Back to Journals » International Journal of Nanomedicine » Volume 21
Organelle-Targeted Nanotherapeutics for Parkinson’s Disease: From Pathogenesis to Preclinical Strategies and Translational Challenges
Authors Liu M, Peng Q, Zhai H ![]() , Wu Y, Ma Z, Tang Z
, Wu Y, Ma Z, Tang Z ![]() , Ouyang H
, Ouyang H ![]() , Chang A, Cao X, Xu Y, Xia Y
, Chang A, Cao X, Xu Y, Xia Y
Received 15 March 2026
Accepted for publication 5 May 2026
Published 13 May 2026 Volume 2026:21 607870
DOI https://doi.org/10.2147/IJN.S607870
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kamakhya Prakash Misra
MaoYu Liu, QiWei Peng, Heng Zhai, Yi Wu, ZhuoRan Ma, ZhiCheng Tang, Haoxuan Ouyang, An Chang, XueBing Cao, Yan Xu, Yun Xia
Department of Neurology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
Correspondence: Yan Xu, Department of Neurology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1277 Jiefang Road, Wuhan, 430022, Hubei, People’s Republic of China, Email [email protected] Yun Xia, Department of Neurology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1277 Jiefang Road, Wuhan, 430022, Hubei, People’s Republic of China, Email [email protected]
Abstract: Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by the loss of dopaminergic neurons and the aggregation of αSynuclein (αSyn). Organelle dysfunction is recognized as a central driver of pathological feature. Current treatments primarily alleviate symptoms but fail to halt disease progression, largely due to the inability to target the underlying subcellular pathology. This narrative review examines the emerging potential of organelle-targeted nanotherapeutics as a precision medicine strategy for PD treatment. We discuss how engineered nanoparticles can be designed to deliver therapeutics specifically to dysfunctional mitochondria, lysosomes, endoplasmic reticulum, Golgi apparatus, and nuclei. These approaches aim to interfere with key pathological mechanisms ameliorating oxidative stress, mitigating protein misfolding, restoring protein homeostasis, and modulating gene expression. We provide a comprehensive overview of recent preclinical advances in nanoparticles design, targeting mechanisms, and therapeutic efficacy. Furthermore, we critically evaluate the current challenges, including delivery efficiency, safety, reproducibility, storage, and large-scale translation before clinical application This review aims to provide a potential route toward disease-modifying nanotherapeutics for PD.
Keywords: nanomedicine, drug delivery system, organelle targeting, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease (NDD). Since the 1980s, the global prevalence of PD has been rising, becoming a significant public health and social burden.1,2 The pathological hallmark of the disease is the progressive loss of dopaminergic neurons in the substantia nigra (SN) of the midbrain and the abnormal aggregation of αSynuclein (αSyn).3,4 These pathological processes are intimately linked to dysfunction of intracellular organelles, particularly mitochondria and lysosomes. Recent years, the endoplasmic reticulum (ER), Golgi apparatus, and nuclei have also been found play an important role in PD pathology.5 Organelles serve as core structural units maintaining cellular homeostasis and function, and their dysfunction has been identified as a critical component in the pathological progression of PD. For instance, abnormalities in mitochondrial energy metabolism, impaired lysosomal degradation, and activation of the endoplasmic reticulum unfolded protein response not only promote abnormal αSyn aggregation but also exacerbate oxidative stress and neuroinflammation, creating a vicious cycle.6–9
Current therapeutic strategies for PD primarily consist of pharmacological interventions and surgical treatments. Traditional drugs face limitations such as low blood-brain barrier (BBB) permeability, poor targeting, and systemic side effects, hindering effective intervention at the site of pathology. Long-term use is often associated with fluctuating efficacy and motor complications. While surgical intervention faces challenges such as inconsistent efficacy, the risk of brain tissue damage, and equipment obsolescence.10 Notably, despite providing partial symptomatic relief, neither treatment can delay or halt disease progression, because they do not address the subcellular pathological mechanisms underlying neuronal loss. In this context, organelle-targeted therapeutic strategies have emerged, aiming to restore neuronal homeostasis by precisely regulating specific subcellular structures, thereby interrupting the pathogenic cascade at its source.11 A major gap in current therapeutic approaches is the inability to precisely target the subcellular sites where pathology originates.
