Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 4
Orphan drugs in development for urea cycle disorders: current perspectives
Authors Häberle J, McCandless S
Received 10 June 2014
Accepted for publication 29 July 2014
Published 30 September 2014 Volume 2014:4 Pages 63—70
DOI https://doi.org/10.2147/ODRR.S44065
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Lise Aagaard
Johannes Häberle,1 Shawn E McCandless2
1Division of Metabolism and Children's Research Center, University Children's Hospital Zurich, Zurich, Switzerland; 2Center for Human Genetics, University Hospitals Case Medical Center, and Department of Genetics and Genome Sciences, Case Western Reserve University, Cleveland, OH, USA
Abstract: The urea cycle disorders are caused by deficiency of one of the six hepatic enzymes or two transporters involved in detoxification of ammonia. The resulting hyperammonemia causes severe brain injury unless aggressive steps are taken to reduce the accumulation of ammonia, which is thought to be the most toxic metabolite. This review describes the current state of chronic management of urea cycle disorders, focusing on new and emerging therapies. Management strategies include the mainstay of treatment, namely dietary protein restriction and supplementation with l-arginine or l-citrulline. Several currently approved medications utilize and enhance alternative pathways of waste nitrogen excretion (sodium benzoate, sodium phenylacetate, sodium phenylbutyrate in several formulations, and glycerol phenylbutyrate), working through conjugation of the drug to either glycine (in the case of benzoate) or glutamine, the products of which are excreted in the urine. Carglumic acid activates the first committed step of conversion of ammonia to urea, carbamoylphosphate synthetase, and thus effectively treats defective synthesis of the endogenous activator, N-acetylglutamate, whether due to genetic defects or biochemical inhibition of the N-acetylglutamate synthase enzyme. Approaches to neuroprotection during episodes of hyperammonemia are discussed, including the use of controlled hypothermia (brain cooling), as well as proposed, but as yet untested, pharmacologic therapies. Finally, cell-based therapies, including liver transplantation, infusion of fresh or cryopreserved hepatocytes, use of stem cells, and new approaches to gene therapy, are reviewed.
Keywords: urea cycle disorders, inherited hyperammonemias, orphan drugs, phenylbutyrate, N-carbamyl-l-glutamate
Introduction
Urea cycle disorders (UCDs) are a group of inherited defects of six enzymes and two transporters that constitute the urea cycle in the periportal liver cells. Defects in any of the enzymes or transporters lead to the respective disorders including arginase 1 deficiency, argininosuccinate lyase (ASL) deficiency, argininosuccinate synthetase deficiency, citrullinemia type 2 (caused by the defect of the citrin transporter), carbamoylphosphate synthetase 1 (CPS1) deficiency, N-acetylglutamate synthase (NAGS) deficiency, ornithine transcarbamylase (OTC) deficiency, and hyperornithinemia-hyperammonemia-homocitrullinuria syndrome (caused by the defect of mitochondrial ornithine transporter) (Figure 1). The main function of the urea cycle is the detoxification of ammonia and the synthesis of arginine. Defects of any of the urea cycle enzymes in the liver can result in metabolic decompensation characterized by hyperammonemia, which is life threatening if the treatment is started late or if the diagnosis is missed.1
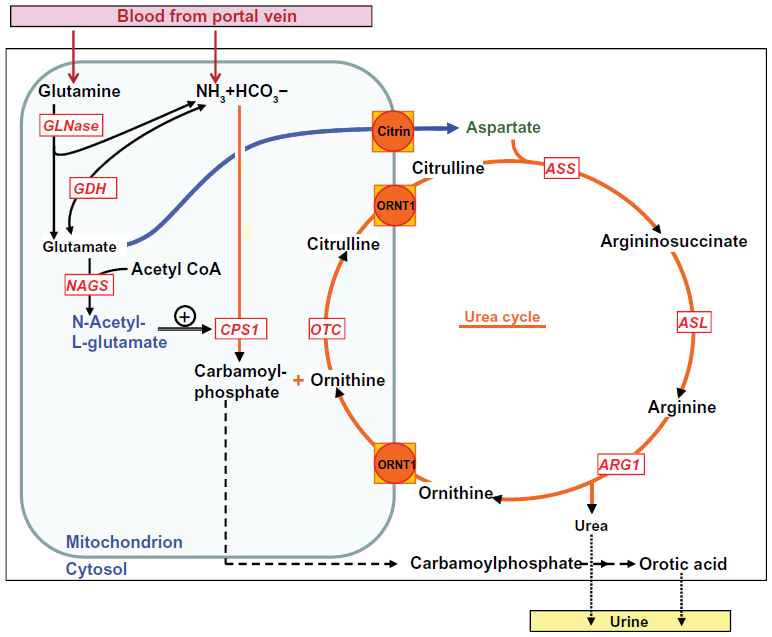 | Figure 1 Enzymes and transporters of the urea cycle. |
UCDs are panethnic with variable prevalence in different populations and an overall incidence of about 1 in 35,000.2 Initial presentation of a UCD can be at any age but about half of the patients present shortly after birth, triggered by postnatal catabolism.3 Other common triggering factors, characterized by endogenous protein catabolism, include viral infections and fasting. Patients with onset of symptoms outside the newborn period are classified as late-onset UCDs.4–6
