Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 5
Orphan drugs in development for Huntington's disease: challenges and progress
Authors Burgunder J
Received 25 September 2014
Accepted for publication 5 November 2014
Published 12 February 2015 Volume 2015:5 Pages 1—9
DOI https://doi.org/10.2147/ODRR.S49198
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Lise Aagaard
Jean-Marc Burgunder1–4
1Swiss Huntington’s Disease Centre, Department of Neurology, University of Bern, Bern, Switzerland; 2Department of Neurology, West China Hospital, Sichuan University, Chengdu, 3Department of Neurology, Xiangya Hospital, Central South University, Changsha, 4Department of Neurology, Sun Yat-sen University, Guangzhou, People’s Republic of China
Abstract: Huntington’s disease is a monogenic disorder encompassing a variable phenotype with progressive cognitive, psychiatric, and movement disorders. Knowledge of the mechanisms involved in this disorder has made substantial advances since the discovery of the gene mutation. The dynamic mutation is the expansion of a CAG (cytosine-adenine-guanine) repeat in the huntingtin (HTT) gene, which is transcribed into an abnormal protein with an elongated polyglutamine tract. Polyglutamine HTT accumulates and is changed in its function in multifaceted ways related to the numerous roles of the normal protein. The protein is expressed in numerous areas of the brain and also in other organs. The major brain region involved in the disease process is the striatum, but it is clear that other systems are involved as well. This accumulated knowledge has now led to the development of treatment strategies based on specific molecular pathways for symptomatic and disease course-modifying treatment. The most proximal way to handle the disturbed protein is to hinder the gene transcription, translation, and/or to increase protein clearance. Other mechanisms now being approached include modulation of energy and intracellular signaling, induction of factors potentially leading to neuroprotection, as well as modulation of glial function. Several clinical trials based on these approaches are now under way, and it is becoming clear that a future disease-modifying therapy will be a combination of several approaches harmonized with symptomatic treatments. In this review, some of the most promising and advanced strategies to develop novel treatments in Huntington's disease are examined.
Keywords: Huntington's disease, symptomatic treatment, disease-modifying therapy
Introduction
Huntington’s disease (HD) is characterized by a complex phenotype including motor, cognitive, and psychiatric symptoms and signs starting at different ages and gradually evolving over the years until death after 15–25 years.1,2 The mutation of this autosomal-dominant inherited disorder is fully penetrant, and HD can be diagnosed well in advance of any symptom, as opposed to other, more frequent degenerative disorders of the brain, including Parkinson’s and Alzheimer’s disease. There is a long presymptomatic phase, during which opportunities for a disease-modifying treatment might be tested and applied, since the molecular diagnosis allows a clear inclusion of people who will develop the disease. No neuroprotective treatment is yet available, and drugs used now treat symptoms only. However, comprehensive care, of which drug treatment is only a part, can be suggested to modify the course of the disorder, specifically through avoiding further complications. A tremendous amount of data on the multifaceted pathophysiology of the brain and systemic disorder included in HD have been accumulated in the last few years.
The increased numbers of a CAG (cytosine-adenine-guanine)-triplet repeat in exon 1 of the huntingtin (HTT) gene located on chromosome 4p16.33 leads after translation to a polyglutamine elongation at the C-terminal of the protein. The polyglutamine tract leads to HTT accumulation in the cell, due to complex mechanisms of protein aggregation through progressive fibril formation.4 Furthermore, HTT mutation impairs a number of cellular functions.5 These include gene transcription and intracellular and synaptic signaling mechanisms. Furthermore, cellular functions like energy metabolism, mitochondrial function, intracellular protein trafficking and dynamic axonal transport mechanisms, and endocytic and vesicular trafficking changes are involved as well. The major region involved in the disease process is the striatum, but other brain areas are involved as well, including the cortex and the hypothalamus. Furthermore, it is increasingly recognized that changes occur in other organs as well. All these molecular mechanisms and systemic involvements represent potential targets for disease-modifying strategies. Furthermore, HTT is involved in a large number of interactions with other proteins and pathways,6 and polyglutamine expansion may modify them.7 This contributes to the complexity of molecular events involved in the disease, but also to the number of potential targets.
