")
Back to Journals » OncoTargets and Therapy » Volume 11
Overexpression of CLEC3A promotes tumor progression and poor prognosis in breast invasive ductal cancer
Authors Ni J, Peng Y, Yang F, Xi X, Huang X, He C
Received 2 January 2018
Accepted for publication 19 March 2018
Published 5 June 2018 Volume 2018:11 Pages 3303—3312
DOI https://doi.org/10.2147/OTT.S161311
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samir Farghaly
Jun Ni, Yun Peng, Fu-Lan Yang, Xun Xi, Xing-Wei Huang, Chun He
Department of Breast and Thyroid Surgery, People’s Hospital of Ganzhou City, Ganzhou, Jiangxi, People’s Republic of China
Introduction: The aim of this study was to evaluate the expression of C-type lectin domain family 3 member A (CLEC3A) and its clinical significance in breast invasive ductal cancer (IDC) as well as its effect on breast cancer (BC) cell proliferation and metastasis. In this study, the level of CLEC3A expression in The Cancer Genome Atlas (TCGA) datasets was analyzed.
Materials and methods: Clinical collected samples and BC cells were measured using quantitative reverse transcription polymerase chain reaction. Its correlations with patients’ clinicopathological characteristics were analyzed by Pearson’s chi-squared test. Overall survival (OS) analysis was performed by the Kaplan–Meier method and Cox’s proportional-hazards model. BC cell proliferation, migration, and invasion by CLEC3A knockdown were assessed using Cell Counting Kit-8 and colony formation assay, wound healing model and transwell assay, respectively, in BT474 cell line. Activities of survival factors and phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) signaling were measured by testing key molecules using Western blot assay.
Results: CLEC3A expression was markedly higher in breast IDC tissues than normal breast tissues or adjacent normal tissue. Patients with high CLEC3A expression related to higher lymph node and poorer OS of breast IDC. CLEC3A knockdown by siRNA could inhibit the BC cells BT474 proliferation, migration, and invasion, together with a decrease in expression of key proteins in survival factors and PI3K/AKT signaling pathway.
Conclusion: Elevated CLEC3A expression may correlate with breast IDC metastatic potential and indicated a poor prognosis in breast IDC. CLEC3A knockdown inhibited BC cell growth and metastasis might be through suppressing PI3K/AKT signaling activity. These findings unravel that CLEC3A is a promising therapeutic target for BC in the future.
Keywords: breast cancer, proliferation, migration, invasion, overall survival, survival factor, PI3K/AKT
Introduction
Breast cancer (BC) is the leading cause of cancer-associated morbidity and mortality among female worldwide, and it affects ~12% of women,1 with an estimated 1,300,000 new cases and 470,000 deaths annually.2 Besides, in China, the number of new BC cases was estimated to be 268,600 and deaths was estimated to be 69,500 in 2015.3 Presently, surgical therapy and radiotherapy and chemotherapy were the major therapeutic strategies.4 Dismally, some of patients having undergone surgical therapy still were died of postoperative metastasis,5 the side-effects caused by radiotherapy were continuous,6,7 drug resistance remained to be a several challenge.8,9 These outcomes was probably attributed to poor understanding of BC pathogenesis or mechanism of drug action. On the basis of these, novel biomarkers or unveiling the underlying mechanism appears to be particularly urgent for the prognosis and effective treatment of BC.
C-type lectin domain family 3 member A (CLEC3A), belonging to the superfamily of C-type lectins, is known to be expressed initially in cartilage.10,11 CLEC3A was selected from the microarray analysis to involve in bone formation.12 Human CLEC3A mRNA has been detected in human BC cell lines and normal breast or BC tissue, and CLEC3A associates with cell adhesion.13 Cell adhesion influenced results in tumor cells proliferation and metastasis.14 Additionally, CLEC3A was reported to activate the plasminogen activation via enhancing tissue plasminogen activator.15 It was found that plasminogen activator system was identified as one of the major mechanisms involved in the processes of cell invasion and metastatic spreading.16,17 However, there is no direct evidence whether CLEC3A associates with clinicopathological significance in BC oncology and cancer progression.
In this study, we analyzed The Cancer Genome Atlas (TCGA) data associated with CLEC3A expression level in breast invasive ductal cancer (IDC) and normal breast tissues, which indicated that high CLEC3A level was found in breast IDC. Based on this, we are forced to hypothesize that CLEC3A possibly plays a significant carcinogenic action in BC. Therefore, the aim of this study is to determine whether the expression level of CLEC3A correlated with the clinical pathological characteristics, prognosis of breast IDC, BC progression, and possible molecular mechanism. Our data showed that CLEC3A was significantly increased in breast IDC patients, with correlation to higher lymph node metastasis and poor prognosis of breast IDC. We further demonstrated that knockdown of CLEC3A decreased BC cells proliferation, migration, and invasion. Finally, we also suggested that activities of survival factors and phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT) were downregulated by CLEC3A knockdown. Collectively, these results indicate that CLEC3A will likely be a drug-targeted marker in further clinic therapy.
Materials and methods
Database and data analysis
The data of CLEC3A RNA-seq were downloaded from the TCGA database. The expression data between BC and normal tissues were compared with the limma package in R.
Patients
This study collected 80 cases of tissue specimens of breast IDC, including breast IDC and adjacent normal tissue (ANT). All specimens were obtained from the Department of Breast and Thyroid Surgery at the Hospital between March 2006 and January 2010. The study was approved by the Review Board of People’s Hospital of Ganzhou City and written informed consent forms were signed by each participant. Pathological characteristics, including age, Pathologic stage, Pathologic-T (tumor status), Pathologic-N (regional lymph node status), and Pathologic-M (metastasis status) were reviewed in all tumor patients. All patients had follow-up records for >5 years. Survival time was measured from the date of primary surgery to the follow-up deadline or the date of death.
