Back to Journals » International Journal of Nanomedicine » Volume 16
Overcoming Radiation Resistance by Iron-Platinum Metal Alloy Nanoparticles in Human Copper Transport 1-Overexpressing Cancer Cells via Mitochondrial Disturbance
Authors Tsai TL ![]() , Lai YH, Chen HHW, Su WC
, Lai YH, Chen HHW, Su WC
Received 23 September 2020
Accepted for publication 11 February 2021
Published 9 March 2021 Volume 2021:16 Pages 2071—2085
DOI https://doi.org/10.2147/IJN.S283147
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ebrahim Mostafavi
Tsung-Lin Tsai,1,2 Yu-Hsuan Lai,3,4 Helen HW Chen,1,2,4 Wu-Chou Su1,2
1Center of Applied Nanomedicine, National Cheng Kung University, Tainan, 701, Taiwan; 2Department of Oncology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, 701, Taiwan; 3Institute of Clinical Medicine, College of Medicine, National Cheng Kung University, Tainan, 701, Taiwan; 4Department of Radiation Oncology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, 701, Taiwan
Correspondence: Wu-Chou Su Rm. 6065, 6F., No. 35, Xiaodong Road, North Dist., Tainan, 704, Taiwan
Tel +886-6-2353535#3120
Fax +886-6-2084112
Email [email protected]
Helen HW Chen No. 138, Sheng Li Road, North Dist., Tainan, 704, Taiwan
Tel +886-6-2353535#2461
Fax +886-6-2359333
Email [email protected]
Background: Radiation therapy remains an important treatment modality in cancer therapy, however, resistance is a major problem for treatment failure. Elevated expression of glutathione is known to associate with radiation resistance. We used glutathione overexpressing small cell lung cancer cell lines, SR3A-13 and SR3A-14, established by transfection with γ-glutamylcysteine synthetase (γ-GCS) cDNA, as a model for investigating strategies of overcoming radiation resistance. These radiation-resistant cells exhibit upregulated human copper transporter 1 (hCtr1), which also transports cisplatin. This study was initiated to investigate the effect and the underlying mechanism of iron-platinum nanoparticles (FePt NPs) on radiation sensitization in cancer cells.
Materials and Methods: Uptakes of FePt NPs in these cells were studied by plasma optical emission spectrometry and transmission electron microscopy. Effects of the combination of FePt NPs and ionizing radiation were investigated by colony formation assay and animal experiment. Intracellular reactive oxygen species (ROS) were assessed by using fluorescent probes and imaged by a fluorescence-activated-cell-sorting caliber flow cytometer. Oxygen consumption rate (OCR) in mitochondria after FePt NP and IR treatment was investigated by a Seahorse XF24 cell energy metabolism analyzer.
Results: These hCtr1-overexpressing cells exhibited elevated resistance to IR and the resistance could be overcome by FePt NPs via enhanced uptake of FePt NPs. Overexpression of hCtr1 was responsible for the increased uptake/transport of FePt NPs as demonstrated by using hCtr1-transfected parental SR3A (SR3A-hCtr1-WT) cells. Increased ROS and drastic mitochondrial damages with substantial reduction of oxygen consumption rate were observed in FePt NPs and IR-treated cells, indicating that structural and functional insults of mitochondria are the lethal mechanism of FePt NPs. Furthermore, FePt NPs also increased the efficacy of radiotherapy in mice bearing SR3A-hCtr1-WT-xenograft tumors.
Conclusion: These results suggest that FePt NPs can potentially be a novel strategy to improve radiotherapeutic efficacy in hCtr1-overexpressing cancer cells via enhanced uptake and mitochondria targeting.
Keywords: FePt nanoparticles, human copper transporter 1, radiation resistance, reactive oxygen species, mitochondrial targeting
Introduction
Radiation therapy (RT) is a very important therapeutic modality to treat cancer and has been used in more than 50% of all cancer patients worldwide, either in curative or palliative aims.1–3 Over the past decades, RT techniques have improved dramatically, thanks to the advancement of imaging and engineering technologies that enable a highly tailored dose delivery to the tumor with maximum sparing of adjacent normal tissues. The overall tumor control rates of RT have improved from about 30% two decades ago to about 80% nowadays in some malignancies.4,5 Despite the improving therapeutic efficacy, many tumors still remain resistant to ionizing radiation.6
Platinum (Pt)-based drugs, especially cisplatin, have been the mainstay of cancer chemotherapy in many different types of human malignancies for the last four decades.7 In particular, the combination of cisplatin and ionizing radiation provides considerable synergistic antitumor activities and is the current standard of first-line treatment in several advanced cancers, such as lung, head and neck, and cervix.8–11 However, many patients eventually relapse and develop resistance to concurrent chemoradiation therapy with cisplatin. One major mechanism associated with resistance to cisplatin is downregulation of the human copper transporter 1 (hCtr1), which is the major copper influx transporter and has been convincingly demonstrated to transport cisplatin and its analogues, resulting in impaired drug intake.12 Although expression of hCtr1 is ubiquitous because all the tissues require copper, studies showed that expression levels of hCtr1 were highly variable among normal tissues and various major human malignancies.13 Studies also found that hCtr1 was overexpressed in nearly 70% of lung cancer and was both predictive and prognostic factors in patients received platinum-based chemotherapy.14
While multiple mechanisms are involved in radiation resistance, one important mechanism is the elevated expression of glutathione (GSH), which is an important physiological regulator of cellular redox conditions.3 Likewise, cisplatin-resistant variants are often overexpressed GSH. However, overexpressed GSH per se does not always result in cisplatin resistance.15 We previously demonstrated that the GSH-overexpressing small cell lung cancer cell lines, SR3A-13, and SR3A-14, established by transfection with γ-glutamylcysteine synthetase (γ-GCS) cDNA which encodes the rate-limiting enzyme for GSH biosynthesis, confer cisplatin sensitization but not resistance. This is because GSH is a copper chelator, which results in cellular Cu(I) depletion. This in turn leads to upregulation of hCtr1, which is also a transporter of cisplatin.16 These findings underscore the complex role of GSH in cisplatin and radiation sensitivities.
Recent advances in nanomedicine have provided a great deal of promising materials to improve the outcome of cancer treatment. Many platinum-based nanoparticles (Pt NPs) have been developed for clinical use,17 and some have shown radiation enhancement properties.18–20 To gain better insights into the roles of Pt NPs in cancer therapy, we synthesized iron-platinum metal alloy nanoparticles (FePt NPs) and demonstrated that FePt NPs exhibited excellent biocompatibility and great contrast ability for both magnetic resonance imaging (MRI) and computed tomography (CT) imaging.21 However, the therapeutic roles of FePt NPs have not been explored. Using the GSH/hCtr1-overproducing cell lines, we report here that radiation resistance in these cells can be overcome by FePt NPs. We further demonstrate that upregulation of hCtr1 alone can confirm radiation sensitivity by FePt NPs, despite the fact that the physical-chemical properties between FePt NPs and cisplatin are drastically different. Therefore, our results provide a previous undiscovered new role of FePt NPs in cancer radiation therapy.