Huntington’s disease: a complex phenotype
Typically, early HD is characterized by the occurrence of subtle cognitive and behavioral changes with involuntary, hyperkinetic, choreatic movements at around 40 years old, but there are large variations in the phenotype and time course, including the sequence of symptom development. Variation of age at onset, usually defined as age at the appearance of clear motor signs, is mainly dependent upon the number of triplet repeats, with a negative correlation and younger age at onset in cases with higher numbers. The course is relentlessly progressive, but with fluctuations, particularly observed for the behavioral symptoms. In the face of such a variation, the importance of using appropriate assessment methods and meaningful end points in clinical trials cannot be stressed enough, as is also the case for symptomatic management.8
Trials can be grouped into symptomatic and disease-modifying treatments; however, the boundary between these is not always absolute. Purely symptomatic modalities may change the course of the disease, eg, by avoiding complications like social isolation. They may also modify adaptive plastic synaptic changes that secondarily participate in the disease process. On the other hand, treatment targeted at long-term disease-course modification may also have an acute symptomatic effect. Issues in clinical trials include the proper choice of age at onset assessment and biomarkers used as end points. Typically, the age at occurrence of first motor symptoms and signs has been considered as defining the age at disease onset. However, experience shows that cognitive and behavioral changes may occur before motor impairment, which is also often subtle during the phenoconversion period. A better method is to capture the history of motor, behavior (depression, irritability, apathy, aggressive behavior, obsession, and psychosis), and cognitive impairment in an open and unbiased way, as is done within the European Huntington’s Disease Network Registry (http://www.euro-hd.net) and the global Enroll-HD (http://studies.enroll-hd.org) studies.
Progress has been made in informing the choice of biomarkers, which has to be tailored to the respective pathway targeted by the intervention under scrutiny. Track-HD, an intensive, 3-year study, has examined of a cohort of HD gene carriers in a premanifest and in an early stage of the disease.9–11 Assessment has included a complex battery, with cognitive, quantitative motor, oculomotor, and neuropsychiatric measures, as well as imaging methods. Magnetic resonance imaging assessments, including whole-brain atrophy, ventricular expansion, caudate atrophy, putamen atrophy, and white-matter atrophy, have turned out to be valuable biomarkers both in the presymptomatic and in the early manifest stages, with cognitive tests, including the symbol digit modality test, the Stroop word-reading and the emotion-recognition tests providing additional information in a subgroup.11
Gene therapy
In order to decrease the expression of the abnormal, long-allele-bearing HTT gene, different types of strategies have been developed. They include the use of interfering ribonucleic acid (RNA) molecules, antisense oligonucleotides (ASOs), and proteins modulating the transcription process. They may be grouped according to specificity, recognizing the elongated allele only in a selective (allele-specific) or nonselective (allele-nonspecific) way. A decrease in HTT expression improves symptoms and prolongs survival in HD mouse models.12,13
ASOs, specifically targeted HTT messenger RNA (mRNA),14 induce degradation by RNase activity and can be modified to make them resistant to exonuclease cleavage, which improves their stability. After injection in the brain of HD animal models, ASOs decrease HTT mRNA levels in the striatum without morphological change, followed after a time lag by a return to previous mutant HTT mRNA levels.15 ASO therapies are in the process of development in other neurodegenerative disorders, and have reached clinical trials in some instances. For example, one such trial that has been started to examine safety, tolerability, and activity of an ASO treatment is presently undergoing in amyotrophic lateral sclerosis caused by SOD1 gene mutations (http://clinicaltrials.gov/show/NCT01041222). There has been a Phase I study in children with spinal muscular atrophy with an ASO targeting SMN2, and the treatment was well tolerated; in an autopsy case, cortical and spinal neuron targeting were reported. Phase II and III trials are planned in spinal muscular atrophy and also in amyotrophic lateral sclerosis. The HD study uses a similar chemical ASO composition. A safety study using ASOs, applied by repetitive monthly lumbar administration with dose escalation in HD patients, is planned in 2015. This will include a large array of detailed cerebrospinal fluid studies, including immunological and chemical assessments and also some exploratory end points.
Small interfering RNA (siRNA) or micro-interfering RNA bind to the abnormal transcript, which undergoes degradation by the RNA-induced silencing complex with a consecutive decrease in mutated protein, and this approach has been found to improve the phenotype in animal models.16 siRNA may be designed specifically to target the mutant allele in a selective way, with conservation of normal wild-type HTT expression.17 A general proof of concept for a siRNA therapy has been delivered in healthy volunteers inoculated with respiratory syncytial virus, in whom it has demonstrated its expected antiviral activity.18 The molecule may be packaged into modified adenoviruses acting as a vector, and stereotactic injections of such constructs have been found to be safe in primates for 6 months.19 This treatment is followed by a significant decrease in HTT mRNA compared with controls. A Phase I study using continuous, pump system-mediated intracerebral application of an RNA-interference therapeutic agent had been announced,20 but its later retraction demonstrates the difficult path along the way of such strategies. The chemical properties of the compounds also play a role, and their modification may improve efficacy and tissue spread.21 Promising, however, is the authorization of gene-silencing compounds for the treatment of cytomegalovirus and of familial hypercholesterolemia, with more trials running and in preparation for other diseases.22
A third potential way to decrease HTT mRNA is the use of factors that could inhibit transcription, eg, zinc-finger protein (ZFP).23 Long ZFPs have been prepared to bind CAG repeats in a selective way and to be expressed in an HD cell-line model, with the effect of a decreased expression of the longer allele, and this was also achieved after striatal delivery using an adenoassociated virus system.24
There are major hurdles in the path toward HTT gene silencing in humans; they include finding the appropriate delivery process, choice of target and tissue volume to be included, and timing of treatment along the course of evolution. Modes of application may include stereotactic intrastriatal delivery in the form of injections or infusions, which can be combined with convection-enhancing processes or with carriers facilitating diffusion, intraventricular injections or continuous delivery with pumps, and viral vectors or other cargo systems with selective brain targeting. Of course, the major size difference between the brains of animal models used so far and the human brain represents a major challenge. Furthermore, the course of long-term effects and side effects in general also need to be better understood. However, these strategies do offer the most upstream option to target the multifaceted molecular pathophysiology of HD in a meaningful way. It is now great news that Phase I trials are on the verge of being launched (Table 1).
