Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 5
Orphan drugs in development for long-chain fatty acid oxidation disorders: challenges and progress
Authors Sun A, Merritt II JL
Received 21 January 2015
Accepted for publication 24 February 2015
Published 28 April 2015 Volume 2015:5 Pages 33—41
DOI https://doi.org/10.2147/ODRR.S63061
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Lise Aagaard
Angela Sun, J Lawrence Merritt II
Department of Pediatrics, University of Washington, Seattle, WA, USA
Abstract: Fatty acid oxidation disorders are inborn errors of metabolism resulting in failure of ß-oxidation within or transport of fatty acids into the mitochondria. The long-chain fatty acid oxidation disorders are characterized by variable presentations ranging from newborn cardiomyopathy, to infantile hypoketotic hypoglycemia resulting from liver involvement, to skeletal myopathy often resulting in rhabdomyolysis in adolescents and adults. Treatments for these long-chain fatty acid oxidation disorders have typically focused upon avoidance of fasting with dietary fat restriction and medium-chain triglyceride supplementation. These treatments have resulted in only a partial response with improvements in hypoglycemia, reduction in frequency of rhabdomyolysis, and improvement in cardiomyopathy with early therapy, but significant risk remains. Recent advances in therapies for long-chain fatty acid oxidation disorders are reviewed in this article. These include sodium D,L-3-hydroxybutyrate, triheptanoin, gene therapy, and bezafibrates. Sodium D,L-3-hydroxybutyrate has shown clinical effect, with improvements in muscle tone, neurological abnormalities, and some cases of cardiomyopathy and leukodystrophy. Triheptanoin has been used as an alternative medium-chain triglyceride in a number of fatty acid oxidation disorders and has shown promising findings in the treatment of cardiomyopathy and hypoglycemia. However, it does not significantly reduce episodes of rhabdomyolysis. Gene therapy has been shown to improve acylcarnitine levels in very-long-chain acyl-coenzyme A dehydrogenase deficiency mouse models, with preservation of glucose levels. Bezafibrates have shown improvements in acylcarnitine concentrations in fibroblast studies, but clinical observations have not demonstrated consistent effects. Together, these treatments have shown some improvements in individual case reports, but there is still a significant need for randomized controlled trials to investigate these therapies, given the ongoing need for improved treatments in these disorders.
Keywords: fatty acid oxidation disorders, 3-hydroxybutyrate, triheptanoin, gene therapy, bezafibrate
Introduction
Fatty acids provide a major source of energy for the body. Beta-oxidation of fatty acids takes place in the mitochondria. Short-chain and medium-chain fatty acids can enter the mitochondria directly, but long-chain fatty acids must be transported in by way of a carnitine shuttle. Once inside the mitochondrial matrix, β-oxidation proceeds in a cycle and results in the sequential cleavage of two-carbon acetyl-coenzyme A (CoA) from the fatty acid chain and the transfer of electrons to the respiratory chain for adenosine triphosphate production. Acetyl-CoA can be used as a substrate in the tricarboxylic acid cycle to generate reducing equivalents for the electron transport chain or it can be used for ketone synthesis, which provides an alternate form of energy for the brain, heart, muscle, kidney, and other tissues.
Fatty acid oxidation (FAO) disorders are a group of recessively inherited disorders caused by deficiency of enzymes required for the β-oxidation process or transport of long-chain fatty acids into the mitochondria (Figure 1). This defect in β-oxidation and the resulting failure to generate acetyl-CoA, nicotinamide adenine dinucleotide, and flavin adenine dinucleotide leads to an energy deficit and produces a wide range of clinical presentations, from mild hypotonia in adults to sudden death.1 As may be expected, the long-chain fatty acid oxidation (LCFAO) disorders are associated with more-severe phenotypes due to the large energy deficit, and affected individuals can manifest with cardiac and skeletal myopathy, arrhythmias, rhabdomyolysis, liver failure, and retinal degeneration (Table 1). Furthermore, the accumulation of long-chain acylcarnitine species has been suggested to be arrhythmogenic and associated with cardiac dysfunction.2
 | Figure 1 Long-chain fatty acid oxidation metabolism. |
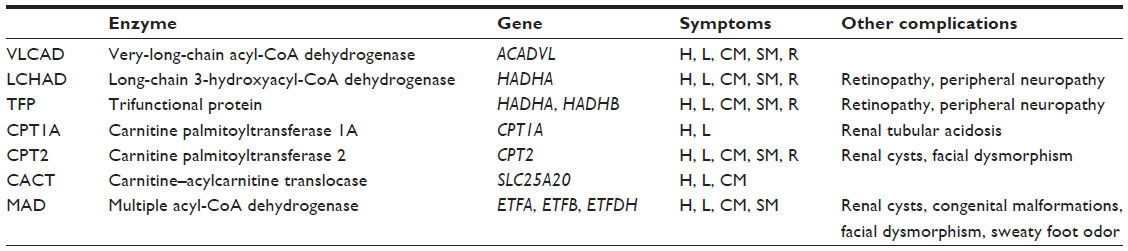 | Table 1 Summary of long-chain fatty acid oxidation disorders |
With early detection and institution of therapy, the initial outcomes are improved for most patients, yet long-term morbidity of affected individuals can be triggered by acute illness or noncompliance with dietary recommendations, resulting in rapid, life-threatening metabolic decompensation. A recent review of all patients with FAO disorders diagnosed symptomatically reported mortality in all forms, with over 60% for most LCFAO disorders including very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency, carnitine palmitoyltransference (CPT) type 2 (CPT2) deficiency, and carnitine–acylcarnitine translocase (CACT) deficiency, despite treatment at the current standard of care.3 In VLCAD patients specifically, the rate of sudden death has been reported at 52% and with an overall mortality of 60%.4 The significant morbidity and mortality rate has led to the development of a number of therapeutic strategies, including novel drugs and gene therapy.