Cell culture
The human breast ductal carcinoma cell lines BT474, MDA-MB-231, MCF-7, and normal human breast cells CCD-1095SK were obtained from American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Roswell Park Memorial Institute (RPMI-1640) (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) (Thermo Fisher Scientific), containing penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37°C in a humidified CO2 incubator.
siRNA transfection
CLEC3A siRNA (si-CLEC3A: 5′-CAG AAG TCA ATG CCT TGA AGG AAA T-3′) and siRNA negative control (si-NC) oligonucleotides were designed and synthesized by Genepharma Co. Ltd. (Shanghai, China). One day before transfection, the cells (1×105) were seeded into 6-well plates. When cells density reached 30%–50%, the medium was replaced with fresh medium without serum and antibiotics. Then, cells were transfected with si-CLEC3A and si-NC using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. After 4–6 h incubation, the media were changed with fresh media containing 10% FBS. The efficiency of CLEC3A knockdown was assessed by Western blot and quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay.
qRT-PCR
Total RNA of tissues, BT474 and CCD-1095SK cells, and BT474 cells (after transfection of 48 h) were extracted with Trizol reagent (Thermo Fisher Scientific) following the manufacturer’s introductions. cDNA was synthesized with PrimeScript™ RT reagent kit (Takara, Dalian, China) in accordance with the manufacturer’s instructions. qRT-PCR was performed using SYBR Premix Ex Taq™ kit (Takara) according to the manufacturer’s instructions. Primers were as follows: CLEC3A: F: 5′-CGA GGC ACT AAA GTT CACAAG A-3′ and R: 5′-CGG AGT TCC TGG GGA TAA CCA-3′.Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (used as the internal control): F: 5′-GGA GCG AGA TCC CTC CAA AAT-3′ and R: 5′-GGC TGT TGT CAT ACT TCT CAT GG-3′. The qRT-PCR results related to CLEC3A relative expression levels normalized to GAPDH after performed in triplicate were analyzed using the 2−ΔΔCT calculation method.
Cell proliferation assay
The viability of BT474 cells was detected by Cell Counting Kit-8 (CCK-8; Beyotime Inc., Shanghai, China) assay after 24 h of transfection in accordance with manufacturer’s introductions. Cells were placed in 96-well plates and divided into 4 groups, then their viabilities were detected at 24, 48, 72, and 96 h. A total of 10 μL of CCK8 reagent was added into each well. Absorbance (optical density, OD) was measured at a wavelength of 450 nm using a spectrophotometer (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Colony formation assay
Twenty-four hours after transfection, 500 BT474 cells from each group were cultured in the 6 cm plate for 7–14 days during this time changing fresh medium every 3 days. When a visible clone in culture vessel was observed, cultivation of cells was terminated. Then, the colonies were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet for 30 min. After washing with water slowly, the plates were air dried. The number of colonies in 10 random view fields was counted under a microscope.
Wound-healing and transwell assay
For in vitro wounds, confluent cells were scratched using a 200 μL pipette tip, and then cell migration into the wound area was photographed under an inverted microscope (Olympus Corporation, Tokyo, Japan) with 100× magnification. Cells invasion and migration assay were also performed using 24-well transwell chamber with or without Matrigel matrix (BD Biosciences, San Jose, CA, USA) following the manufacturer’s instructions. Briefly, for detecting cell invasion, serum-free medium suspension (100 μL) containing a total of 1×105 cells was plated in the Matrigel-coated upper chamber, and the bottom chamber was filled with 600 μL of complete medium (containing 10% FBS). After 24 h incubation, residual cells were wiped with cotton-tipped swabs, invaded cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% of crystal violet for 20 min. Then, the invaded cells were counted under a microscope in 5 random fields per well at 200× magnification. For detecting cell migration, the steps resemble cells invasion detection, except for that Matrigel was not applied.
Western blotting analysis
At 48 h after transfection, total proteins of transfected BT474 cells were extracted by a Radio Immunoprecipitation Assay lysis buffer (Beyotime Inc.) and concentrations of them were quantified by a bicinchoninic acid protein assay kit (Beyotime Inc.). Protein lysates were separated using 8%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membrane. The membrane was blocked with 5% non-fat milk for 1.5 h at room temperature. Then, the membrane was incubated with primary antibodies against PI3K, phosphorylated-(p-)PI3K, AKT, p-AKT, phosphorylated ribosomal p70S6 protein kinase (p-P70S6K), GAPDH (Cell Signaling Technology, Inc., Danvers, MA, USA), and CLEC3A (Sigma-Aldrich Co., St Louis, MO, USA) at the dilution of 1:1,000 at 4°C overnight and were incubated by anti-mouse/rabbit horseradish peroxidase-conjugated secondary antibodies (Proteintech Group, Inc., Wuhan, China) at the dilution of 1:5,000. The bands were visualized by electrochemiluminescence imaging analysis system (Pierce Biotechnology, Inc., Rockford, IL, USA) and were measured with Quantity One software (Bio-Rad Laboratories Inc.).
Statistical analysis
Fold changes (FCs) were calculated as BC versus normal tissue from TCGA database. It would be considered as statistically significant if an FC value was <0.5 or >2 and with the P-value <0.05 in Student’s t-test. Pearson’s chi-squared test (χ2) was applied to calculate the correlation between clinicopathological variables and CLEC3A expression in breast IDC. Kaplan–Meier’s method and the log-rank test were used to evaluate the prognostic value. Univariate and multivariate survival analyses with calculation of hazard ratio (HR) were performed using Cox’s proportional hazards model for the overall survival (OS). qRT-PCR, Western blot, and proliferation results were analyzed by Student’s t-test. A value of P<0.05 was considered statistically significant. All statistical analyses were performed by SPSS version 22.0 (SPSS Inc., Chicago, IL, USA).