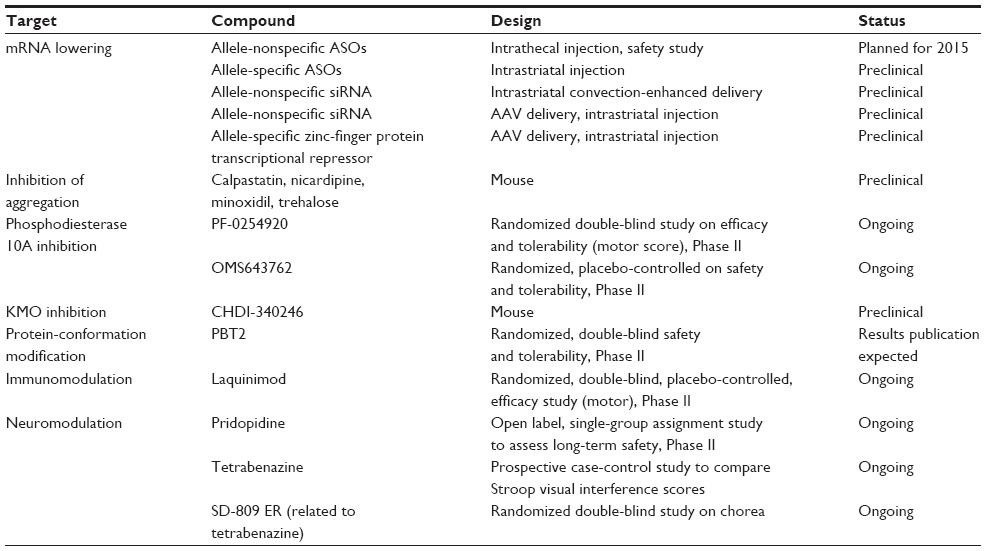 | Table 1 Major planned and ongoing clinical trials in Huntington’s disease |
Targeting the mutated protein
Following CAG-repeat expansion in the HTT gene, a polyglutamine tail is translated in the molecule. HTT is a complex protein involved in a number of posttranslational modifications, including acetylation, phosphorylation, SUMOylation, ubiquitination, palmitoylation, and proteolytic cleavage. This opens the potential to modulate the posttranslational intracellular handling of the molecule with the aim of decreasing levels of abnormal huntingtin,25 while retaining enough of the normal protein to attend to its numerous functions. Such compounds may be easily available and with low toxicity, like polyphenol (2)-epigallocatechin-3-gallate (EGCG), a component of green tea. EGCG inhibits mutated huntingtin aggregation in vitro,26 and a Phase II trial in HD is under way (http://clinicaltrials.gov/show/NCT01357681).
Acetylation of mutated HTT promotes its degradation, and sirtuins are involved in this posttranslational modification of the protein. Inhibition of sirtuin 1-induced deacetylation by selisistat improves a number of parameters in HD models, including cells, Drosophila, and mice, specifically including behavior.27 A randomized, double-blind, placebo-controlled Phase IB trial examining pharmacodynamic handling of the molecule has now been run in HD patients,28 followed by a safety and tolerability study.29 In the latter, 144 patients were randomized to selisistat 50 or 200 mg or placebo once daily for 12 weeks. There were three adverse events in each group; the most common were reversible liver-function test impairments. No change in the Unified Huntington’s Disease Rating Scale (UHDRS) was observed. Levels of soluble mutant huntingtin in peripheral blood mononuclear cells showed a borderline increase in the selisistat arms. So far, these data have only been published in abstract form, but the modulation of HTT levels, if confirmed, is an interesting novel finding.