The most neurotoxic compound metabolites appear to be ammonia itself, which is responsible for most of the clinical symptoms and complications. The exact mechanisms of ammonia neurotoxicity are not completely understood. Different effects of accumulating ammonia on the brain include swelling of astrocytes via glutamine accumulation, energy deficit through mitochondrial dysfunction, enhanced oxidative stress due to increased nitric oxide (NO) production, increased extracellular glutamate stimulating N-methyl-d-aspartate (NMDA) receptors, inhibition of the tricarboxylic acid cycle, and secondary creatine deficiency.7–9
The main goal of treatment is to avoid hyperammonemia in order to prevent neurological sequelae. Scrupulous avoidance of acute and chronic hyperammonemia may be associated with better prognosis for patients with UCDs.10 On the other hand, the prognosis with respect to survival is commonly considered as poor in cases where the ammonia concentration in blood is >1,000 μmol/L (reference <100 in newborns and <50 outside the neonatal period) or with duration of hyperammonemic coma >24 hours.11 Infants who do survive typically have severe and permanent brain injury, thus many specialists recommend against aggressive intensive care when treatment is delayed from the time of initial presentation.
The mainstay of chronic treatment is reduction of dietary protein to the minimum required to support normal endogenous protein synthesis and normal growth, along with supplementation of essential amino acids, vitamins, and trace elements. Other common treatment strategies include the nitrogen scavenging drugs, such as sodium phenylbutyrate (NaPB), licensed in 1996 for oral/enteral use by the United States Food and Drug Administration (FDA) as Buphenyl® in the US, (Hyperion Therapeutics, Brisbane, CA, USA), and Ammonaps® in Europe (Swedish Orphan Biovitrum Ltd, Stockholm, Sweden), the new drug formulation glycerol phenylbutyrate (GPB; FDA-approved as RAVICTI® [Hyperion Therapeutics, Brisbane, CA, USA]), sodium phenylacetic acid (NaPA), and sodium benzoate (SBZ; the combination of phenylacetic acid (PAA) and SBZ is available as an FDA-approved drug for intravenous application, AMMONUL® [Ucyclyd Pharma, Inc., Scottsdale, AZ, USA]), and the urea cycle intermediates l-citrulline (for oral/enteral use only) and l-arginine (for oral/enteral or iv use), the latter being available as free base or hydrochloride solution.11–13 Use of either or both of the latter two compounds, available as chemical compounds only, depends on the specific disorder and aims for excretion of citrulline (containing one waste nitrogen atom) or argininosuccinic acid (containing two waste nitrogen atoms) in the urine.
In spite of these dietary and pharmacological treatment measures, considerable risk of hyperammonemic decompensation remains, especially in the severe UCDs. Thus, liver transplantation, which is currently considered the only curative treatment, is often recommended early in the course of the disease in case of neonatal-onset UCDs or an unstable course in any patient with UCDs.11,14,15 Liver transplantation replaces the complete urea cycle, which is only fully expressed in liver, but other cell types that express individual component enzymes, particularly argininosuccinic acid lyase, are not corrected.
There is certainly a need for novel therapies, both for the treatment of acute hyperammonemia and for the long-term management of UCDs, since the present therapeutic situation is far from ideal. A better understanding of the pathology in ammonia (neuro) toxicity has opened the path for a variety of novel therapeutic approaches, although most have not yet added to the armamentarium of UCD drugs. First, we will describe the few recent products approved for the treatment of UCDs. Second, we will discuss novel therapeutic strategies that provide hope for an improved prognosis for individuals with UCDs in the future.