Nanotechnology offers a promising approach to address these limitations for the properties of superior biocompatibility, controllable release properties, and surface functional plasticity.12 The drug delivery scope of nanoparticles has shifted from broad to specific due to the design of specific targeting ligands. Given that existing therapies fail to address subcellular pathological mechanisms, nanotechnology has been considered as an effective way for achieving precise organelle-level interventions.13,14 While existed reviews have addressed the general application of nanotherapeutics in PD,15 this review specifically focuses on how engineered nanoparticles can be directed toward distinct subcellular organelles to restore their function and alleviate dysfunction-related pathology. Rather than providing a broad overview of nanocarriers or neuroprotective agents, we emphasize organelle-targeted strategies that move beyond conventional drug delivery to restore subcellular function. It is important to acknowledge that the evidence discussed here is predominantly derived from preclinical studies. We primarily include PD-specific research; however, when direct evidence in PD is limited, we selectively incorporate findings from other neurodegenerative diseases to provide mechanistic support. Despite the considerable therapeutic potential, several major challenges continue to hinder clinical translation, including heterogeneity in BBB penetration, inefficient endosomal escape, difficulties in accurately validating subcellular targeting, limited manufacturing reproducibility, and uncertainties regarding long-term biosafety.
This review systematically summarizes the role of dysfunctional organelles in pathogenesis of PD, providing effective organelle-targeting pathways and focusing on recent advances in nanotherapeutic strategies. Furthermore, we discuss current challenges and future prospects for clinical translation. This aims to provide theoretical foundations and technical directions for developing next-generation disease-modifying therapies for PD.
The Overview of Nanotechnology Application in PD
The application of nanotechnology in PD treatment has garnered increasing attention, with its core advantage of facilitating targeted delivery, which mitigates the systemic toxicity associated with conventional administration routes. Leveraging surface modification and small size, nanoparticles (NPs) can effectively penetrate the BBB and cell membranes, significantly enhancing drug accumulation at the lesion site via passive or active targeting mechanisms. Additionally, nanomaterials exhibit outstanding drug-loading capacity and controlled-release properties, effectively improving pharmacokinetics of drugs.16–18 The controlled-release properties of nanocarriers can mitigate motor complications associated with levodopa blood concentration fluctuations, thereby offering considerable potential for clinical application. Diverse nanomaterials enable varied delivery strategies, playing an irreplaceable role particularly in developing novel therapies based on disease-modifying mechanisms. Existing nanocarriers and delivery routes are summarized in Figure 1.
 |
Figure 1 Nanotherapy in CNS disease. Common categories of nanocarriers employed for therapeutic purposes comprise organic, inorganic, and biomimetic nanocarriers. These systems are designed to deliver a diverse range of therapeutic agents, including proteins, nucleic acids, small-molecule drugs, and natural bioactive compounds. To facilitate efficient transport of nanoparticles into the brain, multiple administration routes are utilized, such as oral delivery, intravenous injection, intranasal administration, and localized intracerebral delivery. Following cellular uptake, the nanoparticles undergo lysosomal degradation or structural disassembly, leading to the controlled release of their payloads. These released therapeutics can then target specific intracellular organelles, thereby exerting pharmacological effects and ameliorating pathological conditions. Figure made in BioRender.com. |
In PD treatment, delivering dopamine via nanocarriers has proven to be an efficient and viable strategy. Studies indicate that DA-loaded NPs can effectively penetrate the BBB and improve motor function in animal models.19 Although this targeted approach effectively reduces the accumulation of dopamine in peripheral tissues and minimizes systemic side effects, it provides only symptomatic relief and does not hinder the underlying progression of the disease. Consequently, therapeutic strategies aimed at the pathogenesis of PD have attracted growing attention. Numerous nanosystems targeting oxidative stress, neuroinflammation, αSyn aggregation, impaired autophagy, and neuronal apoptosis are under development.20–22 Such interventions have been shown to improve the survival of dopaminergic neurons and motor function in preclinical models. Nevertheless, their long-term therapeutic efficacy requires further validation.