Materials and Methods
Synthesis of 6 nm FePt Nanoparticles
The synthesis of FePt NPs with 6 nm in diameter was in accordance with the previous study.21 Briefly, platinum acetylacetonate (Pt(acac)2, ACROS, 97%) and iron pentacarbonyl (Fe(CO)5, Aldrich, 99.99%) 1,2-hexadecanediol (Aldrich, 90%), dioctyl ether (ACROS, 90%), oleyl amine (Aldrich, 70%), oleic acid (Aldrich, 90%), cysteamine (Sigma, 95%). Pt(acac)2 (97 mg), 1,2-hexadecanediol (195 mg), dioctyl ether (10 mL), Fe(CO)5 (66 µL), oleyl amine (100 µL), and oleic acid (100 µL) were mixed. The reaction mixture was heated to 240°C at a heating rate of 15°C/min. After 30 minutes under nitrogen, the heating source was removed and the product was cooled to room temperature. Then, the product was precipitated by adding ethanol and separated by centrifugation. For ligand exchange to form water dispersion, FePt NPs (100 mg) were dispersed in ethanol by sonication. Cysteamine (~1 g) was added and dissolved into the solution at room temperature. The mixture was sonicated at 40−50°C overnight and washed by ethanol to remove adsorbent ligands. Finally, the modified particles were collected and stored in bottles filled with N2.
Cell Culture
SR3A was a doxorubicin-resistant cell line established from human small cell lung cancer (SCLC). SR3A and the development of its γ-GCS stably transfected cell lines, SR3A-13 and SR3A-14, were generous gift from Dr. Kuo MT and have been described previously.22 hCtr1-overexpressing (SR3A-hCtr1-WT) cells were established in our lab. All these SR3A series cells used in this study were approved by the ethics committee of National Cheng Kung University. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum at 37°C in 5% of CO2 atmosphere. Additionally, 400 μg/mL G418 (Thermo Fisher, MA, USA) was added for the maintenance of the transfected cell lines.
Western Blot Analysis
Cells plated overnight were washed with 1 x PBS twice and harvested with NP40 cell lysis buffer [50 mM Tris, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate containing 1 x protease inhibitor cocktail]. Equal amounts of protein (50 μg) were loaded to 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred onto PVDF membranes (Millipore Corporation, Billerica, MA, USA). Nonspecific binding sites were eliminated using 5% fat-free milk in TBS with 0.1% Tween 20 (TBST) at room temperature for 1 hour. The membranes were then incubated with each primary antibody, β-actin (43 kDa, MAB1501, Merck Millipore, Darmstadt, Germany), hCtr-1 (25 kDa, NB100-402, Novus Biologicals, LLC, USA), and γ-GCS (73 kDa, sc-166382, Santa Cruz, CA, USA) at 4°C for 16 hours. After incubation, membranes were washed with TBST buffer three times and reacted with the corresponding peroxidase-conjugated anti-rabbit or mouse secondary antibodies at room temperature for 1 hour. The protein signals were detected by chemiluminescence using the HRP Substrate Luminol Reagent (Millipore, Billerica, USA) and visualized using the BioSpectrum Imaging System (UVP, Upland, CA, USA).
Clonogenic Cell Survival Assay
Cells (4 x 105) were seeded into 5-mL flasks and incubated overnight. FePt NPs were added into cells to yield a final concentration of 1 mg/mL for 24 hours before irradiation. Then, cells were irradiated with 6 MV photons for different doses of X-rays (0, 2, 4, 6, and 8 Gy), using a linear accelerator (Clinac iX, Varian Medical Systems, Palo Alto, CA, USA) at the National Cheng Kung University Hospital. Following irradiation, cells were incubated at 37°C for 4 hours before washed with PBS and trypsinized. After exact cell counting, cells of appropriate number were reseeded in triplicates onto 10-cm dishes and left undisturbed for 7 to 14 days (7 days for SR3A, and 14 days for other transfected cells) under normal culture conditions. Then, cells were fixed and stained with crystal violet. Colonies of at least 50 cells were counted manually and surviving fractions were calculated with a correction necessary for plating efficiency. The sensitizer enhancement ratio (SER) was calculated as the radiation dose needed for radiation alone divided by the dose needed for FePt NPs plus radiation at a surviving fraction of 37% (D0 in radiobiology).
Transmission Electronic Microscopy Analysis
Cells were incubated with FePt NPs (1 mg/mL) for 24 hours, washed with cold PBS three times and fixed with 2% paraformaldehyde and 2.5% glutaraldehyde for 30 minutes at room temperature. Cells were then post-fixed with 1% osmium tetraoxide in 0.1 M Na-cacodylate buffer, pH 7.2, for 1 hour, washed and dehydrated in graded concentrations of ethanol (50%, 70%, and 100%) and propylene oxide. Cell samples were then embedded in Epon (Fluka, Buchs, Switzerland) and ultrathin sections were made. Thin sections of 80 nm were collected on copper TEM grids and stained with 5% uranyl acetate for 20 minutes and lead citrate for 10 minutes. The grids were analyzed on a JEOL-1200 Transmitting Electron Microscope at an accelerating voltage of 80 kV.
Cellular Uptake Measurements of FePt NPs
Cells were incubated with 1 mg/mL FePt NPs for 24 hours, collected and neutralized in concentrated nitric acid for 5 minutes on a heating block. Then, the Fe concentrations in the cells in this given volume were measured by inductively coupled plasma optical emission spectrometry (ICP-OES; ThermoFisher iCAPTM 7400, MA, USA).
Flow Cytometry Measurements of ROS
Cells were seeded into 6-well plates overnight and treated with 1 mg/mL FePt NPs for 24 hours, then with or without 6 Gy irradiation. Dihydrorhodamine123 (DHR123), used as a probe, was added at a final concentration of 1 μM for 4 hours. A single-cell suspension was then prepared. Rhodamine123 fluorescence intensity resulting from DHR123 oxidation was measured by a FACS caliber flow cytometry (BD Biosciences, San Jose, CA, USA) with excitation at 488 nm and detection at 525 nm. The final data were analyzed using FlowJo Software. All samples were performed in triplicate to acquire the mean fluorescence intensity (MFI).
Assessment of Mitochondrial Respiration Function by XF24 Cell Analyzer
Cells were seeded with different cell densities according to each experiment purpose in XF Flux-Plates with sensor cartridges (Agilent, Santa Clara, CA, USA) overnight. Then, cells were cultured for 24, 48 and 72 hours in medium with FePt NPs (0.03125 ~ 1 mg/mL) or combined with irradiation. The oxygen consumption rate (OCR) was analyzed by the XF24 cell analyzer (Agilent, Santa Clara, CA, USA) during sequential injection of 1 µM oligomycin, 0.5 µM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), and 1 µM rotenone/antimycin A (Seahorse XF Cell Mito Stress Test Kit 103015–100), respectively, into each group. Dynamic OCR profiles were obtained and converted into the reserved respiratory capacity of mitochondria.
Xenograft Animal Model
SR3A-hCtr1-WT cells (1 × 107 cells) were subcutaneously injected into the dorsal flank of SCID mice (6 to 8 weeks old; National Cheng Kung University Laboratory Animal Center). When the tumor size reached approximately 50 ~ 80 mm3, the mice were randomly assigned to four groups: PBS (control), FePt NPs, 6 Gy X-rays, and FePt NPs plus 6 Gy X-rays treatments (5 mice per group). PBS or FePt NPs (25 mg/kg) were injected into the tail veins of the mice every other day (48 hours apart) for total 2 injections in each group. Approximately 24 hours after the last injection, tumor regions were irradiated with 6 Gy using a linear accelerator with customized lead blocks. Tumor volumes were measured using digital calipers and calculated using the formula: 0.5 × (length × width2). All experiment procedures and handling of the animals were approved by the Institutional Animal Care and Use Committee (IACUC) of National Cheng Kung University (NO.108212) and followed the Guidelines for the Care and Use of Laboratory Animals issued by the Council of Agriculture Executive Yuan, Taiwan.