Chaperones may be used to improve clearance of the abnormal protein, eg, by overexpressing heat-shock proteins. Expression of a human heat-shock protein in HSJ1a in HD mice led to a significant decrease in insoluble HTT with diminished inclusion size.30 This can also be achieved by the application of antibodies specifically to react with mutated huntingtin, eg, with single-chain, single-domain antibodies shown to decrease abnormal HTT in several animal models, which is followed by clinical improvement.31 Another approach to decrease the HTT protein with an elongated polyglutamine tail is based on the fact that the protein accumulation is strongly influenced in vivo by the ratio of normal to elongated HTT,32 and that wild-type HTT has a protective role in HD mice.33 A 23-amino acid peptide within HTT, which prevents aggregation in a Drosophila HD model, has been delivered to HD mice by using a nanostructure-based drug-delivery system applied by buccal or anal administration.34 The peptide was appropriately delivered to the brain, and histological as well as clinical parameters were improved. A recent study has demonstrated the efficacy of such an approach in Drosophila and mice.35 This delivery mechanism is being studied in other disorders, and knowledge accumulating in those studies will be beneficial for the potential development of this concept in HD as well.
Improving neurotransmission
Phosphodiesterase 10A (PDE10A) is a member of a large family of enzymes, which regulate intracellular signaling by hydrolyzing cyclic nucleotides. PDE10A is specifically and highly expressed in medium spiny neurons,36 one of the major neuronal populations affected by the disorder, and cyclic adenosine monophosphate (cAMP) is progressively decreased in HD knock-in mice, even during the presymptomatic phase.37 PDE10A inhibition leads to a restricted accumulation of cyclic guanosine monophosphate and cAMP in the striatum,38 followed by an increase in phosphorylation of a number of proteins involved in signaling, eg, Glu1 subunits39 and extracellular signal-regulated kinase.40 Furthermore, mutated huntingtin protein interacts with CBP/CREB-binding protein,41 leading to the suggestion that increased cAMP could at least partially restore this pathway. Preclinical evidence has accumulated to suggest a potential use in humans as well. Chronic inhibition of PDE10A and PDE10A knockout leads to changes in the expression of selective genes in the striatum, including induction of the expression of genes potentially involved in neuroprotection.42 Acute PDE10A inhibition is followed by a decrease in spontaneous and amphetamine-stimulated locomotor activity,43 and reduces striatal excitotoxicity in a model of HD based on quinolinic acid injection.44 In the R6/2 mouse model of HD, chronic inhibition of PDE10A improves striatal and cortical morphological changes, with improvement of motor function.45 It also improves cognitive function, including spatial and recognition memories.46 One potential issue is the fact that striatal PDE mRNA and protein levels are decreased in transgenic mice;47 however, using positron emission tomography ligands, binding is still measurable in people affected with HD, albeit in much-decreased overall levels.48
Several trials are now under way to examine the potential of chronic PDE10 in people with HD. A randomized, placebo-controlled, double-blind study on the effect of different doses of PF-02545920, a drug with US Food and Drug Administration (FDA) orphan drug designation, has recently been started as a proof of concept (http://clinicaltrials.gov/show/NCT01806896). The trial is going to include 260 patients randomized in groups to receive two doses or placebo, and the total motor score used as a primary outcome. Secondary outcomes include occurrence of abnormal movements, global impression, suicide rate, and hematological parameters. Imaging substudies are also included. The study will last 26 weeks, and final data collection is planned for early 2016. A Phase II trial has been started aimed at enrolling about 120 patients with HD in order to compare the effect of three different doses of OMS643762, another drug that has received FDA orphan drug designation, and placebo on a battery of assessments covering motor, cognitive, and psychiatric symptoms (http://clinicaltrials.gov/show/NCT02074410). Treatment duration is 28 days, and the final data collection for primary outcome assessment should be completed in March 2015. Primary outcome measures include safety assessment, and secondary ones motor function, cognition, behavior, and pharmacokinetic data. Tracers measuring enzyme availability have been developed and tested for the purpose of these studies, and have even been suggested as parameters to measure the striatal effect of other therapies.
Improving mitochondrial function
Impaired mitochondrial function is impaired in HD, as demonstrated by dysfunction of several complexes along the electron-transport chain,49,50 decrease in mitochondrial density, and morphological alteration in brains from HD patients.51 Mutated HTT impairs mitochondrial function by a number of different mechanisms, including permeability transition-pore modulation, decrease of PGCα expression, stimulation of glutamate receptor by increased depolarization, mitochondrial fractionation, and impaired respiratory chain function.52 Several therapeutic approaches have been followed based on this background.