The emergence of novel nanomaterials and advancements in auxiliary targeting technologies have significantly enhanced the in vivo targeting efficiency of NPs. Among these, biomimetic delivery systems and stimulus-responsive nanoparticles hold considerable application potential. Modifying NPs with natural membrane components endows them with biomimetic capabilities, enabling more efficient delivery to biologically active sites. Currently, the most mature biomimetic technologies primarily include cell membrane and exosome biomimicry. Cell membranes serve as multifunctional, biocompatible platforms with potential for surface modification and targeted delivery design. They interact with physiological environments to enhance central nervous system drug delivery while mitigating toxic effects. The targeting ability of cell membrane-coated particles is largely derived from the surface proteins of the source cells.23 Exosomes play a crucial role in intercellular communication. They exhibit high biocompatibility and low immunogenicity, with an inherent ability to traverse the BBB, and selectively interact with specific cells and tissues via surface markers. Exosomes exhibit significant variability due to their different cellular origin. For example, stem cell-derived exosomes are particularly suited for regenerative and neuroprotective applications as they contain unique growth factors, neurotrophic factors, and anti-inflammatory molecules.24 Cell membrane or exosome coated NPs not only retain the benefits of conventional NPs but also enhance biocompatibility, prolong drug circulation time, reduce clearance rates, and improve BBB permeability. These systems are often further modified with targeting motifs to enhance biological specificity. Biomimetic nanotechnology has demonstrated success in multiple PD-related studies, showcasing immense potential as a therapeutic drug delivery vehicle for PD.20,25
Stimuli-responsive NPs represent another major breakthrough in targeted disease therapy. These NPs are engineered to respond to endogenous biological, chemical, or physical stimuli—such as pH, reactive oxygen species (ROS), temperature, or enzyme activity—enabling spatially and temporally controlled drug release at target sites. This strategy enhances therapeutic efficacy while minimizing off-target effects.21,26,27 Meanwhile, integrating controllable-release nanomedicine with optogenetics or sono-chemogenetics can effectively achieve drug release under specific conditions, opening new therapeutic avenues for precise spatiotemporal drug release.28
Furthermore, systemic administration of nanomedicines (eg, intravenous injection) requires traversing the BBB to reach target tissues and cells, significantly reducing bioavailability. Nasal-brain delivery bypasses the BBB, transporting NPs to the brain via olfactory and trigeminal nerve pathways. It offers multiple advantages including ease of use, non-invasiveness, avoidance of hepatic first-pass metabolism, reduced systemic toxicity, prolonged residence time at the target site, and minimized unnecessary drug exposure and safety concerns. However, intranasal administration also presents drawbacks such as potential nasal irritation, tissue toxicity, and local adverse reactions.29–31 Besides, local administration of therapeutics directly into the brain parenchyma can effectively minimize their off-target distribution. However, this method is also associated with challenges such as repeated injections and potential damage to the local brain tissue. To solve this problem, nanoparticles can be integrated with other scaffolds (eg, hydrogels) to form hybrid delivery systems. This approach enables sustained release of therapeutic agents, thereby reducing the frequency of required interventions.32
In summary, current nanotherapies for PD remain at the cellular or tissue level. However, the continuous evolution of nanomaterials and delivery strategies provides a robust foundation for exploring novel therapeutic paradigms. Given the central role of organelles in maintaining cellular homeostasis and regulatory networks, developing organelle-targeted nanotherapeutic strategies holds immense therapeutic potential, offering new avenues for precise intervention in the pathological progression of PD (Figure 2). To navigate the rapidly expanding field of organelle-targeted nanotherapeutics for PD, a systematic comparative lens is essential. As synthesized in Table 1, we compared approaches head-to-head across organelles in terms of delivery route, cargo class, targeting ligand, PD model relevance, mechanistic evidence, and translational readiness. This framework allows for a clear distinction between early proof-of-concept studies and those with genuine disease-modifying potential across different organelles.