Types of LCFAO disorders
Very-long-chain acyl-CoA dehydrogenase deficiency
Deficiency of the VLCAD enzyme has been historically estimated to affect between 1:100,000 and 1:120,000 individuals.5–7 The introduction of acylcarnitine profiling into expanded newborn screening programs in the past two decades has revealed the actual incidence may be as high as 1:42,500.8 VLCAD is highly expressed in liver, heart, and skeletal muscle, and when completely or partially deficient, it causes variable phenotypes from severe cardiomyopathy, neonatal hypoglycemia, and precipitous death, to a milder form presenting as myopathy and/or rhabdomyolysis in adolescence or adulthood.2,9–14 Cardiomyopathy has been reported in 48% of VLCAD cases and arrhythmias in 52%.4 Unfortunately, the diagnosis is all too often delayed until the first critical illness or until postmortem biochemical evaluation.15
Long-chain 3-hydroxyacyl-CoA and trifunctional protein deficiency
The mitochondrial trifunctional protein (TFP) comprises long-chain enoyl-CoA hydratase, LCHAD, and 3-ketoacyl-CoA thiolase. Isolated LCHAD deficiency affects only the dehydrogenase activity of the protein, while deficiency of all three enzymes is termed TFP deficiency. The prevalence of LCHAD deficiency is approximately 1:110,000.16 Some patients have only mild symptoms, but many with severe forms of LCHAD or TFP deficiency die in the first few days of life, with rapidly progressive cardiomyopathy.17,18 In surviving patients, 65% experience skeletal myopathy, 21% develop peripheral neuropathy, and 43% have retinopathy.17 Early diagnosis through newborn screening and timely institution of therapy improves outcomes in some patients but does not entirely prevent morbidity and mortality, especially in those patients with TFP deficiency.18,19 The survival rate is poorer for TFP deficiency than for LCHAD deficiency.17,20
Carnitine palmitoyltransferase type 1 deficiency
The CPT type 1 enzyme (CPT1) has three different isoforms, with CTP1A being expressed in the liver and kidney. CPT1 deficiency either presents in early childhood with attacks of hypoketotic hypoglycemia and liver dysfunction, which may quickly progress to liver failure, or less commonly as an adult-onset skeletal myopathy. A common CPTIA mutation, c.1436C>T (p.P479L), exists in the Inuit, Alaskan native, Canadian First Nations, and Hutterite populations and has been associated with a milder phenotype.21,22 The frequency of this allele in the Inuit population is 0.44.21 With treatment, patients with CPT1 deficiency can have a normal outcome, but some suffer neurological impairment from repeated episodes of metabolic decompensation.23,24 Renal tubular acidosis may occur during attacks.23
Carnitine palmitoyltransferase type 2 deficiency
Infants with the severe, neonatal form of CPT2 deficiency present with hypotonia, cardiomyopathy, arrhythmias, seizures, and multiple congenital anomalies (dysmorphic facies, renal cysts, brain malformations) and die within the first days to months of life.25 However, the majority of affected individuals present as adults in the second or third decade of life with exercise intolerance and attacks of rhabdomyolysis, which can lead to renal failure and death.24 Most of these older patients carry a common mutation that confers a higher degree of residual enzyme activity.26
Carnitine–acylcarnitine translocase deficiency
CACT deficiency is one of the most severe FAO disorders. The most common presentation is sudden death in the neonatal period.20 Despite newborn screening, the mortality rate remains high and the prognosis poor in most infants.20,27 Early treatment can be successful in some cases, but surviving patients may suffer profound developmental delay, seizures, and other complications.28,29 Milder disease associated with higher residual enzyme activity has been reported.30 Enzyme activity does not necessarily correlate with disease severity.31
Multiple acyl-CoA dehydrogenase deficiency
Multiple acyl-CoA dehydrogenase (MAD) deficiency is also known as glutaric acidemia type 2. Patients with severe MAD deficiency may present in the neonatal period with metabolic acidosis, hypoketotic hypoglycemia, and cardiomyopathy. They may have congenital malformations, including polycystic kidneys and dysmorphic facies. Many do not survive beyond the first few weeks to months of life due to rapidly progressing cardiomyopathy that may not be responsive to therapy. Those with the milder, later-onset form are still at risk for episodes of acute decompensation. In individuals identified through newborn screening programs and in whom treatment is initiated early, sudden death or acute, life-threatening events may still occur.32,33
Diagnosis of LCFAO disorder
In many countries, LCFAO disorders are diagnosed through newborn screening programs. For those patients presenting with clinical symptoms, a plasma acylcarnitine profile is the first-line test and can be diagnostic. Urine organic acid analysis may provide additional supporting evidence of abnormal metabolites. Genetic testing is necessary to confirm the diagnosis at a molecular level. If mutations of uncertain significance are identified, enzyme assays may be helpful in determining whether or not an individual is affected.