Coenzyme Q is a lipid-soluble antioxidant molecule with membrane-stabilizing properties involved in oxidative phosphorylation. Neuroprotective effects due to improved mitochondrial function have been tested in HD, with conflicting results. Since higher doses are safe,53 a study has been performed to examine their effects over a longer period of time; however, this has been stopped for futility reasons.
Olesoxime is a small cholesterol-like molecule targeting mitochondrial pores, which has been found to delay muscle denervation, glial changes, and motoneuron degeneration in animal models of motoneuron disorders.54 The drug has been tested in people with amyotrophic lateral sclerosis, but did not change the course of the disorder;55 however, the drug was found to be safe. Olesoxime has demonstrated restorative effects in different models of HD, which were suggested to be due to an improvement of mitochondrial membrane.56 The drug has been tested in a rat model of HD and found to improve biological and clinical parameters.57 Considering the positive tolerance profile found in clinical trials in of other diseases, it would seem reasonable to test it in HD as well.
Modulation of intracellular copper content
Aggregation of HTT protein to form inclusions is stimulated by the addition of copper in the culture medium of cells expressing the glutamine elongated protein, which is reversed by copper chelation.58 PBT2 is a copper chelator with a structure related to clioquinol, which has been tested in Alzheimer’s disease under the assumption that the prevention of β-amyloid aggregation after a decrease in copper binding with the protein would improve symptoms and disease progression. In a mouse HD model, PBT2 improved motor function and prolonged survival.59 PBT2 has been examined in several trials in humans. The drug decreased cerebrospinal fluid amyloid in patients with Alzheimer’s in a dose-dependent way.60 A Phase II study has been run in patients with HD in the US and Australia, including 109 patients in the mild-to-moderate stage of the disease. In a press release, improvement of some cognitive function was mentioned; however, peer review of the publication will be needed to judge it, and a Phase III trial is contemplated.61
Inhibition of excitotoxicity
Quinolinic acid, which had been used to generate HD animal models prior to the establishment of genetic ones, is generated in the brain after degradation of tryptophan, mainly by kynurenine monooxygenase (KMO) in microglial cells.62 Quinolinic acid and 3-hydroxykynurenine are neurotoxic, while kynurenine and kynurenic acid are neuroprotective. The increased levels of quinolinic acid in the brain of animal models and humans with HD suggests that inhibition of this pathway may lead to protection against neurotoxicity in shifting the balance to neuroprotection. Indeed, treatment of HD mice with a KMO inhibitor has led to restoration of markers of synaptic function, decrease in glial hyperactivation, and prolonged animal survival.63 There has been a debate whether this effect occurs centrally or peripherally, and a peripherally acting KMO inhibitor has been reported to increase levels of the neuroprotective quinolinic acid metabolites kynurenic acid and kynurenine in the brain and cerebrospinal fluid of animals.
Modulation of immune function
Immune function is involved in HD, and there is widespread activation of immunological markers in patients and also in asymptomatic gene carriers even long before disease onset.64 These changes mirror findings in animal models of HD, and they have led to the suggestion of a parallel involvement of brain and immune mechanisms underlying the disease process. This immune activation may be mediated by a pathway involving nuclear factor κB, and HTT lowering reverses immune-function impairment induced by this factor.65 Laquinimod, a drug that has been developed for the treatment of multiple sclerosis, decreases nuclear factor κB in astrocytes.66 Laquinimod decreases relapse rate, disability, and brain atrophy in multiple sclerosis,67 and there is also good evidence to suggest a decrease in brain damage.68 Based on this rationale, a proposal has been made to explore the use of this drug in HD as well. The advantage of the substance is that it has been used in quite a large number of patients with multiple sclerosis, and so there is good knowledge of safety profiles. A clinical study of HD patients to assess the efficacy and safety of three oral doses of laquinimod has recently been announced (http://clinicaltrials.gov/show/NCT02215616). The multicenter, international, placebo-controlled study will last 12 months. Primary outcome measures will include change UHDRS total motor score and secondary change from baseline in caudate volume, in the HD cognitive assessment battery, in the UHDRS total functional capacity, and in the Clinician’s Interview-Based Impression of Change.