Results
Elevated expression of CLEC3A mRNA in breast IDC and correlated with clinicopathological features of patients
To evaluate the expression level of CLEC3A in breast IDC and its relation with clinical progression, CLEC3A mRNA expression level between breast IDC tissues and normal breast tissues were compared through TCGA analysis, additionally, we also used qRT-PCR assay to detect CLEC3A mRNA level in IDC tissues and ANT samples. As shown in Figure 1, TCGA data analyzing and qRT-PCR results showed that CLEC3A mRNA expression level was significantly higher in breast IDC tissues than normal or ANT group (both P<0.05). Moreover, Pearson’s chi-squared test was applied to assess the association between CLEC3A expression and clinicopathological features. As shown in Table 1, CLEC3A expression correlated significantly with Pathologic-N (P=0.033), suggesting a relationship with metastatic potential. CLEC3A expression was not significantly correlated with other clinicopathological features, such as age, Pathologic stage, Pathologic-T, and Pathologic-M (All P>0.05).
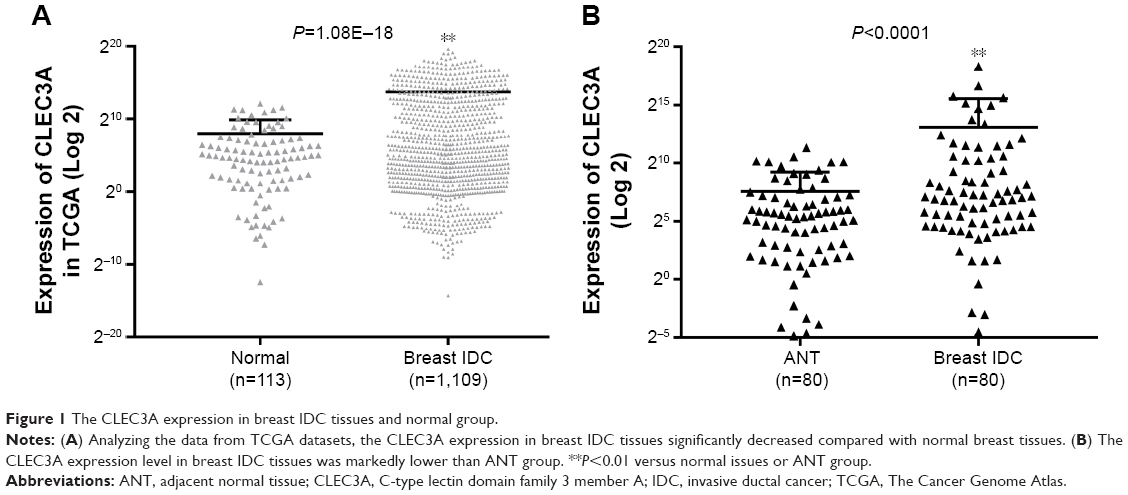 | Figure 1 The CLEC3A expression in breast IDC tissues and normal group. |
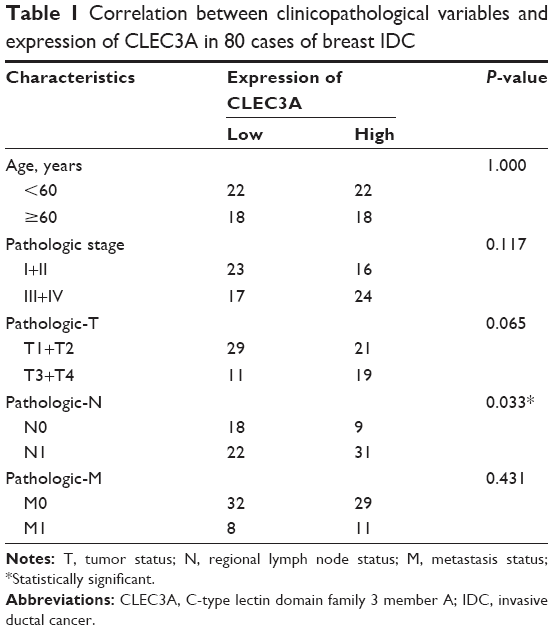 | Table 1 Correlation between clinicopathological variables and expression of CLEC3A in 80 cases of breast IDC |
Prognostic significance of CLEC3A expression in breast IDC
The prognostic impact of CLEC3A expression was analyzed in 80 patients with IDC by Kaplan–Meier OS curves. We found that high CLEC3A expression was associated with poor prognosis (Figure 2). Cox’s proportional hazards regression analysis was also applied to analyze the prognostic relevance to CLEC3A, Pathologic stage, Pathologic-T, Pathologic-M, Pathologic-N, and age in OS. In line with the Kaplan–Meier OS curves, as shown in Table 2, univariate analysis revealed that high CLEC3A (HR=3.603, 95% CI: 1.423–9.121, P=0.007), Pathologic stage (I+II/III+IV) (HR=5.263, 95% CI: 2.129–13.010, P<0.001), Pathologic-T (T1+T2/T3+T4) (HR=4.654, 95% CI: 2.031–10.668, P<0.001), and Pathologic-M (M0/M1) (HR=4.508, 95% CI: 2.004–10.139, P<0.001) were significant predictors of OS. To determine the independent prognostic factors for breast IDC patients in the current cohort, the multivariate Cox’s proportional hazards regression analysis was performed by adopting all the potentially significant variables in univariate analyses. Moreover, the multivariate analysis revealed that high CLEC3A was significantly associated with shorter OS (HR=2.829, 95% CI: 1.045–7.659, P=0.041), while other factors were not significantly correlated with OS in breast IDC patients. Notably, the results suggest that high CLEC3A expression is an independent negative prognostic marker of OS in breast IDC patients.