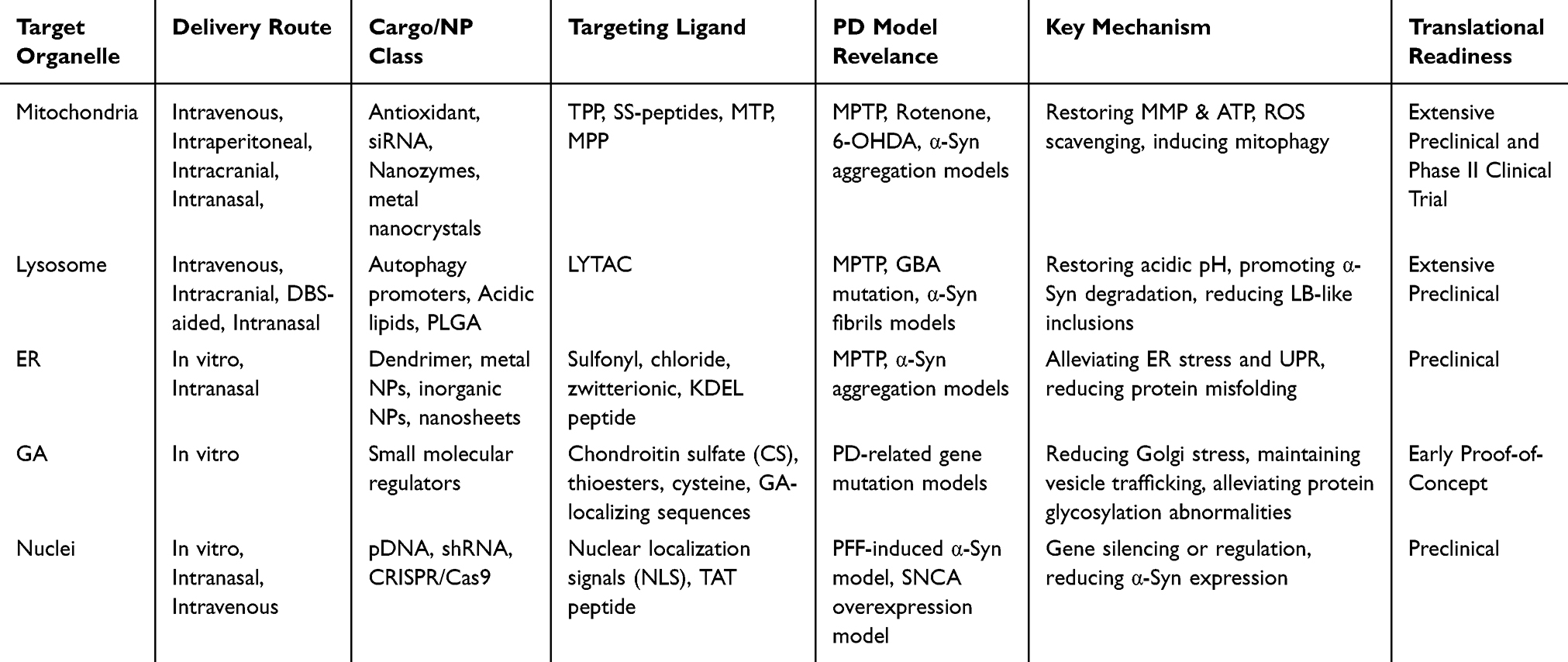 |
Table 1 Comparative Stratification of Evidence and Translational Maturity for Organelle-Targeted Nanotherapeutics in PD |
 |
Figure 2 Organelles-targeted nanotherapy in Parkinson’s Disease. PD is characterized by a central pathological mechanism involving coordinated dysfunction across multiple organelles—including mitochondria, lysosomes, endoplasmic reticulum, Golgi apparatus, and nucleus. This interplay of dysfunctional organelles is a key driver of disease pathogenesis. In response, therapeutic strategies are increasingly focusing on agents designed to precisely target these organelles. (Drugs are distinguish by color: red, PD-established evidence in vivo; orange, PD-established evidence in vitro; green, borrowed proof-of concept from other diseases; yellow, prospective conceptual targets). By restoring organellar integrity and function, such targeted interventions may ameliorate intracellular pathology, and their efficacy can be further enhanced through encapsulation within advanced nanocarrier systems. Figure made in BioRender.com. |
Targeting Mitochondria
Mitochondria in the Pathophysiology of PD
Mitochondria are the core organelles regulating cellular energy production, apoptosis, and metabolism. The brain—due to the high metabolic activity of neurons—becomes one of most energy-demanding organs. As the most crucial organelle for energy metabolism, mitochondria play an immense role in maintaining normal brain function. It has now been confirmed that mitochondrial dysfunction is closely linked to NDDs.33–36 Mitochondrial dysfunction manifests in multiple aspects, including mutations and deletions in mitochondrial DNA (mtDNA), disrupted calcium homeostasis, depolarization of the mitochondrial membrane, abnormal fusion and fission processes, impaired mitochondrial biogenesis, and oxidative stress. These alterations subsequently trigger pathological processes such as neurotransmitter metabolism abnormalities, synaptic dysfunction, and impaired neuronal growth and development.37–41
In PD, neuronal mitochondrial dysfunction is one of the core pathogenic mechanisms. Early evidence emerged from the mitochondrial toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which selectively induces DA neuron death. This mechanism is closely linked to MPTP induced inhibition of mitochondrial complex I, leading to dysfunction in the mitochondrial electron transport chain, reduced adenosine triphosphate (ATP) synthesis, and increased ROS production. Additionally, neurotoxins such as rotenone and 6-OHDA induce PD-like pathology through similar pathways.42,43
Multiple studies indicate direct associations between PD-related gene mutations and mitochondrial dysfunction. For instance, PINK1 and PARKIN mutations are common causes of autosomal recessive familial PD. The absence of which leads to defective mitochondrial autophagy, causing accumulation of damaged mitochondria and subsequent neuronal injury.44 DJ-1, a mitochondrial peroxidase-like enzyme, exhibits mutations that impair mitochondrial antioxidant capacity and heighten cellular sensitivity to oxidative stress. LRRK2 mutations, associated with autosomal dominant PD, are implicated in mitochondrial