Development of symptomatic therapies
Even given the exciting developments of therapies aimed at changing the course of the disease, the search for symptomatic drugs specifically targeted for HD remains important. In this context, the increased knowledge of the disturbed signaling mechanisms in the HD brain allows a further development from a nonspecific approach based on a generalization from studies in disorders with similar symptoms but different etiologies toward more specific ones. Until recently, few high-quality clinical trials had been performed specifically for the treatment of symptoms in HD patients,69 and a wide range of drugs used, due to a lack of consensus.70 Tetrabenazine was the first drug to be approved for symptomatic treatment in HD, in this case chorea. The drug inhibits the vesicular monoamine transporter (VMAT), and at doses up to 100 mg per day UHDRS chorea,71 and improves global clinical impression, with a clear deterioration after withdrawal.72 Tetrabenazine, a compound with 2H substitution of hydrogen to decrease metabolisms, is now being studied to assess safety, tolerability, and efficacy for treating chorea (http://clinicaltrials.gov/show/NCT01795859), and in an open label long-term study to examine long-term safety (http://clinicaltrials.gov/show/NCT01897896). In a single double-blind study specifically devoted to the treatment of cognitive impairment in HD, latrepirdine for 6 months did not improve cognition or function.73
Pridopidine has a state-dependent effect on D2 dopamine receptors, with rapid dissociation kinetics providing interesting properties to regulate striatal function in HD. The drug has been studied in two studies,74,75 which failed to reach primary end points, but did so for a secondary outcome: the total UHDRS motor score. A new study examining a larger range of doses has been launched in a multicenter, multinational, randomized, parallel-group, double-blind, placebo-controlled design to compare safety and efficacy in the treatment of motor impairment (http://clinicaltrials.gov/show/NCT02006472).
Muscle pathology is well documented in HD, with gradual generalized atrophy due to a number of mechanisms, including primary myopathology.76,77 Inhibition of a regulatory pathway linked to myostatin leads to muscle hypertrophy, and this can be performed by several modalities, including receptor decoy and specific antibodies against the myostatin ligand, substances in clinical development for a number of indications. Indeed, in an HD mouse, a profound recovery of muscle loss was induced with an ActRIIB-receptor decoy, and this led to a functional improvement with increased grip strength.78
Conclusion
The field of translational research in HD is a fast-moving area, and has now led to a number of avenues with reasonably valid promise to develop therapies aimed at postponing the start and the course of the disorder. However, given the complex pathophysiology of the disorder, it is very likely that a future therapy will combine a number of drugs targeting different molecular targets in order to both improve symptoms and afford neuroprotection. Such complex therapies are well known in other disorders, such as human immunodeficiency virus, diabetes, and hypertension. Since the single cause of HD is precisely known, and can be confirmed long before onset, this disorder may be considered a paradigm for disease-modifying treatment of other neurodegenerative disorders. This has fostered the interest of researchers, pharmaceutical companies, and sponsors alike.
Disclosure
The author reports no conflicts of interest in this work.
References
Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98. | |
Kim SD, Fung VS. An update on Huntington’s disease: from the gene to the clinic. Curr Opin Neurol. 2014;27(4):477–483. | |
[No authors listed]. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971–983. | |
Perevozchikova T, Stanley CB, McWilliams-Koeppen HP, Rowe EL, Berthelier V. Investigating the structural impact of the glutamine repeat in huntingtin assembly. Biophys J. 15, 2014;107(2):411–421. | |
Munoz-Sanjuan I, Bates GP. The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease. J Clin Invest. 2011;121(2):476–483. | |
Tourette C, Li B, Bell R, et al. A large scale huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. J Biol Chem. 2014;289(10):6709–6726. | |
Ratovitski T, Chighladze E, Arbez N, et al. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle. 2012;11(10):2006–2021. | |
Burgunder JM. Recent advances in the management of choreas. Ther Adv Neurol Dis. 2013;6(2):117–127. | |
Tabrizi SJ, Langbehn DR, Leavitt BR, et al. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8(9):791–801. | |
Tabrizi SJ, Scahill RI, Durr A, et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 2011;10(1):31–42. | |
Tabrizi SJ, Reilmann R, Roos RA, et al. Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol. 2012;11(1):42–53. | |
Harper SQ, Staber PD, He X, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A. 2005;102(16):5820–5825. | |