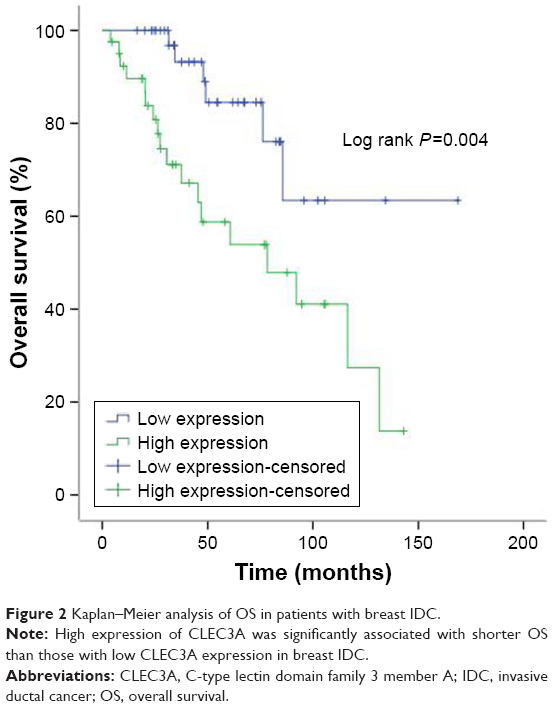 | Figure 2 Kaplan–Meier analysis of OS in patients with breast IDC. |
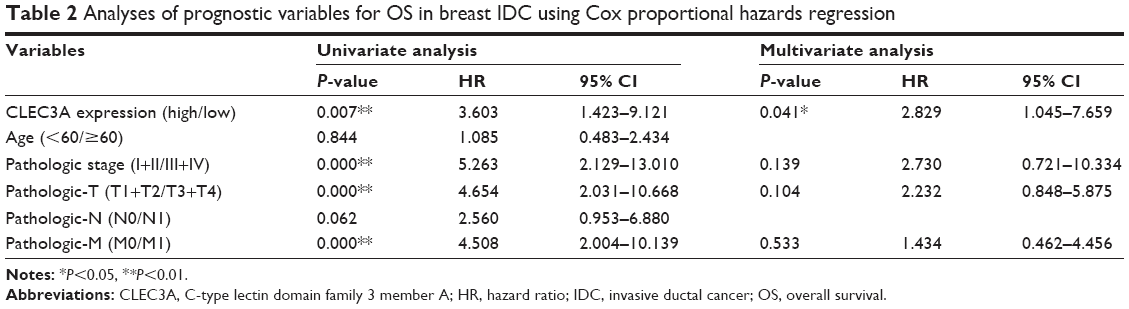 | Table 2 Analyses of prognostic variables for OS in breast IDC using Cox proportional hazards regression |
High expression level of CLEC3A in BC cell and CLEC3A knockdown inhibits BC cells proliferation
First, to explore the expression of CLEC3A in BC cells, CLEC3A at mRNA and protein level in BC cell lines BT474, MDA-MB-231, MCF-7, and normal breast cells CCD-1095Sk were examined. As shown in Figure 3, CLEC3A was obviously upregulated in BC cells, including BT474, MDA-MB-231, MCF-7, in comparison with CCD-1095Sk cells. In addition, the level of CLEC3A in BT474 was the highest in these cell lines; therefore, we used the BT474 cell to perform the subsequent experiments. Second, efficacy of CLEC3A knockdown was assessed using qRT-PCR and Western blot assay. The results showed that the level of CLEC3A both in mRNA and protein was lower evidently in si-CLEC3A group than si-NC group (Figure 4A and B, P<0.01). To examine the proliferation of BT474 cells, we performed CCK-8 assay. The data suggested that the OD value for the si-CLEC3A group was dramatically decreased at 72 h and at 96 h compared with the si-NC group (Figure 5A, P<0.01). Next, to further assess the effect of CLEC3A knockdown on BC cells viabilities, colony formation