Tissue Stain
The tumor tissues were excised and prepared for immunohistochemistry (IHC) tissue stain after 24 hours of administration of PBS or FePt NPs. Briefly. Formalin-fixed paraffin-embedded tissue sections were deparaffinized and rehydrated. The tissue sections were counterstained with hematoxylin and eosin (H&E), Prussian blue and hCtr1 antibody, respectively. Slides were then washed, mounted with coverslip, and examined under a digital microscope.
Statistical Analysis
All measurements were represented as mean ± standard deviation. One-way ANOVA tests were conducted to assess the statistical significance between different groups at the two-tailed significance level of 0.05.
Results
Elevated Expression of GSH in γ-GCS-Transfected Cells Increases hCtr1 Expression and Exhibits Radioresistance Which is Associated with Enhanced FePt NPs Transport
We previously established several stable transfected cell lines by transfecting human full-length γ-GCS cDNA expression recombinant into SR3A cells, designated SR3A-13 and SR3A-14 cells, were used as a cell model in this study. SR3A-13 and SR3A-14 cells exhibited 1.92- and 1.69-fold increases of GSH levels; and 1.85- and 1.81-fold increases of hCtr1 levels compared with those in SR3A cells (Figure 1A). We previously reported that enhanced hCtr1 expression in the GSH-overexpressing cells is due to the fact that GSH chelates cellular Cu ions, resulting in reduced reduction of cellular bioavailable Cu levels and the consequential upregulation of hCtr1 expression by the mechanism of Cu homeostasis regulation.15,16 Intriguingly, SR3A-13 and SR3A-14 cells exhibited significant radiation resistance after 8 Gy X-rays by colony formation assay as compared with their parental cell line SR3A (Figure 1B). This is most likely due to the antioxidant properties of GSH that circumvents radiolysis associated with irradiation.3 Cell survival curves also revealed that SR3A-13 and SR3A-14 were more resistant to X-rays than SR3A (Figure 1C). These results demonstrated that elevated expression of GSH by γ-GCS transfection enhanced hCtr1 expression as well as cellular resistance to X-ray irradiation.
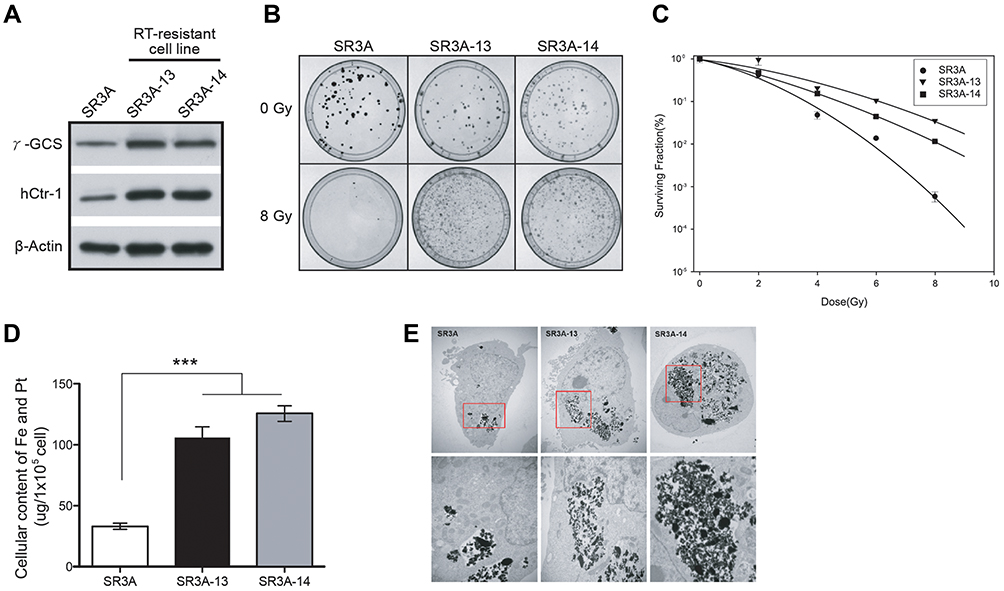 |
Figure 1 γ-GCS overexpressed SR3A-13 and SR3A-14 cells exhibited the radioresistant phenomena and enhanced uptake of FePt NPs. (A) Western blotting analysis of γ-GCS and hCtr1 protein levels in SR3A, SR3A-13 and SR3A-14 cells. β-actin was used as a loading control. (B) Representative colony formation photographs of SR3A, SR3A-13 and SR3A-14 cells irradiated with or without 8 Gy radiation. Noted that number of cells per dish initially plated varied with the dose so that the number of colonies surviving was in the range that could be counted conveniently. (C) Cell survival curves for SR3A series cells exposed to radiation. The surviving fractions of SR3A-13 and SR3A-14 cells were significantly higher than that of the control SR3A cells. (D) Total iron and platinum content determined by ICP-OES (***P < 0.001). Error bars represent ± S.D. (E) Representative TEM images of SR3A, SR3A-13 and SR3A-14 cells treated with FePt NPs. Note that FePt NPs were mainly found in vesicles located in the cytoplasm. Shown in the bottom are high power view of images in red squares shown above. |
hCtr1 has been known to effectively transport cisplatin and carboplatin but not oxaliplatin.23 It is unclear whether enhanced hCtr1 expression could promote FePt NPs transport. To investigate whether hCtr1 facilitated transport of FePt NPs, SR3A, SR3A-13 and SR3A-14 cells were incubated with FePt NPs for 24 hours. Cellular content of FePt NPs were measured by ICP-OES (Figure 1D). We found that cellular content of FePt NPs in SR3A, SA3A-13 and −14 were 33 ± 5.54, 105.4 ± 21.72, and 125.4 ± 15.84 μg per 105 cells, respectively, indicating that uptake of FePt NPs was 3 to 3.5 folds higher in SR3A-13 and SR3A-14, than that of SR3A cells (P <0.001). Higher uptake of FePt NPs was evident in SR3A-13 and SR3A-14 cells by visualizing cellular distribution of the NPs by TEM (Figure 1E). It is noteworthy that these NPs formed large opaque aggregates, mainly located in the cytoplasmic compartment and very few, if any, were observed inside the nucleus. Strikingly, we found that these aggregates were mainly confined in intracellular vacuoles and seemed to be inter-connected (Figure 1E, red squares). This distribution was in drastic contrast with that of intracellular distributions of cisplatin, which is widely distributed in various cellular compartments (eg, mitochondria, Golgi-apparatus, and nuclei) carried by various Cu chaperons once that cisplatin enters the cells.24
The Morphology and Aerobic Respiratory Function of Mitochondria are Altered in SR3A-13 and SR3A-14 Cells After FePt NPs Treatment
The lethal effects of platinum-based antitumor agents are mainly executed on DNA damages inside the nucleus. However, the absence of nuclear localization of FePt NPs prompted us to look for cytoplasmic organelles that may be targets of its cytotoxic effects. Strikingly, TEM revealed that SR3A-13 and SR3A-14 cells exhibited numerous irregularly shaped mitochondria with increased membrane density and absent ridges following cellular uptake/transport of FePt NPs (Figure 2A, red arrowhead), compared with those in SR3A (Figure 2A, black arrow) and untreated control cells (Supplementary Information, Figure S1). We next investigated the mitochondrial respiratory function by Seahorse XF24 analyzer, which measures OCR in living cells. Three drugs were added sequentially. Oligomycin (used in Step 1), an ATP synthase inhibitor, reduces the oxygen consumption needed to assist ATP synthesis.25 Carbonyl cyanide 4-(trifluoromethoxyphenylhydrazone, FCCP) (used in Step 2), behaving like an uncoupling agent for proton reflux, contributes a great amount of mitochondria oxygen consumption which was validated with different concentrations (0.5, 1.0 and 2.0 μM) to optimize the rate of oxygen consumption (Supplementary Information, Figure S2). Thus, the increase in oxygen consumption after the addition of FCCP represents the maximum oxygen consumption capacity of mitochondria.26 Rotenone (used in Step 3), an inhibitor of the mitochondrial aerobic respiratory chain, inhibits mitochondrial oxygen consumption completely.27 Real-time monitoring of mitochondrial OCR in SR3A, SR3A-13 and SR3A-14 cells for different time and concentrations of FePt NPs were investigated. The OCR values in SR3A-13 and SR3A-14 cells treated with FePt NPs were significantly lower than that in SR3A cells. Moreover, a rapid and pronounced reduction in basal and maximal mitochondrial oxygen consumption in a time- and concentration-dependent manner was observed under the same comparison (Figure 2B and C). Taken together, we demonstrated that increased uptake of FePt NPs impacts mitochondrial morphology and related respiratory function, thus resulting in structural and functional damages of mitochondria.