Novel drugs already brought to the market
Glycerol phenylbutyrate (RAVICTI®)
The basis to alternative pathway therapy was recognized 100 years ago with the demonstration that SBZ could divert urea nitrogen to hippurate.16 Shortly thereafter, a similar observation noted that PAA, when given orally, resulted in urinary excretion of phenylacetylglutamine.17 PB, which is converted to PAA in the liver, is now used more frequently for enteral therapy, because it has low odor and is better tolerated by patients. Since its introduction to the treatment of UCDs,18 the use of endogenous pathways for waste nitrogen excretion has become a mainstay of management of patients with UCD.19
Recently, a new formulation of PB was brought to the market. The new drug, GPB, FDA-approved as RAVICTI®, is a pre-prodrug containing three molecules of PB joined to glycerol in ester linkage to form a triglyceride. The pre-prodrug, GPB, is hydrolyzed in the small intestine by pancreatic lipases and releases PB and glycerol. The prodrug, PB, is metabolized in the liver to the active compound PAA. Given the required GPB hydrolysis, gastrointestinal absorption of PB is slower than with NaPB.
When compared with NaPB in a Phase II study in eleven pediatric patients above age 6 years (mean, 10.2 years), GPB was shown to be at least equivalent to NaPB with respect to ammonia control.20 In that crossover study, performed when the blood ammonia was clinically well controlled at baseline, switchover from NaPB to a PB-equimolar GPB dose was well tolerated and resulted in about 25% lower average ammonia exposure (assessed as ammonia 24-hour area under the curve [AUC]). Mean blood ammonia concentrations were mainly <60 μmol/L in the GPB as well as in the NaPB-treated patients, and glutamine levels did not change significantly after switching to GPB.
In a Phase III double-blinded, randomized crossover study in 44 adult patients performed consecutively, treatment with GPB was non-inferior to NaPB if ammonia control was likewise assessed as 24-hour AUC.21 When the results of the latter study were pooled together with data from two similarly designed short-term comparisons of GPB versus NaPB (one in pediatric patients),20 ammonia 24-hour AUC and plasma glutamine were significantly lower with GPB,21 findings that are recently confirmed by other studies in pediatric patients with UCD.22,23 As a preliminary finding from one study,21 executive function in the study population seemed to have improved, although this observation, made after only one year of treatment, must be confirmed in future studies.
In addition to different pharmacokinetics, the main advantage of GPB from the patients’ point of view may be the much improved palatability of GPB. The drug comes as an essentially tasteless liquid. Despite its oily consistency, patients have little difficulty swallowing the drug. In pediatric patients, this may result in better compliance and lessening of the burden of disease. Other potential benefits include markedly reduced volumes of drug to be consumed; one teaspoon of GPB liquid provides an equimolar dose to approximately 13 large tablets of NaPB. The sodium salt of PB also appears to cause significant gastric discomfort, which is absent in the triglyceride (GPB), where the acidic PB is released in the small intestine and more slowly. Finally, the sodium load in therapeutic doses of NaPB is measured in gram quantities, which may contribute to multiple unpleasant effects on fluid balance and blood pressure.
Phenylbutyrate (Pheburane®)
Another formulation of NaPB was introduced to the European market in 2013 (licensed by the European Medicines Agency [EMA] as Pheburane®; Lucane Pharma, Paris, France). The drug consists of small spherical sugar cores, contributing to a sugar content of ~50% of the weight of the drug, which are coated sequentially in two separate layers with sodium PBA and ethylcellulose, the latter being a taste-masking agent for active substances.24 Pheburane®, formulated as tasteless and odor-free granules, is recommended for oral use only and should not be administered by a nasogastric or gastrostomy tube because of its slow dissolution. Importantly, the sodium content of this formulation, 5.4 mmol sodium for each gram of NaPB, is identical to the other NaPB formulations available in the market.
Pharmacodynamic and pharmacokinetic properties are the same as in the active compound, PB, resulting likewise in conversion to PAA and subsequent conjugation with glutamine to form phenylacetylglutamine, which is then excreted in the urine.24 Accordingly, the new formulation reduces the unpleasant taste, which should lead to improved compliance and eventually result in better efficacy of long-term treatment and better outcomes, especially in the pediatric patient population.25
N-Carbamyl-l-glutamate (Carbaglu®)
N-Carbamyl-l-glutamate (carglumic acid; Carbaglu; Orphan Europe Recordati Group, Paris, France) is a licensed drug (both by the FDA and by the EMA) for the treatment of the rare NAGS deficiency.26–28 This defect leads to deficiency of N-acetylglutamate (NAG), the allosteric activator of the first urea cycle enzyme, CPS1. Carglumic acid is a chemical analogue of NAG and has been demonstrated to efficiently enter the mitochondrion. Thus, treatment with carglumic acid effectively substitutes for the NAG deficiency, although the pharmaceutical agent is not regulated by consumption of a protein containing meal in the same way as endogenous NAG. Nevertheless, NAGS deficiency is the only UCD for which an easy and highly efficacious remedy is available, which allows the patients to lead a normal life, with no need for low-protein diet or other drugs except during the periods of illness.11,29,30
In addition, carglumic acid has been licensed in Europe for the treatment of acute hyperammonemia in three of the organic acidurias (methylmalonic aciduria; propionic acidemia; and isovaleric acidemia), which are inherited disorders of branched-chain amino acid degradation.28,31 In these conditions, hyperammonemia results from inhibition of NAGS during states of metabolic decompensation, presumably due to accumulation of specific inhibitory compounds such as methylcitrate or propionyl-CoA.