assay was carried out by measuring the colony formation rate. As shown in Figure 5B, the colony formation rate of si-CLEC3A group was obviously lower than that of the si-NC group (P<0.01), suggesting that CLEC3A knockdown inhibited clonogenesis of BT474 cells. Collectively, these data indicate that knockdown of CLEC3A suppresses BC cells proliferation.
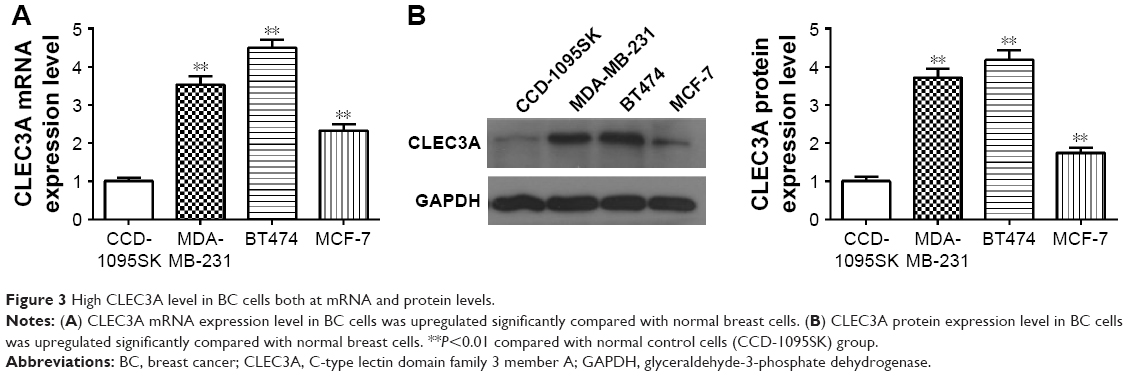 | Figure 3 High CLEC3A level in BC cells both at mRNA and protein levels. |
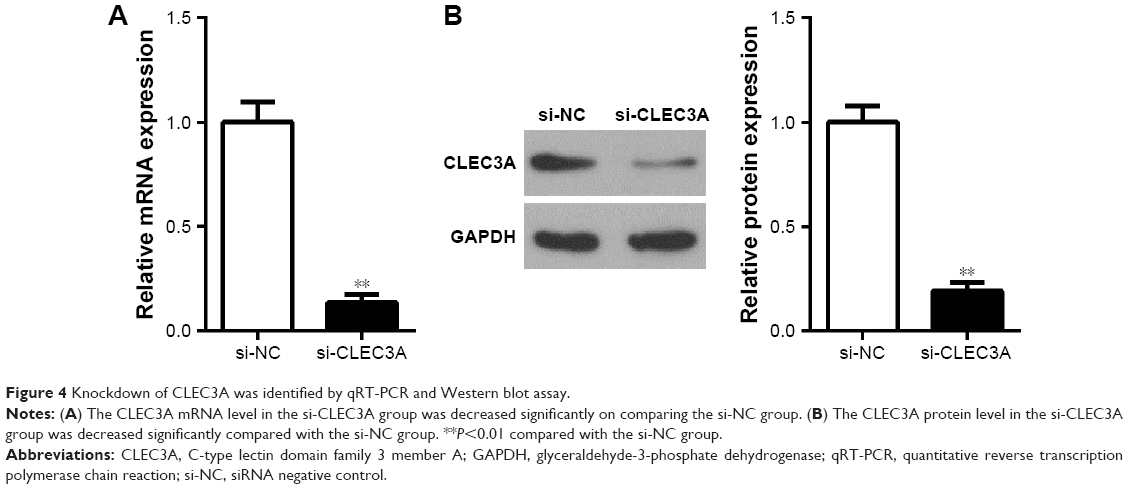 | Figure 4 Knockdown of CLEC3A was identified by qRT-PCR and Western blot assay. |
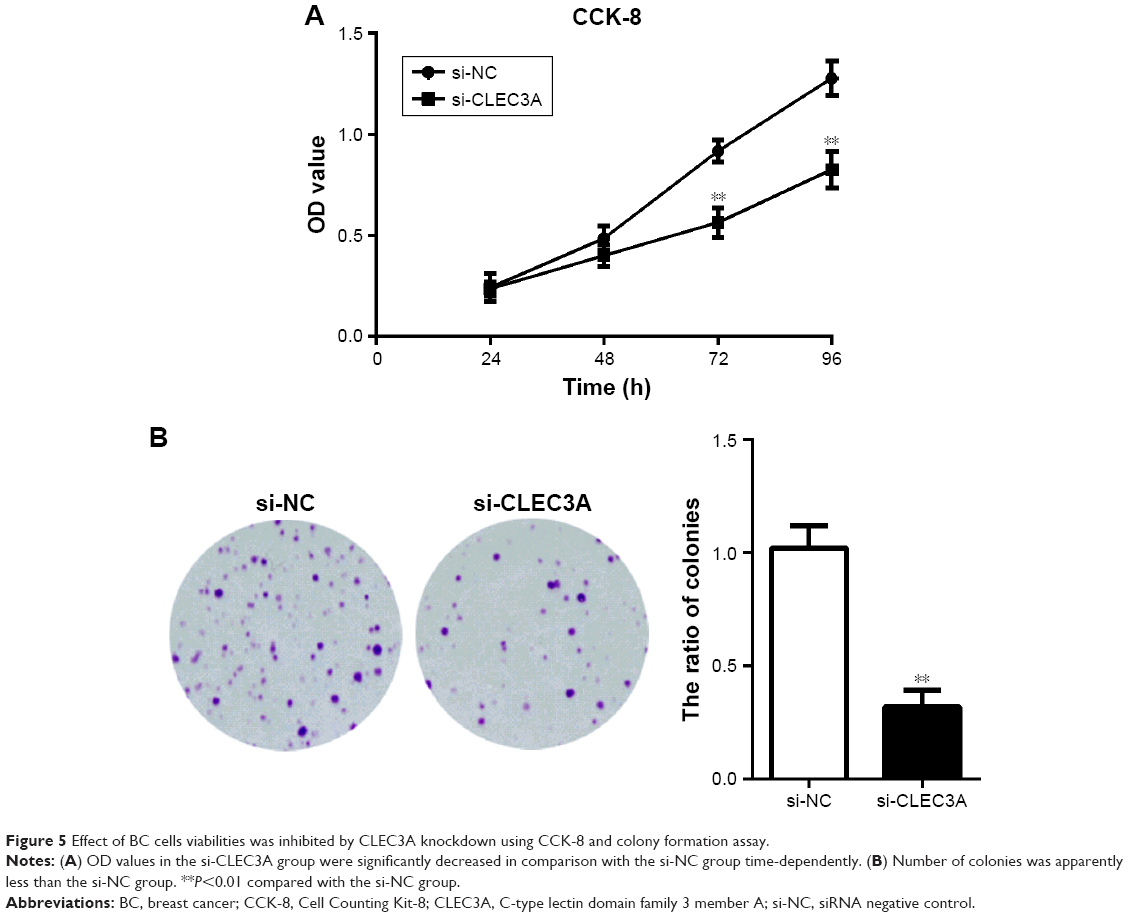 | Figure 5 Effect of BC cells viabilities was inhibited by CLEC3A knockdown using CCK-8 and colony formation assay. |
CLEC3A knockdown prohibits BC cells migration and invasion
To test the effect of the CLEC3A knockdown on BT474 cells migration and invasion, wound-healing and transwell assay were performed. The wound-healing results showed in Figure 6A that CLEC3A knockdown markedly delayed wound closure of BT474 cells. Furthermore, transwell assay results also showed that migrated and invaded BT474 cells number was markedly decreased in BT474 cells after silencing CLEC3A expression in comparison with si-NC group (Figure 6B and C, P<0.01). These results indicate that CLEC3A knockdown represses BC cells proliferation, migration, and invasion.