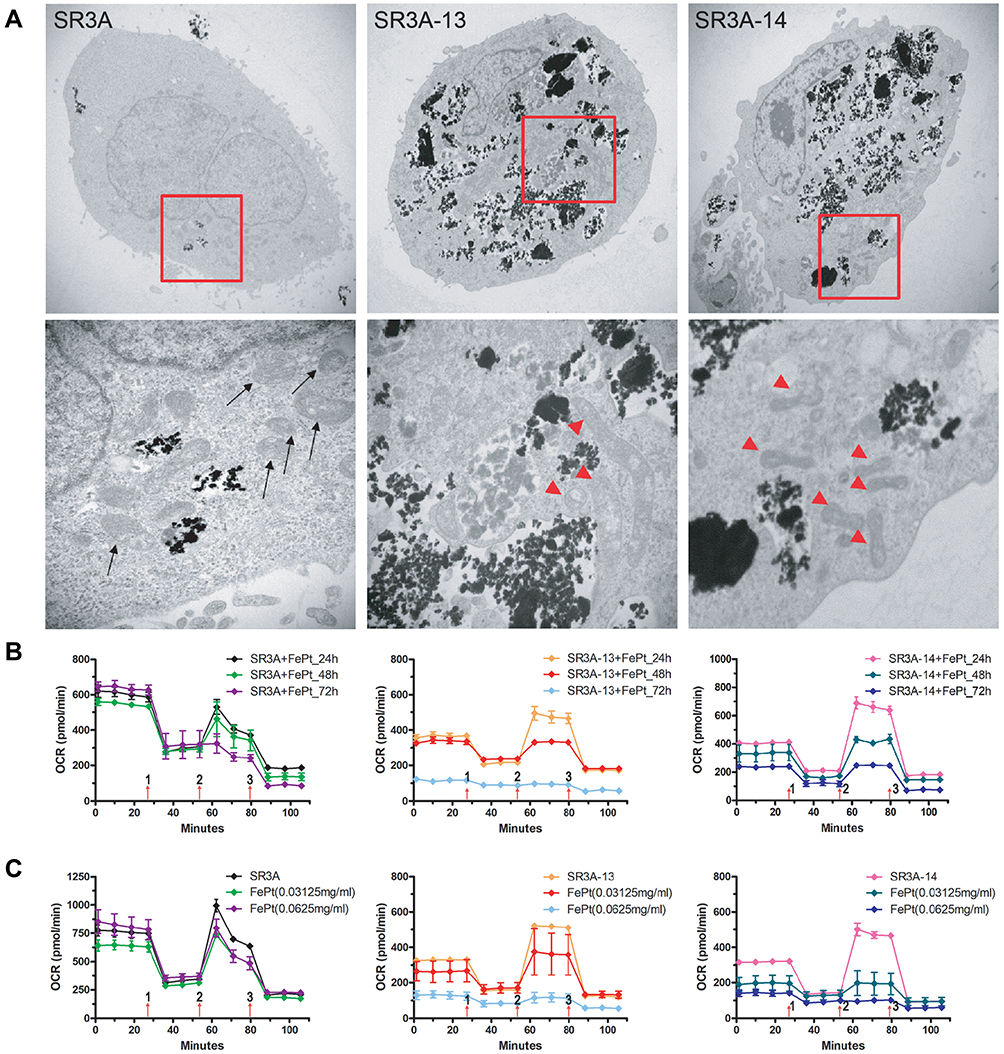 |
Figure 2 Mitochondrial morphology and function were altered in FePt NPs-treated SR3A-13 and SR3A-14 cells. (A) TEM observations of mitochondria in cells as indicated treated with 1 mg/mL FePt NPs for 24 hours. Black arrow, normal mitochondrial; red arrowheads, abnormal mitochondria with increasing membrane density and loss of ridges. (B) Measurements of mitochondrial respiratory function, OCR, by Seahorse XF24 analyzer in SR3A, SR3A-13 and SR3A-14 cells treated with FePt NPs. The OCR values in SR3A-13 and SR3A-14 cells were significantly lower than that in SR3A cells, in a time-dependent manner. (C) Similar findings as in (B), but in concentration-dependent manner. |
Combination Treatment of FePt NPs and X-Rays Enhances ROS Production and Increases Radiation Sensitivity in SR3A-13 and SR3A-14 Cells
Since mitochondria are the powerhouse of cellular metabolism and the major site of ROS production, proper functional mitochondria are essential for cell living. To investigate whether FePt NPs enhance ROS production after irradiation, we examined the amounts of ROS in SR3A, SR3A-13 and SR3A-14 cells treated with FePt NPs and X-rays by analyzing the fluorescence of DHR123 via flow cytometry. As shown in Figure 3A, ROS was increased after X-ray irradiation in all cells as compared with control. But ROS production was more significantly enhanced when combining X-rays with FePt NPs in SR3A-13 and SR3A-14 cells. Moreover, the aerobic respiratory function was drastically abolished by FePt NPs and X-rays in both SR3A-13 and SR3A-14 cells (Figure 3B). Colony formation assay also showed that colony number was much reduced in SR3A-13 and SR3A-14 cells treated with both FePt NPs and X-rays (Figure 3C). The cell survival curve further provided evidence of the radiation sensitizing effect of FePt NPs, with the SER values of 0.9, 2.3 and 2.1 in SR3A, SR3A-13, and SR3A-14 cells, respectively (Figure 3D). These results suggested that FePt NPs induced ROS burst and mitochondrial dysfunction, thus overcoming the radiation resistance of SR3A-13 and SR3A-14 cells.