A further application of carglumic acid, the use in patients with CPS1 deficiency and potentially also in patients with OTC deficiency, has recently been suggested and is currently the subject of a multicenter Phase II study in the US. The rationale for this use is based on the assumption that additional activation of a mutant CPS1, thereby maximizing the flux through the urea cycle, may improve the overall ammonia detoxification capacity. This may be the case, in particular, if mutations affect the allosteric domain of the CPS1 protein, which comprises the C-terminal part of the protein. This theoretical concept has been supported by a number of (mainly single) patient studies in which a beneficial effect was shown.32 More recently, isotope studies performed in a small number of CPS1-deficient patients provide evidence for the efficacy of carglumic acid treatment, demonstrating increased flux through the urea cycle.33 This is further supported by studies in insect cell-derived purified recombinant CPS1, in which carglumic acid exhibited an activity increasing effect in some of the mutants.34 Notably, this effect was not only limited to mutations within the C-terminal part of CPS1 but also found in single missense changes far distant in the secondary structure of CPS1.
Likewise, for males with partial OTC deficiency, ie, with residual enzyme activity, or in female carriers of OTC mutations with hepatic mosaicism due to random X-inactivation, pharmacological upregulation of CPS1 enzyme activity may lead to increased flux through the OTC-deficient enzyme, or in cells expressing the normal enzyme in female carriers.
Drugs with a potential for development
NO supplementation
The recent elucidation of a cytosolic multienzyme complex including argininosuccinate synthetase, ASL, and NO synthase,35,36 and evidence of NO deficiency suggested by reduced nitrosylation of marker compounds in patients with ASL deficiency, suggest an explanation for some of the hitherto poorly understood clinical findings in ASL-deficient patients, namely, arterial hypertension, chronic liver disease, and ammonia-independent neurodevelopmental deterioration.37 Importantly, impaired NO-mediated relaxation of the vasculature has been restored both in ASL-deficient mice and in a single ASL-deficient human with treatment refractory arterial hypertension by NO supplementation.38 Thus, the role of NO supplementation for the subgroup of patients affected by ASL deficiency will need to be determined in clinical trials.
NMDA receptor blockade and other neuroprotective agents
Since ammonia neurotoxicity is in part mediated through activation of the NMDA receptors by glutamate, targeting this receptor by pharmacological blockade is an obvious therapeutic strategy; however, there are no research activities reported that specifically address UCDs. Thus, the reader is referred to literature on other disorders.39
A number of other therapeutic targets have been inferred from research into ammonia neurotoxicity, as reviewed by Braissant et al,8 unfortunately, none of them have yet been evaluated in clinical trials. Nevertheless, they are listed in Table 1 to illustrate the large variety of potential therapeutic targets that may, at least in part, come into clinics in the future. Other nitrogen scavenger substances, such as l-ornithine-phenylacetate or l-ornithine-l-aspartate, have also been proposed, although possible advantages over currently existing treatments are not clear.