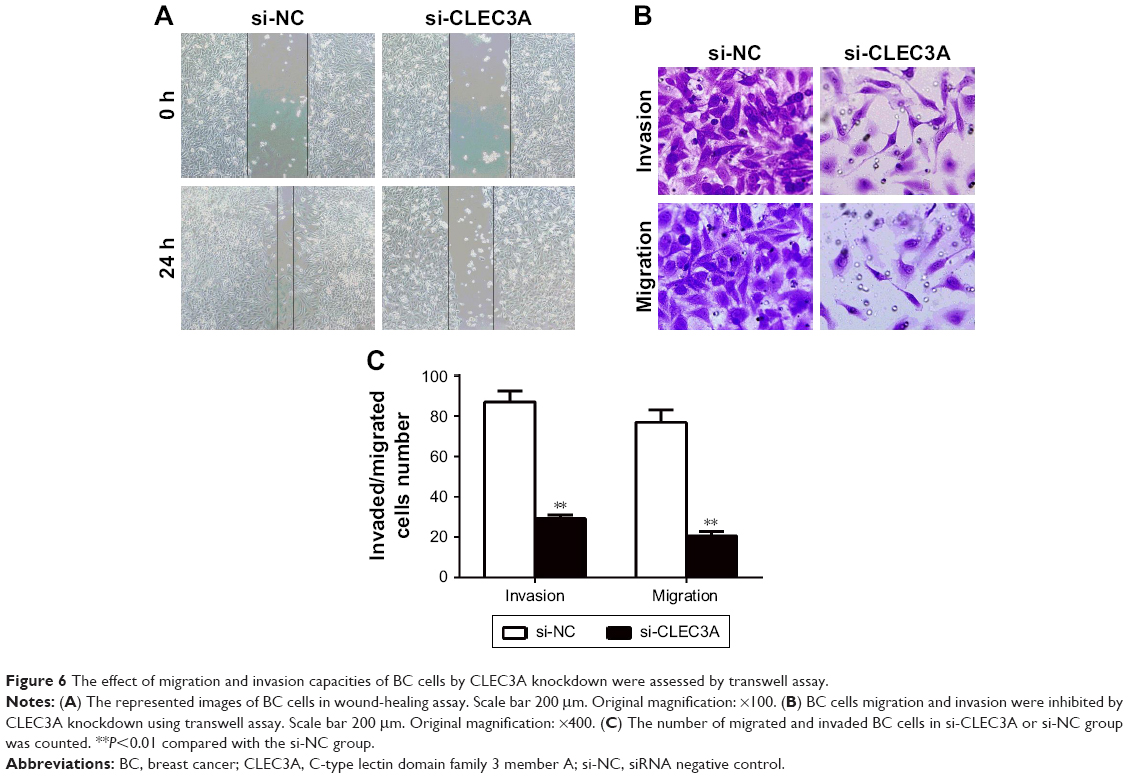 | Figure 6 The effect of migration and invasion capacities of BC cells by CLEC3A knockdown were assessed by transwell assay. |
Effect of CLEC3A on survival factors
Having established that cell survival regulator genes are involved in the proliferation, cell cycle progression, cell apoptosis, and cellular transformation, in an effort to investigate whether CLEC3A affect survival factors, c-Myc, Cyclin D1, and Bcl-2 were examined by Western blot analysis. The result showed that c-Myc, Cyclin D1, and Bcl-2 proteins levels were markedly reduced in the si-CLEC3A group compared with the si-NC group (Figure 7, P<0.01). The result suggests that these cell survival regulatory molecules are involved in suppressive effect on cell proliferation and metastasis by CLEC3A silencing.
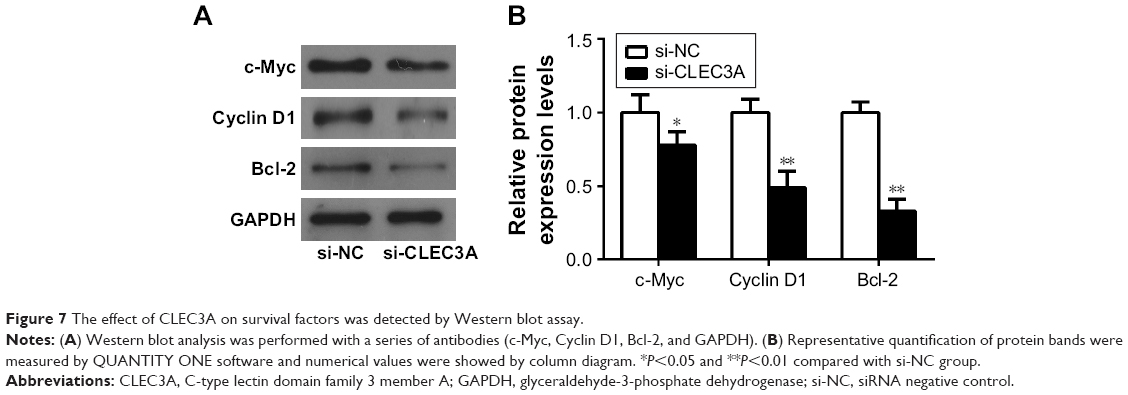 | Figure 7 The effect of CLEC3A on survival factors was detected by Western blot assay. |
CLEC3A knockdown decreases the activity of PI3K/AKT signaling
Previous studies30,31 have suggested that the PI3K/AKT signaling pathway was closely related to cancer progression. To explore the underlying mechanism, we measured expression levels of PI3K/AKT signaling-related marker PI3K/p-PI3K, AKT/p-AKT and p-P70S6K in both proteins levels using the Western blot assay. The results showed that si-CLEC3A transfection induced downregulated expressions of p-PI3K, p-AKT, and p-P70S6K while no detectable observations of PI3K and AKT expression levels (Figure 8). These data suggest that inactivated PI3K/AKT signaling may be involved in the inhibitory effects of BC cells proliferation, migration, and invasion induced by CLEC3A silencing.
