. Silencing of Livin inhibits tumorigenesis and metastasis via VEGF and MMPs pathway in lung cancer. Int J Oncol. 2015;47(2):657–667. | ||
Ghebeh H, Al-Khaldi S, Olabi S, et al. Fascin is involved in the chemotherapeutic resistance of breast cancer cells predominantly via the PI3K/Akt pathway. Br J Cancer. 2014;111(8):1552–1561. | ||
Li F, Yin X, Luo X, et al. Livin promotes progression of breast cancer through induction of epithelial-mesenchymal transition and activation of AKT signaling. Cell Signal. 2013;25(6):1413–1422. | ||
Cao H, Xu E, Liu H, Wan L, Lai M. Epithelial-mesenchymal transition in colorectal cancer metastasis: a system review. Pathol Res Pract. 2015;211(8):557–569. | ||
Baum B, Settleman J, Quinlan MP. Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol. 2008;19(3):294–308. | ||
Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. | ||
Sanchez-Tillo E, Liu Y, de Barrios O, et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69(20):3429–3456. | ||
Foroni C, Broggini M, Generali D, Damia G. Epithelial-mesenchymal transition and breast cancer: role, molecular mechanisms and clinical impact. Cancer Treat Rev. 2012;38(6):689–697. | ||
Huber MA, Beug H, Wirth T. Epithelial-mesenchymal transition: NF-kappaB takes center stage. Cell Cycle. 2004;3(12):1477–1480. | ||
Huber MA, Azoitei N, Baumann B, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114(4):569–581. | ||
Kojima M, Morisaki T, Sasaki N, et al. Increased nuclear factor-kB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer Res. 2004;24(2B):675–681. | ||
Zubair A, Frieri M. Role of nuclear factor-kB in breast and colorectal cancer. Curr Allergy Asthma Rep. 2013;13(1):44–49. | ||
Chen F, Yang D, Wang S, et al. Livin regulates prostate cancer cell invasion by impacting the NF-kappaB signaling pathway and the expression of FN and CXCR4. IUBMB Life. 2012;64(3):274–283. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.