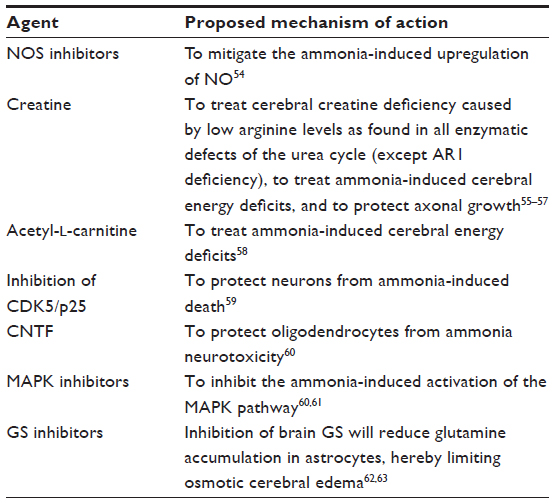 | Table 1 Potential neuroprotective agents |
Other therapeutic strategies
UCDs cannot be cured by drugs alone. It is clear that additional or alternative strategies are needed. Currently, liver transplantation by restoring the defective enzyme in the liver is the only correction for the ineffective urea cycle; thus, it can be reasonably recommended for all severe UCDs, unless there are already neurological sequelae from previous episodes of hyperammonemic decompensation rendering the extent of preservation of cognitive functions small or irrelevant.11,15,40 Even then, liver transplant may be considered to ease the burden of care and enhance the quality of life for affected individuals, mainly by normalizing the diet and removing the need for expensive scavenger medications and nutritional supplements, and for their families by reducing the anxiety of the constant risk of rapid metabolic decompensation. It is also possible that liver transplantation may be less expensive over the course of a lifetime than the current standard medical care. It should be remembered that liver transplant does not replace those component enzymes of the urea cycle that are expressed in non-hepatic tissues, especially argininosuccinic acid lyase, which is involved in NO synthesis. In addition, the life-long need for immunosuppression must be considered.
Because cadaveric liver transplantation is not immediately available for most patients and is technically challenging in newborns and small infants, several bridging therapies are currently being considered or already evaluated in clinical trials.
Hepatocyte transplantation
As a less-invasive but potentially effective approach, transplantation of (fresh or cryopreserved) human hepatocytes was suggested in the 1990s.41 Most researchers regard this as a potential bridging therapy for newborns with severe initial hyperammonemic decompensation at significant risk of further ammonia neurotoxicity if recurrent crises were to occur.42–44 This is a particularly attractive alternative for newborns in whom full organ transplantation carries high risks. Another advantage is increased availability of cells, as the cryopreserved cells can be stored until needed. It should be noted that, as in liver transplantation, there is a need for immunosuppression.
Concerns have been raised about the viability of cryopreserved hepatocytes, not only because these cells may be derived from organ donors not suitable for full organ transplantation because of long ischemia times, but also because of concerns toward mitochondrial damage due to cryopreservation.45 The latter concern could be overcome by the use of freshly isolated cells that may have better viability.46 At least two clinical trials are currently underway to address this approach to therapy. An ongoing multicenter, open-label clinical trial using historical controls in the US and Europe evaluating safety and efficacy of cryopreserved hepatocyte transplantation is in progress (Clinicaltrials.gov identifier NCT01195753). An additional, single-center study using freshly isolated hepatocytes transplanted into an irradiated liver to enhance engraftment and expansion of the grafted cells is also in progress (Clinicaltrials.gov identifier NCT01345578).
Stem cell transplantation
Another alternative approach, in principle with the same rationale as the aforementioned hepatocyte transplantation approach, is the proposed transfusion of stem cells.47–49 These cells may exhibit advantages over mature hepatocytes in terms of efficacy and may have the additional benefit of being able to be produced in large quantities in vitro.49
Gene therapy
Gene therapy is a research focus for several inherited metabolic diseases including UCDs. In fact, in the most common UCD, OTC deficiency, a dose-escalating study was performed as early as in 1999. Unfortunately, this trial was stopped due to overwhelming, and ultimately fatal, viral sepsis from the vector in one of the subjects,50 which put the entire field at hold for many years. Recent advances in vector biology and development have brought gene therapy back to the list of possible future therapies for UCDs,51 although the suitability of adenoviral-associated vectors for treatment of newborns remains the subject of debate. A recent review article on this topic describes the current status of, and prospects for, gene therapy for UCD treatment.52
Therapeutic hypothermia
Since damage to the brain during hyperammonemia is the most relevant complication in UCDs, neuroprotective measures are warranted. Among these, therapeutic hypothermia has been proposed as potentially beneficial as it has been observed in other conditions, including traumatic brain injury or hypoxic–ischemic encephalopathy.
A recent pilot study in six newborns with UCDs, and one case with isovaleric acidemia, a classical organic acidemia, all affected by severe hyperammonemia, has proven the feasibility of adjunct therapeutic hypothermia.53 Currently, a randomized multicenter study is being planned in US and European centers. This study is designed to ascertain whether treatment with hypothermia during the initial hyperammonemic decompensation can improve the long-term neurological outcome of patients presenting with hyperammonemic crisis.