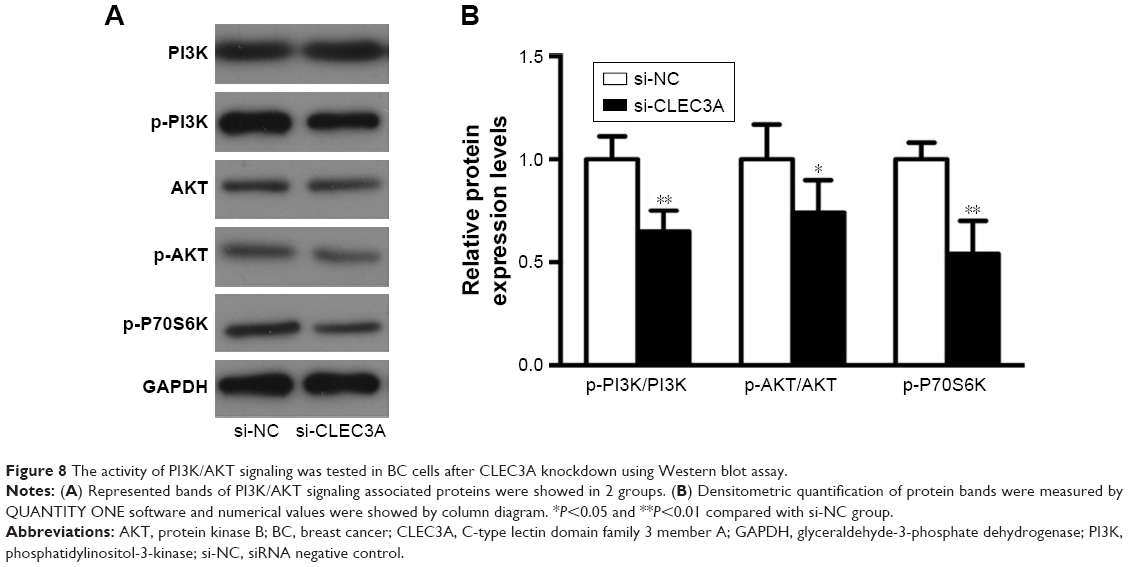 | Figure 8 The activity of PI3K/AKT signaling was tested in BC cells after CLEC3A knockdown using Western blot assay. |
Discussion
In this study, the expression of CLEC3A was higher in IDC patients. Additionally, high CLEC3A expression notably correlated with higher lymph node metastasis and the poor prognosis of breast IDC. Moreover, it was observed that knockdown of CLEC3A by RNAi approach impeded the proliferation, migration, and invasion of BC cells; furthermore, the activities of survival factor and PI3K/AKT signaling were downregulated by CLEC3A knockdown, demonstrating that CLEC3A is a potential gene target in IDC therapy.
BC is the most common neoplasm diagnosed and mortal among women worldwide. Its pathogenesis mechanism remains to be probed. In this study, it was demonstrated that CLEC3A expression level was higher CLEC3A in breast IDC than ANT samples, besides, elevation of it associated with higher lymph node and negative postoperative prognosis of breast IDC patients. As is known to all, tumor metastasis has been the challenge of therapy in numerous cancers. Thus, it was pointed out by our finding to some extent that CLEC3A would be a therapeutic breach for BC. It was determined that high CLEC-2 expression was correlated with unfavorable prognosis of renal cell carcinoma.18 Moreover, we further found that knockdown of CLEC3A inhibited BC cells proliferation, invasion, and migration. It was identified that CLEC-2 promoted hematogenous tumor metastasis in mice in the previous study.19 CLEC-2 was also recognized to influence the tumor cell migratory and invasive properties.20 CLEC-2 activation was thought to promote efficient motility of dendritic cell and trigger cell spreading.21 Activity of CLEC-2 was inhibited, which was related to anti-metastatic potency of cancer.22 Our obtained data, in line with the previous investigations, might indicate that CLEC3A could be a predictive marker for BC cancer progression.
It has been established that cell survival factors are responsible for cell growth, apoptosis, and migration, which play a crucial role in various types of cancers, including BC.23,24 Previous studies demonstrated that Cyclin D1 was found to be upregulated in human BCs, which predicted poor outcome of BC patient.25–27 Prior studies had assessed that c-Myc was a strong proto-oncogene and it was often found to be upregulated in many types of tumors.28,29 Bcl-2, identified as the best-characterized regulators of apoptosis, has been critical in determining how cells respond to apoptotic or survival signals. In the current work, we detected c-Myc, Cyclin D1, and Bcl-2 expression levels and found that these survival factors were decreased by knockdown of CLEC3A. These observations indicate that CLEC3A functions as a potential oncogenic action in BC progression.
To further investigate the mechanism underlying drives the anti-proliferative and anti-metastatic effect of CLEC3A knockdown on BC, we assessed the activity of PI3K/AKT signaling. It is well known that aberrant PI3K/AKT signaling is recognized in multitude of cancers and has been demonstrated to be mediated in carcinoma cells proliferation, survival, migration, and invasion.30,31 PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol 3,4,5-triphosphate that binds to AKT, a serine-threonine protein kinase identified as the central mediator of PI3K pathway, leading to the phosphorylation of Akt. P-AKT elicits the activation of the mammalian target of rapamycin complex, and thus is responsible for the regulation of p-P70S6K, which modulates the cell growth.32,33 Hereby, PI3K/p-PI3K, p-AKT/AKT, and p-P70S6K were detected in this study, their relative expression levels were obviously decreased in BC cells after silencing CLEC3A in comparison with si-NC transfection group. It was identified that the activation of this pathway was associated with enhanced cell growth and clinical outcome, and its overexpression correlated with a worse prognosis of tumor, which proposed it might be a therapeutic potential strategic point for BC.34,35 Aforementioned evidence demonstrates that PI3K/AKT signaling may be involved in the action of CLEC3A knockdown inducing anti-proliferation and anti-metastasis potency of BC. In spite of this, limitation of the current work should be acknowledged. Only one cell line was used and animal model was absent. Besides, in-depth mechanistic verification would be warranted in the future study.