Current therapies
Treatment of FAO disorders is primarily aimed at maintaining a constant energy supply through frequent meals and fasting avoidance. Spiekerkoetter et al34 has recommended the following safe fasting time periods for different age groups: 1) a neonate should fast no longer than 3 hours; 2) between 6 and 12 months, infants may fast up to 4 hours during the day and 6–8 hours during the night; and 3) children older than 12 months can fast 4 hours during the day and 10–12 hours at night. If the patient will be fasting for surgery, it is important to provide intravenous fluids with 10% dextrose in order to maintain a continuous energy supply.
The diet of patients with LCFAO deficiencies should be high in carbohydrates and low in fats, with restriction of long-chain fats. The strictness of the diet may vary depending on disease severity. For example, in patients with severe VLCAD deficiency, total fat is limited to 25%–30% of total calories, with long-chain fatty acids comprising approximately one third, and infants may not breastfeed due to the high fat content in breast milk.34 Infants with mild cases of VLCAD deficiency, such as those detected through newborn screening programs, may be allowed a more liberal goal of 30%–40% of total calories from fat, and some breastfeeding is permitted. Levels of essential fatty acids must be monitored as they may become deficient, and supplementation of essential fatty acids is often necessary. Use of a higher protein diet has been investigated in patients with LCHAD and TFP deficiency due to the increased incidence of overweight and obesity observed by some groups. The higher protein diet was found to be safe and well tolerated over the short term and resulted in lowered energy intake and increased energy expenditure as compared to the standard high-carbohydrate diet.35 Further studies are needed to clarify the long-term safety and efficacy of this diet.
MCT (medium-chain triglyceride) oil is used to provide a substrate for the β-oxidation process that bypasses the block created by the LCFAO disorders. In general, MCT oil should provide 20%–25% of total energy.34 In a study of LCHAD patients on a low-fat diet and MCT oil supplementation, plasma hydroxyacylcarnitine species decreased and most participants remained healthy without episodes of metabolic decompensation, although they required essential fatty acid and fat-soluble vitamin supplementation.36 In patients with LCHAD and TFP deficiency, MCT oil “loading” prior to exercise was shown to reduce the concentration of long-chain hydroxyacylcarnitines by 30% and raise β-hydroxybutyrate levels three-fold compared to patients who were not given MCT oil.37 MCT supplementation has been associated with a reversal of cardiomyopathy in case reports of patients with CACT and VLCAD deficiency.28,38,39 However, reports have noted that even highly compliant VLCAD patients still had significant muscle weakness, muscle pain, or myoglobinuria.17
Although secondary carnitine deficiency is common in the LCFAO disorders, carnitine supplementation is controversial. There is no published evidence that carnitine supplementation is beneficial in the long-term treatment of LCFAO disorders.34 It has been hypothesized that long-chain hydroxyacylcarnitines exert a toxic effect by inducing arrhythmias.40 In a study of VLCAD knockout mice, carnitine supplementation induced production of acylcarnitines but did not replenish low free-carnitine levels.41 Conflicting results were seen in LCAD knockout mice, in which carnitine supplementation did not induce accumulation of long-chain acylcarnitines or affect cardiac function but was shown to lower myocardial triglyceride levels.42
In the specific case of MAD deficiency, treatment may include riboflavin. Specific mutations in ETFDH confer a milder phenotype, and individuals with at least one of these mutations often respond to pharmacological doses of riboflavin.43 In some cases, clinical and biochemical parameters can be completely corrected after riboflavin treatment.43
Despite early detection through newborn screening and advances in clinical care, dietary modification and supplementation with MCT oil and L-carnitine are unable to address significant potential long-term risks of this disorder.1,44–46 Therefore, additional therapies are currently in development.