division, leading to morphological and functional abnormalities that ultimately precipitate PD.43,45 Although no PD-associated mtDNA mutations have been definitively identified, most studies indicate that mtDNA mutations, deletions, and rearrangements disrupt the mitochondrial respiratory chain, resulting in reduced cytochrome c oxidase (COX) activity and complex I defects. Artificial disruption of mtDNA induces PD onset, demonstrating the critical importance of mtDNA integrity in maintaining healthy mitochondrial function.43
Mitochondrial quality control serves as a vital mechanism for maintaining intracellular mitochondrial homeostasis. This process encompasses monitoring, identifying, repairing, and/or eliminating abnormal or misfolded mitochondrial proteins, as well as dysfunctional mitochondria.46 In PD, disrupted mitochondrial quality control manifests primarily as impaired mitochondrial dynamics (fusion and fission), defective mitochondrial autophagy, and abnormal accumulation of mitochondrial αSyn. Mitochondrial fusion proteins (MFN1 and MFN2) and Opa1 (retinitis pigmentosa 1) mediate fusion of the mitochondrial outer and inner membranes, respectively. Opa1 mutations are associated with PD and dementia, and reduced Opa1 expression increases mitochondrial fragmentation and autophagy. Various PD-associated neurotoxic molecules in vitro induce increased mitochondrial fusion-fission imbalance, where disrupted fission leads to synaptic loss and neuronal death. Impairment of the PINK1/PARKIN pathway causes mitochondrial autophagy dysfunction, preventing timely clearance of damaged mitochondria and subsequently triggering cellular energy metabolism disorders and increased oxidative stress. Although studies suggest mitochondrial dysfunction precedes αSyn accumulation, αSyn preferentially binds to mitochondria, inhibiting mitochondrial protein import and depolarizing the mitochondrial membrane. Concurrently, αSyn binds to the electron transport chain, disrupting ATP production, increasing ROS, and impairing mitochondrial respiration, further exacerbating the pathological state. αSyn can also interact with mitochondrial membrane proteins, activating the mitochondrial permeability transition pore to release mitochondrial contents and trigger inflammatory responses. These imbalanced processes collectively drive the pathological progression of PD.47,48
Ferroptosis, an iron-dependent form of cell death, is closely associated with mitochondrial dysfunction. It has been demonstrated as a prevalent cell death pathway in PD. Mitochondria, as iron-rich organelles, utilize iron ions for iron-sulfur cluster synthesis or store them as mitochondrial ferritin. Disruption of mitochondrial iron homeostasis leads to excessive intracellular iron accumulation, triggering the Fenton reaction. This generates ROS that react with fatty acids to form peroxides, further damaging mitochondrial structure and exacerbating oxidative stress, ultimately causing DA neuron death. In PD models, increased iron influx through the mitochondrial iron channel mitoferrin-2 is observed, and significant iron deposition is present in the SN of PD patients.49,50