Summary
Treatment of UCDs is still very challenging and there were only few new developments in pharmacological therapy in recent years. An improvement not only of palatability but probably also of pharmacological properties is the new formulation of glycerol phenylbutyrate that acts as an alternative pathway of waste nitrogen excretion. Carglumic acid, able to activate CPS1 as the first committed step of ammonia conversion to urea, can be useful for genetic defects or biochemical inhibition of the NAGS enzyme. Other therapeutic strategies that are not yet available but subject of studies include approaches to neuroprotection (eg, the use of controlled hypothermia aiming for brain cooling), cell-based therapies (eg, infusion of fresh or cryopreserved hepatocytes, use of stem cells), and new approaches to gene therapy. Overall, novel pharmacological developments for treating patients with UCDs are much desired as the currently available treatment options are still far from being optimal. As all UCDs have low incidences, such novel therapies will require the joint interest and enthusiasm of scientists and industry dedicated to the field of rare diseases.
Acknowledgments
This review was stimulated by a recent workshop held during the fourth International Symposium on Urea Cycle Disorders in Barcelona, Spain. This workshop was jointly organized by the “Urea Cycle Disorders Consortium”, UCDC, an NIH-sponsored network and registry in the US, and by the “European registry and network for intoxication type metabolic diseases” (E-IMD, EAHC no 2010 12 01), which has received funding from the European Union, in the framework of the Health Programme. The work on urea cycle disorders is supported by the Swiss National Science Foundation (grants 310030_127184 and 310030_153196 to JH).
Disclosure
The authors report no conflicts of interest in this work.
References
Brusilow S, Horwich A. Urea cycle enzymes. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:1909–1963. | |
Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110(1–2):179–180. | |
Leonard JV, Morris AA. Urea cycle disorders. Semin Neonatol. 2002;7(1):27–35. | |
Klaus V, Vermeulen T, Minassian B, et al. Highly variable clinical phenotype of carbamylphosphate synthetase 1 deficiency in one family: an effect of allelic variation in gene expression? Clin Genet. 2009; 76(3):263–269. | |
Rüegger CM, Lindner M, Ballhausen D, et al. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J Inherit Metab Dis. 2014;37(1):21–30. | |
Summar ML, Barr F, Dawling S, et al. Unmasked adult-onset urea cycle disorders in the critical care setting. Crit Care Clin. 2005; 21(Suppl 4):S1–S8. | |
Braissant O. Current concepts in the pathogenesis of urea cycle disorders. Mol Genet Metab. 2010;100(Suppl 1):S3–S12. | |
Braissant O, McLin VA, Cudalbu C. Ammonia toxicity to the brain. J Inherit Metab Dis. 2013;36(4):595–612. | |
Butterworth RF. Effects of hyperammonaemia on brain function. J Inherit Metab Dis. 1998;21(Suppl 1):6–20. | |
Kido J, Nakamura K, Mitsubuchi H, et al. Long-term outcome and intervention of urea cycle disorders in Japan. J Inherit Metab Dis. 2012;35(5):777–785. | |
Häberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. | |
Batshaw ML. Sodium benzoate and arginine: alternative pathway therapy in inborn errors of urea synthesis. Prog Clin Biol Res. 1983;127:69–83. | |
Batshaw ML, Brusilow SW. Treatment of hyperammonemic coma caused by inborn errors of urea synthesis. J Pediatr. 1980;97(6):893–900. | |
Morioka D, Kasahara M, Takada Y, et al. Current role of liver transplantation for the treatment of urea cycle disorders: a review of the worldwide English literature and 13 cases at Kyoto University. Liver Transpl. 2005;11(11):1332–1342. | |
Meyburg J, Hoffmann GF. Liver, liver cell and stem cell transplantation for the treatment of urea cycle defects. Mol Genet Metab. 2010; 100(Suppl 1):S77–S83. | |
Lewis HB. Studies in the synthesis of hippuric acid in the animal organism. J Biol Chem. 1914;18:225–231. | |
Shiple GJ, Sherwin CP. Synthesis of amino acids in animal organisms. I. Synthesis of glycocol and glutamine in the human organism. J Am Chem Soc. 1922;44:618–624. | |