Therapies currently in development
Sodium D,L-3-hydroxybutyrate
As FAO disorders result in the failure of energy production, ketone body replacement therapy has been proposed as a supplement to standard therapy, but evidence has been limited to case reports. Sodium D,L-3-hydroxybutyrate (Special Products Limited, Surrey, UK) was first reported in a 1999 abstract to replace endogenous energy production in MAD deficiency.47 In this case report, administration of sodium D,L-3-hydroxybutyrate at doses up to 10 grams/day was associated with improvements in muscle tone and feeding as well as decreased concentrations of free fatty acids, creatine kinase, and plasma acylcarnitines. Van Hove et al48 reported the use of D,L-3-hydroxybutyrate in three patients, with initiation of treatment ranging between 2 months to 2 years and with duration of therapy up to 2 years. All patients showed measureable detection of ketone bodies in plasma and cerebral spinal fluid. No adverse effects were reported. Patient 1 had improvements in motor symptoms (including independent walking), neurological abnormalities, liver size, and leukodystrophy as seen on magnetic resonance imaging. Patient 2 was given 430 mg/kg/day and had improvements in muscle tone so that she could walk and speak two words; over 14 months she had normalization of severe dilated cardiomyopathy. Similar improvements in motor development and cardiomyopathy were also seen in Patient 3, at an initial dose of 700 mg/kg/day. In a more recent case report, a much higher dose of 2,600 mg/kg/day was required to see an improvement in clinical outcomes and cardiac markers.49 Improvements in extensive leukodystrophy in a Portuguese patient without cardiomyopathy have also been reported, with a dose of 900 mg/kg/day.50 While these cases are promising, this literature shows the clear need for larger, systematic studies to investigate the full effectiveness of D,L-3-hydroxybutyrate.
Triheptanoin
Triheptanoin (Ultragenyx Pharmaceutical Inc., Novato, CA, USA) used as an anaplerotic therapy was first reported by Roe et al in 2002.51 Triheptanoin is a triglyceride of three seven-carbon fatty acids that is hydrolyzed to three molecules of heptanoate that then enter the β-oxidation cycle to produce acetyl-CoA and pentanoyl-CoA, which then can generate five-carbon ketone bodies, β-hydroxypentaonate, and β-ketopenanoate in the liver. These five-carbon ketone bodies can be taken up in peripheral tissues and then additionally generate a second acetyl-CoA and propionyl-CoA through another cycle of β-oxidation. Propionyl-CoA then can be used in anaplerosis in the citric acid cycle.52 Additionally, triheptanoin may suppress lipolysis and accumulation of toxic metabolite production in LCFAO disorders.
The initial report by Roe et al51 studied three VLCAD patients with hepatomegaly, hypoglycemia, cardiomyopathy, and muscle pain and weakness while continuing to receive standard treatment. These patients had rapid improvement within the first month of treatment, and longer-term follow-up showed continued improvement in cardiac symptoms, muscle weakness, fatigue, hypoglycemia, and hepatomegaly, although improvement of rhabdomyolysis was not as dramatic.53 Similar effects were seen in patients with LCHAD, TFP, CPT2, and CPT1 deficiency, with the exception of retinopathy in LCHAD deficiency.53 A more-recent study of 22 patients with VLCAD (n=11), LCHAD (n=5), TFP (n=2), CPT2 (n=3), and CACT (n=1) deficiency in a compassionate-use protocol has shown triheptanoin to be well tolerated and to have resulted in reduced hospitalizations, myopathy, and hypoglycemia, but one patient demonstrated continued cardiomyopathy.54 Additional clinical studies, including a double-blind US Food and Drug Administration Phase II study, are underway.54 The most recent report is of a male child with LCHAD deficiency diagnosed at 20 months of age following liver disease and cardiomyopathy. He was placed on a fat-defined diet (40% total energy intake) containing 50% MCT supplementation but had recurrent episodes of creatine kinase elevations. Following initiation of heptanoate at 0.6 grams/kg/day equaling 15% of total energy intake, the boy displayed more stability with no further episodes over the next 10 years. During an endurance exercise test, he had no significant changes in blood glucose, blood lactate, acylcarnitine profiles, or urine organic acids.55 While the use of triheptanoin shows promise, detailed clinical outcomes and long-term randomized studies comparing triheptanoin to standard therapy, including supplementation with MCT oil, are needed to investigate the safety and benefit of triheptanoin.