 |
Figure 3 Enhancements of ROS production and radiosensitivity in SR3A-13 and SR3A-14 cells treated with 1 mg/mL FePt NPs for 24 hours and then irradiated with X-rays. (A) Flow cytometry analysis of ROS. Note that the combination treatment groups had significantly enhanced ROS levels than X-rays alone or control group especially in SR3A-13 and SR3A-14 cells. The result of MFI was presented as mean ± SD of three dependent experiments. ***p < 0.01. (B) Mitochondrial OCR by Seahorse XF24 analyzer showing mitochondrial functions were nearly totally abolished in SR3A-13 and SR3A-14 cells treated with FePt NPs and X-rays. (C) Representative photographs of colony formations of SR3A-13 and SR3A-14 cells treated with FePt NPs without (top row) or with (bottom) radiation (6 Gy). Noted that cell numbers seeded were different in 0 Gy and 6 Gy groups (500 vs 15,000 cells). (D) Cell survival curves of SR3A, SR3A-13 and SR3A-14 cells exposed to radiation with or without FePt NPs. |
Elevated hCtr1 Expression Confers Enhanced Uptake/Transport of FePt NPs Accompanied with Mitochondria Morphology Alteration and Aerobic Respiratory Function Loss
All the above experiments were performed using the γ-GCS-cDNA-transfected cells which overexpress GSH, a potent copper chelator, which then consequently depletes the bioavailable copper pool. Reduced copper levels lead to up-regulation of hCtr1 to homeostatically maintain proper cellular copper levels.16 To further demonstrate the causal link between increased expression of hCtr1 and enhanced uptake/transport of FePt NPs, we used wild-type hCtr1-transfected SR3A cells. Several stable clones were selected and identified by western bolts and ICP-OES for hCtr1 expression and related uptake of FePt NPs. Among which, clone #3 was picked and named as SR3A-hCtr1-WT for further experiments (Supplementary Information, Figure S3A). These cells exhibited higher levels of hCtr1 expression (Figure 4A) and FePt NPs uptake (Figure 4B and Supplementary Information, Figure S3B) as compared with those in the un-transfected SR3A cell. ICO-OES results show that uptake/transport capabilities of FePt NPs in SR3A-hCtr1-WT cells were enhanced 4.9 ± 1.9 fold higher than that in SR3A cells (Figure 4B). No significant effect for γ-GCS expression was observed in the SR3A-hCtr1-WT cells, indicating that hCtr1 and γ-GCS are not mutually regulated. TEM imaging again shows a large amount of FePt NPs inside the SR3A-hCtr1-WT cells, and the internalized FePt NPs were cytoplasmically localized in vacuoles and appeared in clusters (Figure 4C). In addition, mitochondrial morphological changes were evident, including variably shrunken or vanished cristae, and increased membrane density (Figure 4C, red arrowhead). Cellular OCR in SR3A-hCtr1-WT cells treated with different concentrations of FCCP was determined by Seahorse XF24 analyzer first (Supplementary Information, Figure S4). OCR in FePt NPs treated SR3A-hCtr1-WT cells began to reduce after 48 hours of treatment and with a minimum concentration at 0.0625 mg/mL (Figure 4D). These results collectively are reminiscent of those found in SR3A-13 and SR3A-14 cells, demonstrating the important roles of hCtr1 expression in regulating FePt NPs uptake/transport activity and the resulting mitochondrial abnormalities.
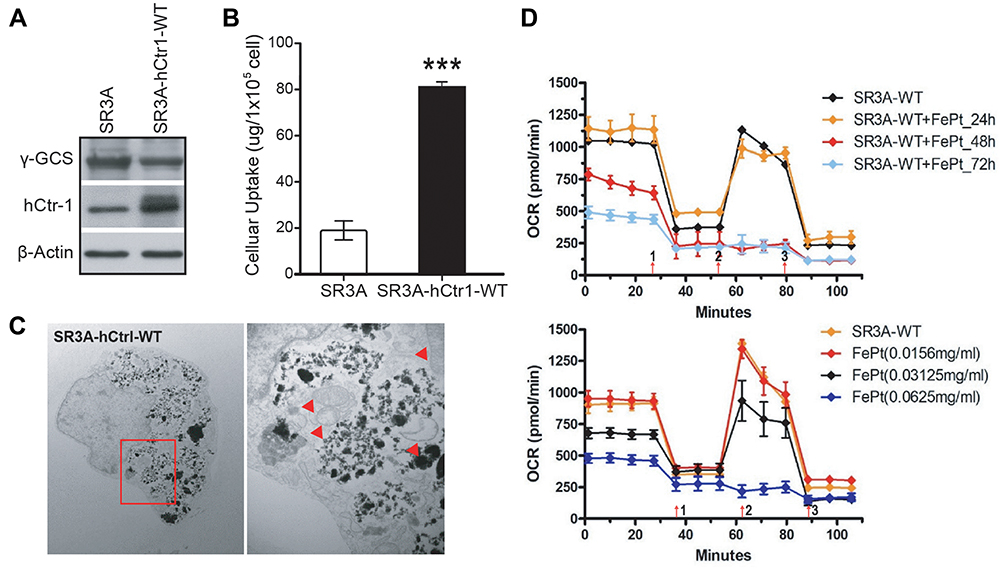 |
Figure 4 Elevated hCtr1 expression confers enhanced uptake/transport activity of FePt NPs and induces mitochondria dysfunction. (A) Western blotting analysis of γ-GCSh and hCtr1 protein levels in SR3A-hCtr1-WT cells. β-actin was used as a loading control. (B) The uptake/transport of 1 mg/mL FePt NPs for 24 hrs was significantly increased in SR3A-hCtr1-WT cells as compared with SR3A cells by ICP-OES measurement (***P < 0.001). Error bars represent ± S.D. (C) Representative TEM images of SR3A-hCtr1-WT cells treated with FePt NPs (left). Shown in the right is high-power view of image in red square demonstrating abnormal mitochondria in SR3A-hCtr1-WT cells with increasing membrane density and losing ridges after FePt NPs treatment (red arrowheads). (D) The OCR levels were significantly decreased in SR3A-hCtr1-WT cells treated with FePt NPs, in a time (left)- and concentration (right)-dependent manner. |
hCtr1 Expression Significantly Enhances FePt NPs-Induced Radiosensitivity
Whether FePt NPs can affect radiation therapy efficacy by regulating hCtr1 expression was investigated here. Clonogenic cell survival assay revealed that treating SR3A-hCtr1-WT cells with FePt NPs or X-rays irradiation alone slightly reduced the surviving colonies, but substantial further reduction was attained by the combination treatment (Figure 5A, left). Overall, the surviving fractions in these treatments were 9.96 ± 3.75%, 34.7 ± 7.19%, and <1%, respectively, in reference to the untreated cells (100%) (Figure 5A, right). These results indicated that by regulating hCtr1 expression alone, the synergistic cell-killing effect between the FePt NPs and ionizing radiation is achieved.