Conclusion
The data of the current study showed that CLEC3A was elevated in IDC patients, and overexpression of CLEC3A was associated with higher lymph node metastasis and poor prognosis of IDC. Furthermore, our present study suggested that knockdown of CLEC3A by RNAi approach efficiently inhibited BC cells proliferation, migration, and invasion, which may be mediated by PI3K/AKT signaling pathway. Our findings unveiled the anti-cancer effect of CLEC3A knockdown in IDC as well as possibly the underlying mechanism, implying CLEC3A would be a promising biological target for IDC treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
McGuire A, Brown JA, Malone C, McLaughlin R, Kerin MJ. Effects of age on the detection and management of breast cancer. Cancers (Basel). 2015;7(2):908–929. | ||
Eccles SA, Aboagye EO, Ali S, et al. Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer. Breast Cancer Res. 2013;15(5):R92. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Kanbayashi C, Iwata H. Current approach and future perspective for ductal carcinoma in situ of the breast. Jpn J Clinl Oncol. 2017;47(8):671–677. | ||
Stuart KE, Houssami N, Taylor R, Hayen A, Boyages J. Long-term outcomes of ductal carcinoma in situ of the breast: a systematic review, meta-analysis and meta-regression analysis. BMC Cancer. 2015;15:890. | ||
Adar A, Canyilmaz E, Kiris A, et al. Radiotherapy induces development of fragmented QRS in patients with breast cancer. Breast Care (Basel). 2015;10(4):277–280. | ||
Obi N, Eulenburg C, Seibold P, et al. Associations between adjuvant radiotherapy and different causes of death in a German breast cancer cohort. Breast. 2017;38:75–80. | ||
Kalra R, Chavada B, Madhani NR, Purohit B, Tripathi CB. Cyclophosphamide and/or anthracyclines induced Epiphora in breast cancer patients: a rare side-effect. Curr Drug Saf. 2018;13(1):62–64. | ||
Ojo D, Wu Y, Bane A, Tang D. A role of SIPL1/SHARPIN in promoting resistance to hormone therapy in breast cancer. Biochim Biophys Acta. 2018;1864(3):735–745. | ||
Zelensky AN, Gready JE. The C-type lectin-like domain superfamily. FEBS J. 2005;272(24):6179–6217. | ||
Neame PJ, Young CN, Treep JT. Primary structure of a protein isolated from reef shark (Carcharhinus springeri) cartilage that is similar to the mammalian C-type lectin homolog, tetranectin. Protein Sci. 1992;1(1):161–168. | ||
Karlsson C, Dehne T, Lindahl A, et al. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage. 2010;18(4):581–592. | ||
Tsunezumi J, Higashi S, Miyazaki K. Matrilysin (MMP-7) cleaves C-type lectin domain family 3 member A (CLEC3A) on tumor cell surface and modulates its cell adhesion activity. J Cell Biochem. 2009;106(4):693–702. | ||
Boguslawska J, Rodzik K, Poplawski P, et al. TGF-beta1 targets a microRNA network that regulates cellular adhesion and migration in renal cancer. Cancer Lett. 2018;412:155–169. | ||
Lau D, Elezagic D, Hermes G, et al. The cartilage-specific lectin C-type lectin domain family 3 member A (CLEC3A) enhances tissue plasminogen activator-mediated plasminogen activation. J Biol Chem. 2018;293(1):203–214. | ||
Duffy MJ, McGowan PM, Harbeck N, Thomssen C, Schmitt M. uPA and PAI-1 as biomarkers in breast cancer: validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014;16(4):428. | ||
Resmini G, Rizzo S, Franchin C, et al. HMGA1 regulates the Plasminogen activation system in the secretome of breast cancer cells. Sci Rep. 2017;7(1):11768. | ||
Xiong Y, Liu L, Xia Y, et al. High CLEC-2 expression associates with unfavorable postoperative prognosis of patients with clear cell renal cell carcinoma. Oncotarget. 2016;7(39):63661–63668. | ||
Shirai T, Inoue O, Tamura S, et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J Thromb Haemost. 2017;15(3):513–525. | ||
Lowe KL, Navarro-Nunez L, Watson SP. Platelet CLEC-2 and podoplanin in cancer metastasis. Thromb Res. 2012;129(Suppl 1):S30–S37. | ||
Acton SE, Astarita JL, Malhotra D, et al. Podoplanin-rich stromal networks induce dendritic cell motility via activation of the C-type lectin receptor CLEC-2. Immunity. 2012;37(2):276–289. | ||
Chang YW, Hsieh PW, Chang YT, et al. Identification of a novel platelet antagonist that binds to CLEC-2 and suppresses podoplanin-induced platelet aggregation and cancer metastasis. Oncotarget. 2015;6(40):42733–42748. | ||
Sun YS, Zhao Z, Yang ZN, et al. Risk factors and preventions of breast cancer. Int J Biol Sci. 2017;13(11):1387–1397. | ||
Xu H, Yu S, Liu Q, et al. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J Hematol Oncol. 2017;10(1):97. | ||
Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005;23(18):4215–4224. | ||
Peurala E, Koivunen P, Haapasaari KM, Bloigu R, Jukkola-Vuorinen A. The prognostic significance and value of cyclin D1, CDK4 and p16 in human breast cancer. Breast Cancer Res. 2013;15(1):R5. | ||
Pestell TG, Jiao X, Kumar M, et al. Stromal cyclin D1 promotes heterotypic immune signaling and breast cancer growth. Oncotarget. 2017;8(47):81754–81775. | ||
Wang C, Tai Y, Lisanti MP, Liao DJ. c-Myc induction of programmed cell death may contribute to carcinogenesis: a perspective inspired by several concepts of chemical carcinogenesis. Cancer Biol Ther. 2011;11(7):615–626. | ||
Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014;26(2):262–272. | ||
Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142(2):164–175. | ||
Tapia O, Riquelme I, Leal P, et al. The PI3K/AKT/mTOR pathway is activated in gastric cancer with potential prognostic and predictive significance. Virchows Arch. 2014;465(1):25–33. | ||
Shawky NM, Segar L. Sulforaphane inhibits platelet-derived growth factor-induced vascular smooth muscle cell proliferation by targeting mTOR/p70S6kinase signaling independent of Nrf2 activation. Pharmacol Res. 2017;119:251–264. | ||
Li Y, Wu D, Wang P, Li X, Shi G. miR-195 regulates proliferation and apoptosis through inhibiting the mTOR/p70s6k signaling pathway by targeting HMGA2 in Esophageal Carcinoma Cells. 2017;2017:8317913. | ||
Bahrami A, Khazaei M, Shahidsales S, et al. The therapeutic potential of PI3K/Akt/mTOR inhibitors in breast cancer: rational and progress. J Cell Biochem. 2018;119(1):213–222. | ||
Delaloge S, DeForceville L. Targeting PI3K/AKT pathway in triple-negative breast cancer. Lancet Oncol. 2017;18(10):1293–1294. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.