Future therapies
Gene therapy strategies have been developed for medium-chain and short-chain acyl-CoA dehydrogenase-deficient mouse models.56–58 There has been more research into developing a strategy for VLCAD deficiency using adeno-associated virus (AAV) vectors. Tail-vein injection of a recombinant AAV serotype 8 expressing human ACADVL in a VLCAD mouse model resulted in expression of the human VLCAD enzyme in liver and heart, with lower expression in skeletal muscle, up to 104 days postinjection. Biochemical testing revealed improvements in long-chain acylcarnitines and fasting-induced hypoglycemia.59 Using a recombinant pseudotyped AAV2/9-VLCAD vector, Keeler et al60 were able to show long-term expression up to 20 weeks as evidenced by maintenance of euglycemia and survival by maintaining body temperature greater than 20°C following a fasting cold-challenge. This was an important physiological marker for correction of the mouse phenotype. These studies, although promising in the preclinical setting, underscore the need to develop long-term therapy for VLCAD deficiency.
Bezafibrate is a drug used in treatment of hyperlipidemia that has been found to be an agonist of peroxisome proliferator-activated receptor and promotes transcription of many genes encoding FAO enzymes. In 2009, Bonnefont et al61 reported reductions in episodes of rhabdomyolysis and creatine kinase levels and improvements in measures of quality of life in patients with CPT2 deficiency treated with bezafibrate. A study by Gobin-Limballe et al62 investigated the differences between different genotype groups of bezafibrate-incubated fibroblasts from VLCAD patients. All groups demonstrated increased VLCAD mRNA levels. Group 1 demonstrated no significant changes in β-oxidation flux, which correlated with genotypes associated with more severe, neonatal-onset disease. Significant improvements in flux were seen in Groups 2 and 3, whose genotypes corresponded to milder disease, with Group 3 achieving rates similar to controls. Bezafibrate was shown to improve FAO flux and protein levels as measured by Western blot using rabbit polyclonal anti-VLCAD.63 Another trial of bezafibrate in a case of CACT deficiency reported an in vitro response but failed to demonstrate any short-term clinical effect.64
Yamaguchi et al64 described the effect of bezafibrate in multiple FAO disorders, including medium-chain acyl-CoA dehydrogenase deficiency, VLCAD, CPT2, CACT, TFP, and MAD deficiency. They described clinical improvement in a child with MAD deficiency, including motor and social development. However, in a study by Ørngreen et al,65 patients with CPT2 and VLCAD deficiency did not show improvement of clinical symptoms or FAO during exercise, giving rise to concern for lack of clinical effectiveness.
Other therapeutic strategies have been described that are in early stages of development. Studies of resveratrol and stilbenes have shown improvements in vitro in FAO flux in CPT2 and VLCAD fibroblasts.66,67 While the regulatory mechanism is not fully clear, these findings provide further evidence of the effects of FAO enzyme gene transcription as a potential therapy.
Chaperone therapy has become a significant area of investigation for novel treatments. A study in 2006 reported on the effect of various mutations in SLC22A5 upon carnitine transporter maturation.68 Incubation with phenylbutyrate, quinidine, and verapamil showed stimulated carnitine transport in all cells, but this was only significant with some mutations. An additive effect was seen with the simultaneous use of phenylbutyrate and quinidine, showing the promise of this strategy.
Summary
The LCFAO disorders are associated with significant morbidity and mortality. Affected individuals suffer from acute hepatic dysfunction during intercurrent illnesses, cardiomyopathy, skeletal myopathy, rhabdomyolysis, and other complications. Current treatment involves dietary modification, supplementation with MCT oil, and consideration of L-carnitine supplementation, although the latter remains controversial. These measures, while significantly improving quality of life and survival in many cases, do not completely treat these disorders. Furthermore, early diagnosis and initiation of therapy through newborn screening programs does not prevent severe complications and death, and not all disorders are screened for by all countries.
Part of the difficulty in treating the LCFAO disorders is that the underlying pathophysiology is not completely understood. The energy deficit created by the block in fatty acid β-oxidation accounts for some of the clinical effects, but accumulation of long-chain fatty acids or long-chain acylcarnitines, secondary carnitine deficiency, generalized mitochondrial dysfunction, or other mechanisms could also play a role.
Several novel therapies are in development that could potentially add to the armamentarium of treatment options for the LCFAO disorders. Ketone body replacement therapy has been shown to have positive effects on a wide range of symptoms, from motor development to liver size to leukodystrophy. Triheptanoin not only bypasses the metabolic block but also has an anaplerotic effect. Patients on triheptanoin experience an improvement in biochemical abnormalities and overall symptoms, in particular cardiomyopathy and hypoglycemia. Other potential therapies under investigation include gene therapy and bezafibrates, the latter being found to promote transcription of FAO genes. These novel therapies highlight the need for larger randomized controlled therapeutic trials to validate current data and to implement future therapies. These trials then should be coupled with long-term clinical follow-up studies similar to those ongoing with urea cycle disorders through the Rare Disease Consortium69 in order to correlate these therapies with long-term outcomes for all FAO disorders.70
Disclosure
The authors have no disclosures of any relevant financial relationships or conflicts of interest to resolve related to the content of this manuscript.