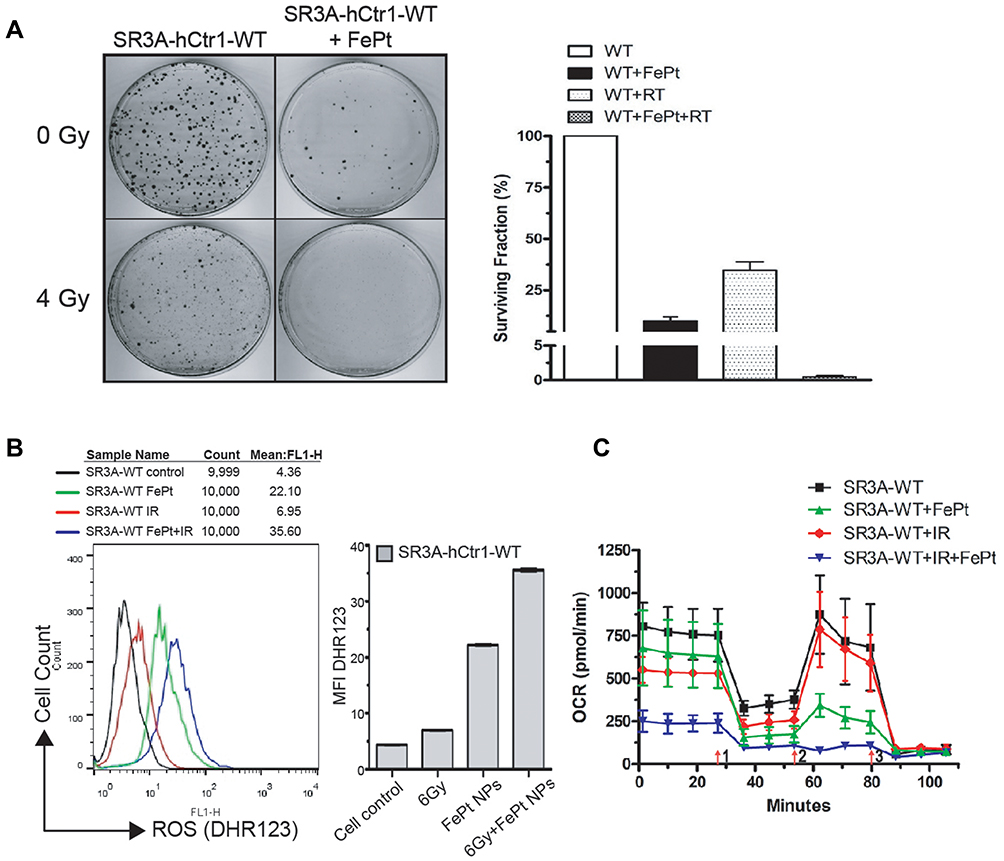 |
Figure 5 hCtr1 expression significantly enhances FePt NPs-induced radiosensitivity. (A) SR3A-hCtr1-WT cells were treated with 1 mg/mL FePt NPs for 24 hours then irradiated with or without X-rays. Clonogenic assay shows significant decrease of surviving colony numbers in the combination treatment (left). Surviving fractions in these treatments shown (right). (B) ROS was considerably increased after combined treatment of FePt NPs and X-rays in SR3A-hCtr1-WT cells. (C) OCR measured by Seahorse XF24 analyzer was markedly attenuated in SR3A-hCtr1-WT cells treated with FePt NPs and X-rays. |
To have a better understanding of the effect FePt NPs on X-ray irradiation in SR3A-hCtr1-WT cells, the ROS production and the function of aerobic respiration were also assayed. The intracellular level of ROS in SR3A-hCtr1-WT cells was measured by flow cytometry with DHR123 staining. The total intracellular ROS level was higher in the combination treatment with FePt NPs and X-rays than those in the control, FePt NPs or X-rays alone groups (Figure 5B). Moreover, OCR was also attenuated in the same combination treatment as well (Figure 5C). These results demonstrated that FePt NPs enhanced radiation sensitivity through ROS production and the lost function of mitochondria and was impacted by hCtr1 overexpression, which further strengthened the important role of hCtr1 in FePt NPs-induced radiation sensitivity leading to cell killing.
FePt NPs Increases the Efficacy of Radiotherapy in SR3A-hCtr1-WT-Bearing Mice
The SR3A-hCtr1-WT cells xenograft tumor-bearing mouse model was used to evaluate the therapeutic efficacy of the FePt NPs as a radiation sensitizer in vivo. Figure 6A outlines the experimental treatment schedule. SR3A-hCtr1-WT cells were first injected into the flanks of SCID mice at day 0. When the tumor size reached approximately 50 ~ 80 mm3, animals were randomly divided into four groups: groups (i) and (iii) were injected with PBS, whereas groups (ii) and (iv) were intravenously injected with FePt NPs dispersed in PBS solution. X-ray irradiation (6 Gy) was given to groups (iii) and (iv) 24 hours later, whereas groups (i) and (ii) were untreated. The tumor tissues were collected and evaluated by using H&E, iron and hCtr1 staining. As expected, hCtr1 in the tumor tissues was highly expressed in both PBS and FePt NPs treated groups. However, the signal of ferric iron was only detectable in tissues from the FePt NPs-treated animals and correlated with hCtr1 expression (Figure 6B). In order to evaluate the effect of FePt NPs on tumor growth, we measured the volume of tumor and the body weight of mice daily for 24 days. At the end of experiment, tumor volume was increased about 19-fold in the PBS-treated mice, whereas in the FePt NPs alone- and X-rays alone-treated groups, tumor growth was modestly reduced. Tumor volume was most significantly reduced in the group of combined treatment with FePt NPs and X-rays, as compared with that in the untreated control (***P <0.01, Figure 6C). Moreover, since body weight is a common parameter for evaluating the treatment-associated toxicity to the animals, we found no significant change in body weight in all groups over the course (Figure 6D). The treatments were well tolerated by the animals. These results demonstrated that FePt NPs can enhance the efficacy of radiotherapy in animal model.
 |
Figure 6 Enhancement of radiation therapy efficacy by utilizing FePt NPs in SR3A-hCtr1-WT-bearing mice under various treatments. (A) Schematic drawing of experimental design for assessing the efficacy of FePt NPs and irradiation (6 Gy) in vivo. (B) Representative H&E and immunostainings of hCtr1 and iron in tumor tissues of SR3A-hCtr1-WT-bearing mice, square was the region magnified 400X in each tumor sections. (C) Tumor growth inhibition of SR3A-hCtr1-WT-subcutaneous xenograft. Growth reduction was seen in the FePt NPs- and X-rays irradiation-treated groups (*p < 0.05; **p < 0.01), but greater reduction was seen in the group of combined treatment with FePt NPs and X-rays (***P<0.01). Error bars represent ± S.D. (D) No significant changes of body weights of the mice among all the treatment groups. |
Discussion
In this study, we found the therapeutic value of FePt NPs as a radiation sensitizer in cancer cells, particularly in cells that overexpress hCtr1. hCtr1 overexpression confers enhanced uptake/transport of FePt NPs, accompanied with mitochondria morphology alteration and aerobic respiratory function loss. We further demonstrated that FePt NPs can increase the efficacy of radiotherapy through hCtr1 in SR3A-hCtr1-WT-bearing mice. Our current findings of overcoming radiation resistance by FePt NPs through hCtr1 and mitochondrial disturbance may provide a novel strategy to improve the efficacy of radiation therapy, as illustrated in Scheme 1.
 |
Scheme 1 The schematic illustration of the GSH-overexpressed small-cell lung cancer cells exhibit elevated expression of hCtr1 and are resistant to X-rays irradiation. Radiation resistance can be overcome by enhanced uptake/transport of FePt NPs due to the overexpressed hCtr1 through the mechanisms of ROS outburst and mitochondria dysfunction. |
FePt NPs have recently been revealed to be significant multifunctional materials. Investigations of the potential applications in the diagnostic and therapeutic fields of cancer have been carried out due to their excellent physicochemical characteristics. Several studies were conducted investigating FePt NPs as dual MRI/CT imaging contrast,21,28 drug delivery system,29 photothermic or hyperthermia agent,30 and chemo-radiation sensitizers.31,32 Our findings that FePt NPs can overcome radioresistance further support their roles in radiotherapy of cancer treatments. It was proposed that FePt NPs enhance the effect of radiation therapy by triggering apoptosis pathway through the augmentation of intracellular ROS accumulation.32 Our results suggest that the induced ROS may cause overwhelming mitochondrial damages that lead to cell death.