Boudreau RL, McBride JL, Martins I, et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17(6):1053–1063. | |
Sah DW, Aronin N. Oligonucleotide therapeutic approaches for Huntington disease. J Clin Invest. 2011;121(2):500–507. | |
Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–1044. | |
DiFiglia M, Sena-Esteves M, Chase K, et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. 2007;104(43):17204–17209. | |
Pfister EL, Kennington L, Straubhaar J, et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol. 2009;19(9):774–778. | |
DeVincenzo J, Lambkin-Williams R, Wilkinson T, et al. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc Natl Acad Sci U S A. 2010;107(19):8800–8805. | |
Grondin R, Kaytor MD, Ai Y, et al. Six-month partial suppression of huntingtin is well tolerated in the adult rhesus striatum. Brain. 2012;135(Pt 4):1197–1209. | |
Kaplan A, Stockwell BR. Therapeutic approaches to preventing cell death in Huntington disease. Prog Neurobiol. 2012;99(3):262–280. | |
Yu D, Pendergraff H, Liu J, et al. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell. 2012;150(5):895–908. | |
Kay C, Skotte NH, Southwell AL, Hayden MR. Personalized gene silencing therapeutics for Huntington disease. Clin Genet. 2014;86(1):29–36. | |
Papworth M, Kolasinska P, Minczuk M. Designer zinc-finger proteins and their applications. Gene. 2006;366(1):27–38. | |
Garriga-Canut M, Agustín-Pavón C, Herrmann F, et al. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc Natl Acad Sci U S A. 2012;109(45):E3136–E3145. | |
Ehrnhoefer DE, Sutton L, Hayden MR. Small changes, big impact: posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist. 2011;17(5):475–492. | |
Ehrnhoefer DE, Duennwald M, Markovic P, et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum Mol Genet. 2006;15(18):2743–2751. | |
Smith MR, Syed A, Lukacsovich T, et al. A potent and selective sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum Mol Genet. 2014;23(11):2995–3007. | |
Süssmuth S, Landwehrmeyer G, Tabrizi S, et al. A randomised, double-blind, placebo-controlled phase IB pharmacodynamic study with selisistat (SEN0014196) in HD patients. J Neurol Neurosurg Psychiatry. 2012;83(Suppl 1):A55. | |
Reilmann R, Squitieri F, Priller J, et al. Safety and tolerability of selisistat for the treatment of Huntington’s disease: results from a randomized, double-blind, placebo-controlled phase II trial. Neurology. 2014;82(Suppl 10):S47.004. | |
Labbadia J, Novoselov SS, Bett JS, et al. Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain. 2012;135(Pt 4):1180–1196. | |
Butler DC, McLear JA, Messer A. Engineered antibody therapies to counteract mutant huntingtin and related toxic intracellular proteins. Prog Neurobiol. 2012;97(2):190–204. | |
Leavitt BR, Guttman JA, Hodgson JG, et al. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. 2001;68(2):313–324. | |
Mugat B, Parmentier ML, Bonneaud N, Chan HY, Maschat F. Protective role of engrailed in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2008;17(22):3601–3616. | |
Bonneaud N, Paucard A, Bauer C, et al. Systemic delivery of P42 peptide to fight Huntington’s disease. J Neurol Neurosurg Psych. 2014;85(Suppl 1):A98. | |
Marelli C, Bonneaud N, Paucard A, Maschat F. Efficacy of the peptide P42B in HeLa cells and in the HD Drosophila model. J Neurol Neurosurg Psych. 2014;85(Suppl 1):A97. | |
Coskran TM, Morton D, Menniti FS, et al. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J Histochem Cytochem. 2006;54(11):1205–1213. | |
Gines S, Seong IS, Fossale E, et al. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum Mol Genet. 2003;12(5):497–508. | |
Schmidt CJ, Chapin DS, Cianfrogna J, et al. Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther. 2008;325(2):681–690. | |
Grauer SM, Pulito VL, Navarra RL, et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther. 2009;331(2):574–590. | |
Siuciak JA, Chapin DS, Harms JF, et al. Inhibition of the striatum-enriched phosphodiesterase PDE10A: a novel approach to the treatment of psychosis. Neuropharmacology. 2006;51(2):386–396. | |
Steffan JS, Kazantsev A, Spasic-Boskovic O, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97(12):6763–6768. | |
Kleiman RJ, Kimmel LH, Bove SE, et al. Chronic suppression of phosphodiesterase 10A alters striatal expression of genes responsible for neurotransmitter synthesis, neurotransmission, and signaling pathways implicated in Huntington’s disease. J Pharmacol Exp Ther. 2011;336(1):64–76. | |
Siuciak JA, McCarthy SA, Chapin DS, et al. Genetic deletion of the striatum-enriched phosphodiesterase PDE10A: evidence for altered striatal function. Neuropharmacology. 2006;51(2):374–385. | |
Giampa C, Patassini S, Borreca A, et al. Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of Huntington’s disease. Neurobiol Dis. 2009;34(3):450–456. | |
Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS One. 2010;5(10):e13417. | |
Giralt A, Saavedra A, Carreton O, et al. PDE10 inhibition increases GluA1 and CREB phosphorylation and improves spatial and recognition memories in a Huntington’s disease mouse model. Hippocampus. 2013;23(8):684–695. | |