References
Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64:477–502. | |
Bonnet D, Martin D, de Lonlay P, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100(22):2248–2253. | |
Baruteau J, Sachs P, Broué P, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2013;36(5):795–803. | |
Baruteau J, Sachs P, Broué P, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study from 187 patients. Complementary data. J Inherit Metab Dis. 2014;37(1):137–139. | |
Zytkovicz TH, Fitzgerald EF, Marsden D, et al. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem. 2001;47(11):1945–1955. | |
Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348(23):2304–2312. | |
Chace DH, Kalas TA, Naylor EW. The application of tandem mass spectrometry to neonatal screening for inherited disorders of intermediary metabolism. Annu Rev Genomics Hum Genet. 2002;3:17–45. | |
Spiekerkoetter U, Tenenbaum T, Heusch A, Wendel U. Cardiomyopathy and pericardial effusion in infancy point to a fatty acid b-oxidation defect after exclusion of an underlying infection. Pediatr Cardiol. 2003;24(3):295–297. | |
Vianey-Saban C, Divry P, Brivet M, et al. Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta. 1998; 269(1):43–62. | |
Hoffman JD, Steiner RD, Paradise L, et al. Rhabdomyolysis in the military: recognizing late-onset very long-chain acyl Co-A dehydrogenase deficiency. Mil Med. 2006;171(7):657–658. | |
Bertrand C, Largillière C, Zabot MT, Mathieu M, Vianey-Saban C. Very long chain acyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid oxidation in fibroblasts. Biochim Biophys Acta. 1993;1180(3):327–329. | |
Bonnet D, de Lonlay P, Gautier I, et al. Efficiency of metabolic screening in childhood cardiomyopathies. Eur Heart J. 1998;19(5):790–793. | |
de Lonlay-Debeney P, Fournet JC, Bonnet D. Fatty acid beta-oxidation deficiency masquerading as fulminant myocarditis. Int J Cardiol. 1998;65(3):287–289. | |
Kluge S, Kühnelt P, Block A, et al. A young woman with persistent hypoglycemia, rhabdomyolysis, and coma: recognizing fatty acid oxidation defects in adults. Crit Care Med. 2003;31(4):1273–1276. | |
Chace DH, DiPerna JC, Mitchell BL, Sgroi B, Hofman LF, Naylor EW. Electrospray tandem mass spectrometry for analysis of acylcarnitines in dried postmortem blood specimens collected at autopsy from infants with unexplained cause of death. Clin Chem. 2001;47(7):1166–1182. | |
Das AM, Illsinger S, Lücke T, et al. Isolated mitochondrial long-chain ketoacyl-CoA thiolase deficiency resulting from mutations in the HADHB gene. Clin Chem. 2006;52(3):530–534. | |
Spiekerkoetter U, Lindner M, Santer R, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32(4):488–497. | |
Sperk A, Mueller M, Spiekerkoetter U. Outcome in six patients with mitochondrial trifunctional protein disorders identified by newborn screening. Mol Genet Metab. 2010;101(2–3):205–207. | |
Sander J, Sander S, Steuerwald U, et al. Neonatal screening for defects of the mitochondrial trifunctional protein. Mol Genet Metab. 2005;85(2):108–114. | |
Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis. 2010;33(5):501–506. | |
Collins SA, Sinclair G, McIntosh S, et al. Carnitine palmitoyltransferase 1A (CPT1A) P479L prevalence in live newborns in Yukon, Northwest Territories, and Nunavut. Mol Genet Metab. 2010;101(2–3):200–204. | |
Prasad C, Johnson JP, Bonnefont JP, et al. Hepatic carnitine palmitoyl transferase 1 (CPT1 A) deficiency in North American Hutterites (Canadian and American): evidence for a founder effect and results of a pilot study on a DNA-based newborn screening program. Mol Genet Metab. 2001;73(1):55–63. | |
Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med. 2004;25(5–6):495–520. | |
Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C(2):77–85. | |
Albers S, Marsden D, Quackenbush E, Stark AR, Levy HL, Irons M. Detection of neonatal carnitine palmitoyltransferase II deficiency by expanded newborn screening with tandem mass spectrometry. Pediatrics. 2001;107(6):E103. | |
Wieser T, Deschauer M, Olek K, Hermann T, Zierz S. Carnitine palmitoyltransferase II deficiency: molecular and biochemical analysis of 32 patients. Neurology. 2003;60(8):1351–1353. | |
Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33(5):527–532. | |
Pierre G, Macdonald A, Gray G, Hendriksz C, Preece MA, Chakrapani A. Prospective treatment in carnitine-acylcarnitine translocase deficiency. J Inherit Metab Dis. 2007;30(5):815. | |
Al-Sannaa NA, Cheriyan GM. Carnitine-acylcarnitine translocase deficiency. Clinical course of three Saudi children with a severe phenotype. Saudi Med J. 2010;31(8):931–934. | |
Rubio-Gozalbo ME, Bakker JA, Waterham HR, Wanders RJ. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol Aspects Med. 2004;25(5–6):521–532. | |
Lopriore E, Gemke RJ, Verhoeven NM, et al. Carnitine-acylcarnitine translocase deficiency: phenotype, residual enzyme activity and outcome. Eur J Pediatr. 2001;160(2):101–104. | |
Angle B, Burton BK. Risk of sudden death and acute life-threatening events in patients with glutaric acidemia type II. Mol Genet Metab. 2008;93(1):36–39. | |
Singla M, Guzman G, Griffin AJ, Bharati S. Cardiomyopathy in multiple acyl-CoA dehydrogenase deficiency: a clinico-pathological correlation and review of literature. Pediatr Cardiol. 2008;29(2):446–451. | |
Spiekerkoetter U, Lindner M, Santer R, et al. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009;32(4):498–505. | |
Gillingham MB, Purnell JQ, Jordan J, Stadler D, Haqq AM, Harding CO. Effects of higher dietary protein intake on energy balance and metabolic control in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2007;90(1):64–69. | |
Gillingham MB, Connor WE, Matern D, et al. Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2003;79(2):114–123. | |
Gillingham MB, Scott B, Elliott D, Harding CO. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2006;89(1–2):58–63. | |
Pervaiz MA, Kendal F, Hegde M, Singh RH. MCT oil-based diet reverses hypertrophic cardiomyopathy in a patient with very long chain acyl-coA dehydrogenase deficiency. Indian J Hum Genet. 2011;17(1):29–32. | |
Sharef SW, Al-Senaidi K, Joshi SN. Successful treatment of cardiomyopathy due to very long-chain acyl-CoA dehydrogenase deficiency: first case report from Oman with literature review. Oman Med J. 2013;28(5):354–356. | |
DaTorre SD, Creer MH, Pogwizd SM, Corr PB. Amphipathic lipid metabolites and their relation to arrhythmogenesis in the ischemic heart. J Mol Cell Cardiol. 1991;23 Suppl 1:11–22. | |
Primassin S, Ter Veld F, Mayatepek E, Spiekerkoetter U. Carnitine supplementation induces acylcarnitine production in tissues of very long-chain acyl-CoA dehydrogenase-deficient mice, without replenishing low free carnitine. Pediatr Res. 2008;63(6):632–637. | |
Bakermans AJ, van Weeghel M, Denis S, Nicolay K, Prompers JJ, Houten SM. Carnitine supplementation attenuates myocardial lipid accumulation in long-chain acyl-CoA dehydrogenase knockout mice. J Inherit Metab Dis. 2013;36(6):973–981. | |
Olsen RK, Olpin SE, Andresen BS, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130(Pt 8):2045–2054. | |
Vockley J, Whiteman DA. Defects of mitochondrial beta-oxidation: a growing group of disorders. Neuromuscul Disord. 2002;12(3):235–246. | |
Solis JO, Singh RH. Management of fatty acid oxidation disorders: a survey of current treatment strategies. J Am Diet Assoc. 2002;102(12):1800–1803. | |
Straussberg R, Harel L, Varsano I, Elpeleg ON, Shamir R, Amir J. Recurrent myoglobinuria as a presenting manifestation of very long chain acyl coenzyme A dehydrogenase deficiency. Pediatrics. 1997; 99(6):894–896. | |
Bonham JR, Tanner MS, Polliu RJ, et al. Oral sodium 3-hydroxybutyrate, a novel adjunct to treatment for multiple acyl-CoA dehydrogenase deficiency. Society for the Study of Inborn Errors of Metabolism (SSIEM) 37th Annual Symposium. September 7–10, 1999; Genova, Italy. J Inherit Metab Dis. 1999;22(Suppl 1):101. | |
Van Hove JL, Grünewald S, Jaeken J, et al. D,L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD). Lancet. 2003;361(9367):1433–1435. | |
Van Rijt WJ, Heiner-Fokkema MR, du Marchie Sarvaas GJ, et al. Favorable outcome after physiologic dose of sodium-D,L-3-hydroxybutyrate in severe MADD. Pediatrics. 2014;134(4):e1224–e1228. | |