Another important observation described here is that elevated transport of FePt NPs is associated with mitochondrial damages and ROS burst that may serve as its major cellular lethal effects, particularly in conjunction with ionizing radiation. It has been reported that Fe2+ is produced by FePt NPs at lysosome during endocytosis which induces cellular stress and increases mitochondrial ROS to induce cell death.33 Likewise, exposure to ionizing radiation also causes abundant cell stress directly or indirectly, due to the generation of ROS in mitochondria.34 Excessive levels of ROS can alter mitochondrial membrane permeability to disrupt the electron transport chain and cause imbalance of the intracellular redox system.35–37 Therefore, combination of NPs with ionizing radiation triggering ROS burst that leads to mitochondrial dysfunction may be an effective approach for cancer treatment using FePt NPs/radiation strategy.38,39 Given that technical advancement of modern radiation therapy uses imaging guidance during radiation therapy to improve the precision and accuracy of treatment delivery, this is particularly intriguing in light that FePt NPs are a perfect and potential duo-imagining agent that can be used in both MRI/CT-guided radiation therapy.
We took the advantage of using GSH-overproducing cells to investigate the roles of ROS in the effect of FePt NPs on radiation therapy in this study. GSH is the most abundant antioxidant found in living organisms to maintain cellular redox homeostasis. Elevated levels of GSH also confer resistance of chemotherapy and radiation in cancer cells.40,41 Studies revealed that cellular damages by radiation-induced ROS can be reduced by GSH.42 We used this cell model to entail that the radioresistance in these cells was contributed by the overexpressed GSH.43 But because these GSH-overproducing cells also upregulate hCtr1 expression due to the copper chelating function of GSH, this allows us to make an important finding that hCtr1 plays an important role in increasing FePt NPs uptake. Using cells transfected with hCtr1 recombinant alone conferred similar effects, which further supported this finding.
While we observed that hCtr1 is responsible for the enhanced FePt NPs transport, however, we do not believe that FePt NPs enter cells through the channel as described above for the following reasons: (i) the size of FePtNPs is apparently too big (6 nm) to pass through hCtr1 pore (about 8Å),44 and (ii) the unique intracellular behaviors and distributions of FePt NPs observed by TEM are also not consistent with the channel-related transport mechanism. Internalization of FePt NPs by hCtr1-mediated endocytotic mechanism might result in its cytoplasmic confinement.23 Nonetheless, detailed mechanisms by which hCtr1-regulated FePt NPs delivery still require further investigation.
Finally, our present results that elevated hCtr1 can enhance combination therapy of FePt NPs and radiation may have important clinical implications. Our previous studies demonstrating that overexpression of hCtr1 was observed in about 70% of lung cancer tumors and hCtr1 was a predictor of platinum-based chemotherapy response suggest that combination of FePtNPs and radiation may be suitable for treating human lung cancers.14 Moreover, many copper depletors have been approved for treating copper-deficient diseases such as Wilson’s disease. Recent clinical studies have shown promising results using Cu chelator (trientine) to upregulate hCtr1 in Pt-based cancer chemotherapy.45,46 These Cu chelators may improve the treatment efficacy of FePtNPs/radiation through enhanced hCtr1 expression. Overall, our results have potential for improving human cancer treatments.
Conclusion
In this study, we found that FePt NPs can be a radiation sensitizer in vitro and in vivo, particularly in tumors, which overexpress hCtr1. Overexpression of hCtr1 was responsible for the increased uptake of FePt NPs as demonstrated by using hCtr1-transfected parental SR3A cells. We also observed increased ROS and mitochondria morphology alteration with substantial reduction of oxygen consumption rate in FePt NPs and IR-treated cells, indicating that structural and functional insults of mitochondria are the lethal mechanism of FePt NPs. Our results bear important implications that FePt NPs can potentially be a novel strategy to improve radiotherapeutic efficacy in hCtr1-overexpressing cancer cells via enhanced uptake and mitochondria targeting.
Abbreviations
GSH, Glutathione; γ-GCS, γ-glutamylcysteine synthetase; hCtr1, Human copper transporter 1; Pt, Platinum; FePt NPs, Iron-platinum nanoparticles; ROS, Reactive oxygen species; OCR, Oxygen consumption rate; SCLC, Small cell lung cancer; IHC, Immunohistochemistry; H&E, Hematoxylin and eosin; DHR123, Dihydrorhodamine123; ICP-OES, Inductively coupled plasma optical emission spectrometry; SER, Sensitizer enhancement ratio; TEM, Transmission electronic microscopy; MFI, Mean fluorescence intensity.
Acknowledgments
This work was supported by the Center of Applied Nanomedicine, National Cheng Kung University from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan; and the Ministry of Science and Technology, Taiwan [grant number MOST-108-2314-B-006-019-MY3, grant number MOST-106-2313-B-006-018-MY2]. Finally, the authors thank Drs. Macus Tien Kuo, in Department of Translational Molecular Pathology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA, and Hao-Chen Wang, in Clinical Medicine Research Center of National Cheng Kung University Hospital, for providing cell lines, consulting services and manuscript editing.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Delaney G, Jacob S, Featherstone C, et al. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005;104:1129–1137. doi:10.1002/cncr.21324
2. Orth M, Lauber K, Niyazi M, et al. Current concepts in clinical radiation oncology. Radiat Environ Biophys. 2014;53:1–29.
3. Chen HHW, Kuo MT. Improving radiotherapy in cancer treatment: promises and challenges. Oncotarget. 2017;8:62742–62758. doi:10.18632/oncotarget.18409
4. Baumann M, Krause M, Overgaard J, et al. Radiation oncology in the era of precision medicine. Nat Rev Cancer. 2016;16:234–249.
5. Le QT, Shirato H, Giaccia AJ, et al. Emerging treatment paradigms in radiation oncology. Clin Cancer Res. 2015;21:3393–3401. doi:10.1158/1078-0432.CCR-14-1191
6. Kelley K, Knisely J, Symons M, et al. Radioresistance of brain tumors. Cancers (Basel). 2016;8:42. doi:10.3390/cancers8040042
7. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi:10.1038/nrc2167
8. Curran WJ, Paulus R, Langer CJ, et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: randomized Phase III trial RTOG 9410. J Natl Cancer Inst. 2011;103:1452–1460. doi:10.1093/jnci/djr325
9. Stehman FB, Ali S, Keys HM, et al. Radiation therapy with or without weekly cisplatin for bulky stage 1B cervical carcinoma: follow-up of a Gynecologic Oncology Group trial. Am J Obstet Gynecol. 2007;197:
10. Al-Sarraf M, LeBlanc M, Giri PG, et al. Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer: phase III randomized Intergroup study 0099. J Clin Oncol. 1998;16:1310–1317. doi:10.1200/JCO.1998.16.4.1310
11. Cooper JS, Pajak TF, Forastiere AA, et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med. 2004;350:1937–1944. doi:10.1056/NEJMoa032646
12. Song IS, Savaraj N, Siddik ZH, et al. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol Cancer Ther. 2004;3:1543–1549.