Hebb AL, Robertson HA, Denovan-Wright EM. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience. 2004;123(4):967–981. | |
Ahmad R, Bourgeois S, Postnov A, et al. PET imaging shows loss of striatal PDE10A in patients with Huntington disease. Neurology. 2014;82(3):279–281. | |
Benchoua A, Trioulier Y, Zala D, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17(4):1652–1663. | |
Browne SE, Bowling AC, MacGarvey U, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41(5):646–653. | |
Kim J, Moody JP, Edgerly CK, et al. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet. 2010;19(20):3919–3935. | |
Costa V, Scorrano L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012;31(8):1853–1864. | |
Hyson HC, Kieburtz K, Shoulson I, et al. Safety and tolerability of high-dosage coenzyme Q10 in Huntington’s disease and healthy subjects. Mov Disord. 2010;25(12):1924–1928. | |
Sunyach C, Michaud M, Arnoux T, et al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology. 2012;62(7):2346–2352. | |
Lenglet T, Lacomblez L, Abitbol JL, et al. A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur J Neurol. 2014;21(3):529–536. | |
Eckmann J, Clemens LE, Eckert SH, et al. Mitochondrial membrane fluidity is consistently increased in different models of Huntington disease: restorative effects of olesoxime. Mol Neurobiol. 2014;50(1):107–118. | |
Clemens L, Weber J, Wlodkowski TT, et al. Olesoxime treatment inhibits the formation of mHtt fragments through suppression of calpain activity, and leads to behavioral and neurological improvements in the BACHD rat. J Neurol Neurosurg Psych. 2014;85(Suppl 1):A62. | |
Hands SL, Mason R, Sajjad MU, Giorgini F, Wyttenbach A. Metallothioneins and copper metabolism are candidate therapeutic targets in Huntington’s disease. Biochem Soc Trans. 2010;38(2):552–558. | |
Chernya R, Aytona S, Finkelsteina D, Busha A, McColla G, Massac S. PBT2 reduces toxicity in a C. elegans model of polyQ aggregation and extends lifespan, reduces striatal atrophy and improves motor performance in the R6/2 mouse model of Huntington’s disease. J Huntingtons Dis. 2012;1(2):211–219. | |
Lannfelt L, Blennow K, Zetterberg H, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIA, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7(9):779–786. | |
Sampaio C, Borowsky B, Reilmann R. Clinical trials in Huntington’s disease: interventions in early clinical development and newer methodological approaches. Mov Disord. 2014;29(11):1419–1428. | |
Vécsei L, Szalárdy L, Fülöp F, Toldi J. Kynurenines in the CNS: recent advances and new questions. Nat Rev Drug Discov. 2013;12(1):64–82. | |
Zwilling D, Huang SY, Sathyasaikumar KV, et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. 2011;145(6):863–874. | |
Björkqvist M, Wild EJ, Thiele J, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205(8):1869–1877. | |
Träger U, Andre R, Lahiri N, et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFκB pathway dysregulation. Brain. 2014;137(Pt 3):819–833. | |
Brück W, Pförtner R, Pham T, et al. Reduced astrocytic NF-κB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol. 2012;124(3):411–424. | |
Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366(11):1000–1009. | |
Filippi M, Rocca MA, Pagani E, et al. Placebo-controlled trial of oral laquinimod in multiple sclerosis: MRI evidence of an effect on brain tissue damage. J Neurol Neurosurg Psychiatry. 2014;85(8):851–858. | |
Bonelli RM, Hofmann P. A systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin Pharmacother. 2007; 8(2):141–153. | |
Burgunder JM, Guttman M, Perlman S, Goodman N, van Kammen DP, Goodman L. An International survey-based algorithm for the pharmacologic treatment of chorea in Huntington’s disease. PLoS Curr. 2011;3:RRN1260. | |
Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006; 66(3):366–372. | |
Frank S, Ondo W, Fahn S, et al. A study of chorea after tetrabenazine withdrawal in patients with Huntington disease. Clin Neuropharmacol. 2008;31(3):127–133. | |
HORIZON Investigators of the Huntington Study Group and European Huntington’s Disease Network. A randomized, double-blind, placebo-controlled study of latrepirdine in patients with mild to moderate Huntington disease. Arch Neurol. JAMA Neurol. 2013;70(1):25–33. | |
de Yebenes JG, Landwehrmeyer B, Squitieri F, et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD):a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011;10(12):1049–1057. | |
Huntington Study Group HART Investigators. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov Disord. 2013;28(10):1407–1415. | |
Kosinski CM, Schlangen C, Gellerich FN, et al. Myopathy as a first symptom of Huntington’s disease in a marathon runner. Mov Disord. 2007;22(11):1637–1640. | |
Zielonka D, Piotrowska I, Marcinkowski JT, Mielcarek M. Skeletal muscle pathology in Huntington’s disease. Front Physiol. 2014;5:380. | |
Mielcarek M, Rattray I, Osborne G, et al. Myostatin inhibition as a novel approach to targeting muscle pathology in HD. J Neurol Neurosurg Psych. 2014;85(Suppl 1):A97. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.