Gautschi M, Weisstanner C, Slotboom J, Nava E, Zürcher T, Nuoffer JM. Highly efficient ketone body treatment in multiple acyl-CoA dehydrogenase deficiency-related leukodystrophy. Pediatr Res. 2015;77(1-1):91–98. | |
Roe CR, Sweetman L, Roe DS, David F, Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 2002;110(2):259–269. | |
Deng S, Zhang GF, Kasumov T, Roe CR, Brunengraber H. Interrelations between C4 ketogenesis, C5 ketogenesis, and anaplerosis in the perfused rat liver. J Biol Chem. 2009;284(41):27799–27807. | |
Roe CR, Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis. 2006;29(2–3):332–340. | |
Barone AR, DeWard SJ, Payne N, Goldstein A, Vockley J. Triheptanoin therapy for inherited disorders of fatty acid oxidation. Program for Society for Inherited Medical Disorders Annual Meeting; March 31–April 3, 2012; Charlotte, NC. Mol Genet Metab. 2012;105(3):304. | |
Karall D, Mair G, Albrecht U, et al. Sports in LCHAD Deficiency: Maximal incremental and endurance exercise tests in a 13-year-old patient with long-chain 3-hydroxy acyl-CoA dehydrogenase deficiency (LCHADD) and heptanoate treatment. JIMD Rep. 2014;17:7–12. | |
Schowalter DB, Matern D, Vockley J. In vitro correction of medium chain acyl CoA dehydrogenase deficiency with a recombinant adenoviral vector. Mol Genet Metab. 2005;85(2):88–95. | |
Conlon TJ, Walter G, Owen R, et al. Systemic correction of a fatty acid oxidation defect by intramuscular injection of a recombinant adeno-associated virus vector. Hum Gene Ther. 2006;17(1):71–80. | |
Beattie SG, Goetzman E, Conlon T, et al. Biochemical correction of short-chain acyl-coenzyme A dehydrogenase deficiency after portal vein injection of rAAV8-SCAD. Hum Gene Ther. 2008;19(6):579–588. | |
Merritt JL 2nd, Nguyen T, Daniels J, Matern D, Schowalter DB. Biochemical correction of very long-chain acyl-CoA dehydrogenase deficiency following adeno-associated virus gene therapy. Mol Ther. 2009;17(3):425–429. | |
Keeler AM, Conlon T, Walter G, et al. Long-term correction of very long-chain acyl-coA dehydrogenase deficiency in mice using AAV9 gene therapy. Mol Ther. 2012;20(6):1131–1138. | |
Bonnefont JP, Bastin J, Behin A, Djouadi F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. 2009;360(8):838–840. | |
Gobin-Limballe S, Djouadi F, Aubey F, et al. Genetic basis for correction of very-long-chain acyl-coenzyme A dehydrogenase deficiency by bezafibrate in patient fibroblasts: toward a genotype-based therapy. Am J Hum Genet. 2007;81(6):1133–1143. | |
Gobin-Limballe S, McAndrew RP, Djouadi F, Kim JJ, Bastin J. Compared effects of missense mutations in Very-Long-Chain Acyl-CoA Dehydrogenase deficiency: Combined analysis by structural, functional and pharmacological approaches. Biochim Biophys Acta. 2010;1802(5):478–484. | |
Yamaguchi S, Li H, Purevsuren J, et al. Bezafibrate can be a new treatment option for mitochondrial fatty acid oxidation disorders: evaluation by in vitro probe acylcarnitine assay. Mol Genet Metab. 2012;107(1–2):87–91. | |
Ørngreen MC, Madsen KL, Preisler N, Andersen G, Vissing J, Laforêt P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: a randomized clinical trial. Neurology. 2014;82(7):607–613. | |
Bastin J, Lopes-Costa A, Djouadi F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum Mol Genet. 2011;20(10):2048–2057. | |
Aires V, Delmas D, Le Bachelier C, et al. Stilbenes and resveratrol metabolites improve mitochondrial fatty acid oxidation defects in human fibroblasts. Orphanet J Rare Dis. 2014;9:79. | |
Amat di San Filippo C, Pasquali M, Longo N. Pharmacological rescue of carnitine transport in primary carnitine deficiency. Hum Mutat. 2006;27(6):513–523. | |
Rare Clinical Diseases Research Network: Initiative of the National Center for Advancing Translational Sciences (NCATS) [homepage on the Internet]. Bethesda, MD: National Institutes of Health; 2015. Available from: http://www.rarediseasesnetwork.org. Accessed February 28, 2015. | |
Seminara J, Tuchman M, Krivitzky L, et al. Establishing a consortium for the study of rare diseases: The Urea Cycle Disorders Consortium. Mol Genet Metab. 2010;100 Suppl 1:S97–S105. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.