Brusilow SW, Valle DL, Batshaw M. New pathways of nitrogen excretion in inborn errors of urea synthesis. Lancet. 1979;2(8140):452–454. | |
Niemi AK, Enns G. Sodium phenylacetate and sodium benzoate in the treatment of neonatal hyperammonemia. NeoReviews. 2006; 7(9):e486–e495. | |
Lichter-Konecki U, Diaz GA, Merritt JL 2nd, et al. Ammonia control in children with urea cycle disorders (UCDs); phase 2 comparison of sodium phenylbutyrate and glycerol phenylbutyrate. Mol Genet Metab. 2011;103(4):323–329. | |
Diaz GA, Krivitzky LS, Mokhtarani M, et al. Ammonia control and neurocognitive outcome among urea cycle disorder patients treated with glycerol phenylbutyrate. Hepatology. 2013;57(6):2171–2179. | |
Smith W, Diaz GA, Lichter-Konecki U, et al. Ammonia control in children ages 2 months through 5 years with urea cycle disorders: comparison of sodium phenylbutyrate and glycerol phenylbutyrate. J Pediatr. 2013;162(6):1234. e1221. | |
Berry SA, Lichter-Konecki U, Diaz GA, et al. Glycerol phenylbutyrate treatment in children with urea cycle disorders: Pooled analysis of short and long-term ammonia control and outcomes. Mol Genet Metab. 2014; 112(1):17–24. | |
Guffon N, Kibleur Y, Copalu W, Tissen C, Breitkreutz J. Developing a new formulation of sodium phenylbutyrate. Arch Dis Child. 2012; 97(12):1081–1085. | |
Rieder M. How sweet it isn’t: a new formulation of sodium phenylbutyrate and the challenge of palatability for medicines for children. Arch Dis Child. 2012;97(12):1080. | |
Ah Mew N, Caldovic L. N-Acetylglutamate synthase deficiency: an insight into genetics, epidemiology, pathophysiology, and treatment. Appl Clin Genet. 2011;4:127–135. | |
Häberle J. Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther Clin Risk Manag. 2011;7:1–6. | |
Häberle J. Carglumic acid for the treatment of N-acetylglutamate synthase deficiency and of acute hyperammonemia. Expert Rev Endocrinol Metab. 2012;7:263–271. | |
Van Leynseele A, Jansen A, Goyens P, et al. Early treatment of a child with NAGS deficiency using N-carbamyl glutamate results in a normal neurological outcome. Eur J Pediatr. Epub November 14, 2013. | |
Gessler P, Buchal P, Schwenk HU, Wermuth B. Favourable long-term outcome after immediate treatment of neonatal hyperammonemia due to N-acetylglutamate synthase deficiency. Eur J Pediatr. 2010; 169(2):197–199. | |
Ah Mew N, McCarter R, Daikhin Y, Nissim I, Yudkoff M, Tuchman M. N-Carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics. 2010;126(1):e208–e214. | |
Williams M, Huijmans JG, van Diggelen OP, van der Louw EJTM, De Klerk JBC, Häberle J. Carbamoylphosphate synthase I (CPS 1) deficiency: treatment with carglumic acid (Carbaglu) (abstract). J Inherit Metab Dis. 2010;33:S118. | |
Ah Mew N, Daikhin E, Payan I, et al. N-Carbamylglutamate increases ureagenesis in patients with CPSI deficiency (abstract). Mol Genet Metab. 2010;99:207. | |
Pekkala S, Martínez AI, Barcelona B, et al. Understanding carbamoyl-phosphate synthetase I (CPS1) deficiency by using expression studies and structure-based analysis. Hum Mutat. 2010;31(7):801–808. | |
Erez A, Nagamani SC, Lee B. Argininosuccinate lyase deficiency-argininosuccinic aciduria and beyond. Am J Med Genet C Semin Med Genet. 2011;157(1):45–53. | |
Erez A, Nagamani SC, Shchelochkov OA, et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat Med. 2011;17(12):1619–1626. | |
Brunetti-Pierri N, Erez A, Shchelochkov O, Craigen W, Lee B. Systemic hypertension in two patients with ASL deficiency: a result of nitric oxide deficiency? Mol Genet Metab. 2009;98(1–2):195–197. | |
Nagamani SC, Campeau PM, Shchelochkov OA, et al. Nitric-oxide supplementation for treatment of long-term complications in argininosuccinic aciduria. Am J Hum Genet. 2012;90(5):836–846. | |
Niesters M, Dahan A. Pharmacokinetic and pharmacodynamic considerations for NMDA receptor antagonists in the treatment of chronic neuropathic pain. Expert Opin Drug Metab Toxicol. 2012;8(11):1409–1417. | |
Meyburg J, Hoffmann GF. Liver transplantation for inborn errors of metabolism. Transplantation. 2005;80(Suppl 1):S135–S137. | |