13. Holzer AK, Varki NM, Le QT, et al. Expression of the human copper influx transporter 1 in normal and malignant human tissues. J Histochem Cytochem. 2006;54:1041–1049. doi:10.1369/jhc.6A6970.2006
14. Chen HH, Yan JJ, Chen WC, et al. Predictive and prognostic value of human copper transporter 1 (hCtr1) in patients with stage III non-small-cell lung cancer receiving first-line platinum-based doublet chemotherapy. Lung Cancer. 2012;75:228–234. doi:10.1016/j.lungcan.2011.06.011
15. Chen HH, Kuo MT. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met Based Drugs. 2010;2010:430939. doi:10.1155/2010/430939
16. Chen HH, Song IS, Hossain A, et al. Elevated glutathione levels confer cellular sensitization to cisplatin toxicity by up-regulation of copper transporter hCtr1. Mol Pharmacol. 2008;74:697–704. doi:10.1124/mol.108.047969
17. Johnstone TC, Suntharalingam K, Lippard SJ. The next generation of platinum drugs: targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem Rev. 2016;116:3436–3486. doi:10.1021/acs.chemrev.5b00597
18. Kuncic Z, Lacombe S. Nanoparticle radio-enhancement: principles, progress and application to cancer treatment. Phys Med Biol. 2018;63:02TR01. doi:10.1088/1361-6560/aa99ce
19. Bonvalot S, Rutkowski PL, Thariat J, et al. NBTXR3, a first-in-class radioenhancer hafnium oxide nanoparticle, plus radiotherapy versus radiotherapy alone in patients with locally advanced soft-tissue sarcoma (Act.In.Sarc): a multicentre, Phase 2–3, randomised, controlled trial. Lancet Oncol. 2019;20:1148–1159. doi:10.1016/S1470-2045(19)30326-2
20. Verry C, Dufort S, Lemasson B, et al. Targeting brain metastases with ultrasmall theranostic nanoparticles, a first-in-human trial from and MRI perspective. Sci Adv. 2020;6:eaay5279. doi:10.1126/sciadv.aay5279
21. Chou SW, Shau YH, Wu PC, et al. In vitro and in vivo studies of FePt nanoparticles for dual modal CT/MRI molecular imaging. J Am Chem Soc. 2010;132:13270–13278. doi:10.1021/ja1035013
22. Yamane Y, Furuichi M, Song R, et al. Expression of multidrug resistance protein/GS-X pump and gamma-glutamylcysteine synthetase genes is regulated by oxidative stress. J Biol Chem. 1998;273:31075–31085. doi:10.1074/jbc.273.47.31075
23. Chen HH, Chen WC, Liang ZD, et al. Targeting drug transport mechanisms for improving platinum-based cancer chemotherapy. Expert Opin Ther Targets. 2015;19:1307–1317. doi:10.1517/14728222.2015.1043269
24. Lai YH, Kuo C, Kuo MT, et al. Modulating chemosensitivity of tumors to platinum-based antitumor drugs by transcriptional regulation of copper homeostasis. Int J Mol Sci. 2018;19:1486. doi:10.3390/ijms19051486
25. Symersky J, Pagadala V, Osowski D, et al. Structure of the c(10) ring of the yeast mitochondrial ATP synthase in the open conformation. Nat Struct Mol Biol. 2012;19:485–491. doi:10.1038/nsmb.2284
26. Brennan JP, Southworth R, Medina RA, et al. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res. 2006;72:313–321. doi:10.1016/j.cardiores.2006.07.019
27. Li N, Ragheb K, Lawler G, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi:10.1074/jbc.M210432200
28. Liang S, Zhou Q, Wang M, et al. Water-soluble L-cysteine-coated FePt nanoparticles as dual MRI/CT imaging contrast agent for glioma. Int J Nanomedicine. 2015;10:2325–2333. doi:10.2147/IJN.S75174
29. Fuchigami T, Kawamura R, Kitamoto Y, et al. Ferromagnetic FePt-nanoparticles/polycation hybrid capsules designed for a magnetically guided drug delivery system. Langmuir. 2011;27:2923–2928. doi:10.1021/la1041019
30. Chen CL, Kuo LR, Lee SY, et al. Photothermal cancer therapy via femtosecond-laser-excited FePt nanoparticles. Biomaterials. 2013;34:1128–1134. doi:10.1016/j.biomaterials.2012.10.044
31. Zheng Y, Tang Y, Bao Z, et al. FePt nanoparticles as a potential X-ray activated chemotherapy agent for HeLa cells. Int J Nanomedicine. 2015;10:6435–6444. doi:10.2147/IJN.S88458
32. Sun Y, Miao H, Ma S, et al. FePt-Cys nanoparticles induce ROS-dependent cell toxicity, and enhance chemo-radiation sensitivity of NSCLC cells in vivo and in vitro. Cancer Lett. 2018;418:27–40. doi:10.1016/j.canlet.2018.01.024
33. Mazumder V, Chi M, More KL, et al. Core/shell Pd/FePt nanoparticles as an active and durable catalyst for the oxygen reduction reaction. J Am Chem Soc. 2010;132:7848–7849. doi:10.1021/ja1024436
34. Tulard A, Hoffschir F, de Boisferon FH, et al. Persistent oxidative stress after ionizing radiation is involved in inherited radiosensitivity. Free Radic Biol Med. 2003;35:68–77. doi:10.1016/S0891-5849(03)00243-0
35. Kam WW, Banati RB. Effects of ionizing radiation on mitochondria. Free Radic Biol Med. 2013;65:607–619. doi:10.1016/j.freeradbiomed.2013.07.024
36. Kim W, Youn H, Kang C, et al. Inflammation-induced radioresistance is mediated by ROS-dependent inactivation of protein phosphatase 1 in non-small cell lung cancer cells. Apoptosis. 2015;20:1242–1252. doi:10.1007/s10495-015-1141-1
37. Kim W, Lee S, Seo D, et al. Cellular stress responses in radiotherapy. Cells. 2019;8:1105. doi:10.3390/cells8091105
38. Yu Z, Sun Q, Pan W, et al. A near-infrared triggered nanophotosensitizer inducing domino effect on mitochondrial reactive oxygen species burst for cancer therapy. ACS Nano. 2015;9:11064–11074. doi:10.1021/acsnano.5b04501
39. Li N, Yu L, Wang J, et al. A mitochondria-targeted nanoradiosensitizer activating reactive oxygen species burst for enhanced radiation therapy. Chem Sci. 2018;9:3159–3164. doi:10.1039/C7SC04458E
40. Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217:2291–2298. doi:10.1083/jcb.201804161
41. Miura M, Sasaki T. Role of glutathione in the intrinsic radioresistance of cell lines from a mouse squamous cell carcinoma. Radiat Res. 1991;126:229–236.
42. Brouazin-Jousseaume V, Guitton N, Legué F, et al. GSH level and IL-6 production increased in Sertoli cells and astrocytes after gamma irradiation. Anticancer Res. 2002;22:257–262.
43. Janowiak BE, Griffith OW. Glutathione synthesis in Streptococcus agalactiae. One protein accounts for gamma-glutamylcysteine synthetase and glutathione synthetase activities. J Biol Chem. 2005;280:11829–11839. doi:10.1074/jbc.M414326200
44. Ren F, Logeman BL, Zhang X, et al. X-ray structures of the high-affinity copper transporter Ctr1. Nat Commun. 2019;10:1386. doi:10.1038/s41467-019-09376-7
45. Huang YF, Kuo MT, Liu YS, et al. A dose escalation study of trientine plus carboplatin and pegylated liposomal doxorubicin in women with a first relapse of epithelial ovarian, tubal, and peritoneal cancer within 12 months after platinum-based chemotherapy. Front Oncol. 2019;9:437. doi:10.3389/fonc.2019.00437
46. Fu S, Hou MM, Wheler J, et al. Exploratory study of carboplatin plus the copper-lowering agent trientine in patients with advanced malignancies. Invest New Drugs. 2014;32:465–472. doi:10.1007/s10637-013-0051-8
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.