Strom SC, Fisher RA, Rubinstein WS, et al. Transplantation of human hepatocytes. Transplant Proc. 1997;29(4):2103–2106. | |
Stephenne X, Najimi M, Smets F, Reding R, de Ville de Goyet J, Sokal EM. Cryopreserved liver cell transplantation controls ornithine transcarbamylase deficient patient while awaiting liver transplantation. Am J Transplant. 2005;5(8):2058–2061. | |
Dhawan A, Mitry RR, Hughes RD. Hepatocyte transplantation for liver-based metabolic disorders. J Inherit Metab Dis. 2006;29(2–3):431–435. | |
Puppi J, Tan N, Mitry RR, et al. Hepatocyte transplantation followed by auxiliary liver transplantation – a novel treatment for ornithine transcarbamylase deficiency. Am J Transplant. 2008;8(2):452–457. | |
Stéphenne X, Najimi M, Ngoc DK, et al. Cryopreservation of human hepatocytes alters the mitochondrial respiratory chain complex 1. Cell Transplant. 2007;16(4):409–419. | |
Puppi J, Strom SC, Hughes RD, et al. Improving the techniques for human hepatocyte transplantation: report from a consensus meeting in London. Cell Transplant. 2012;21(1):1–10. | |
Sokal EM. From hepatocytes to stem and progenitor cells for liver regenerative medicine: advances and clinical perspectives. Cell Prolif. 2011;44(Suppl 1):39–43. | |
Sandhaus RA. Gene therapy meets stem cells. N Engl J Med. 2012;366(6):567–569. | |
Sokal EM. Treating inborn errors of liver metabolism with stem cells: current clinical development. J Inherit Metab Dis. Epub March 26, 2014. | |
Raper SE, Chirmule N, Lee FS, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80(1–2):148–158. | |
Wang L, Morizono H, Lin J, et al. Preclinical evaluation of a clinical candidate AAV8 vector for ornithine transcarbamylase (OTC) deficiency reveals functional enzyme from each persisting vector genome. Mol Genet Metab. 2012;105(2):203–211. | |
Viecelli HM, Thöny B. Challenges of experimental gene therapy for urea cycle disorders. J Pediatr Biochem. 2014;4(1):65–73. | |
Lichter-Konecki U, Nadkarni V, Moudgil A, et al. Feasibility of adjunct therapeutic hypothermia treatment for hyperammonemia and encephalopathy due to urea cycle disorders and organic acidemias. Mol Genet Metab. 2013;109(4):354–359. | |
Kosenko E, Kaminsky Y, Lopata O, et al. Nitroarginine, an inhibitor of nitric oxide synthase, prevents changes in superoxide radical and antioxidant enzymes induced by ammonia intoxication. Metab Brain Dis. 1998;13(1):29–41. | |
Braissant O. Ammonia toxicity to the brain:effects on creatine metabolism and transport and protective roles of creatine. Mol Genet Metab. 2010;100(Suppl 1):S53–S58. | |
Braissant O, Henry H, Villard AM, et al. Ammonium-induced impairment of axonal growth is prevented through glial creatine. J Neurosci. 2002;22(22):9810–9820. | |
Braissant O, Cagnon L, Monnet-Tschudi F, et al. Ammonium alters creatine transport and synthesis in a 3D culture of developing brain cells, resulting in secondary cerebral creatine deficiency. Eur J Neurosci. 2008;27(7):1673–1685. | |
Ratnakumari L, Qureshi IA, Maysinger D, Butterworth RF. Developmental deficiency of the cholinergic system in congenitally hyperammonemic spf mice: effect of acetyl-l-carnitine. J Pharmacol Exp Ther. 1995;274(1):437–443. | |
Cagnon L, Braissant O. Role of caspases, calpain and cdk5 in ammonia-induced cell death in developing brain cells. Neurobiol Dis. 2008;32(2):281–292. | |
Cagnon L, Braissant O. CNTF protects oligodendrocytes from ammonia toxicity: intracellular signaling pathways involved. Neurobiol Dis. 2009;33(1):133–142. | |
Agusti A, Cauli O, Rodrigo R, Llansola M, Hernandez-Rabaza V, Felipo V. p38 MAP kinase is a therapeutic target for hepatic encephalopathy in rats with portacaval shunts. Gut. 2011;60(11):1572–1579. | |
Tanigami H, Rebel A, Martin LJ, et al. Effect of glutamine synthetase inhibition on astrocyte swelling and altered astroglial protein expression during hyperammonemia in rats. Neuroscience. 2005;131(2):437–449. | |
Willard-Mack CL, Koehler RC, Hirata T, et al. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience. 1996;71(2):589–599. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.