Mitochondria play a crucial role in maintaining intracellular calcium homeostasis. In PD neuronal models with PINK1 defects, reduced NCLX (Na+/Ca2+ exchanger)-dependent mCa2+ efflux leads to mitochondrial calcium overload. This triggers the opening of the mitochondrial permeability transition pore, resulting in mitochondrial swelling and apoptosis. Concurrently, mitochondrial dysfunction increases ROS production, which directly damages intracellular biomolecules including proteins, lipids, and DNA. Reduced glutathione levels in the SN of PD patients weaken antioxidant defenses, and mtDNA damage further degrades mitochondrial quality.50,51
Given mitochondrial dysfunction’s pivotal role in PD pathogenesis, mitochondrial-associated biomarkers hold potential diagnostic and therapeutic value. For instance, circulating cell-free mitochondrial DNA (cfc-mtDNA) levels may be altered in PD patients, potentially serving as a biomarker for disease diagnosis and progression monitoring.52 Additionally, elevated levels of mitochondrial proteins like TOMM40 in cerebrospinal fluid may indicate disease progression.42 However, research on these potential biomarkers remains preliminary, requiring further validation of their clinical utility.
Although therapeutic strategies targeting mitochondrial dysfunction face numerous challenges in disease-modifying clinical trials, the ongoing development and maturation of nanotechnology hold promise for targeted mitochondrial therapies in PD.
Mitochondria-Targeting Nano-Delivery Strategies
Mitochondria are semi-autonomous organelles composed of an inner membrane and an outer membrane, forming a natural biological membrane barrier. The outer mitochondrial membrane (OMM) acts as a lipophilic barrier, while the inner mitochondrial membrane (IMM) is rich in phosphatidylserine, exhibiting a higher protein-to-lipid ratio and a strong negative mitochondrial membrane potential (MMP).53 To effectively deliver drugs into mitochondria, these barriers need to be overcome. Current effective approaches include: membrane potential-driven, affinity-driven, and nanotechnology-driven methods.54,55
First, the mitochondrial-targeted delivery of nanoparticles could be achieved by exploiting the membrane potential difference. The covalent modification of compounds is an important modification method for mitochondrial-targeted carriers, and its chemical synthesis is simple and has a high targeting efficiency. Common lipophilic cations include alkyl-triphenylphosphine cations (TPP+), rhodamine, cyanine cations, and cationic peptides, etc. TPP+ is currently the most studied cation for targeting mitochondria due to its high chemical stability, ease of modification and strong membrane-penetrating ability and was initially used as a probe to monitor MMP.53–56 Nowadays, it is widely applied in decorating NPs for targeted delivery to mitochondria. For instance, Yuan et al developed a TPP-PEG-PCL-based carrier that significantly enhances the mitochondrial accumulation of its drug payload.57 TPP-modified quercetin NPs (TQCN) delivered intranasally effectively penetrate brain regions and localize to mitochondria to relieve oxidative stress.55 Concurrently, novel cationic compounds targeting mitochondria continue to emerge. For instance, Schlichtmann et al developed a TPP derivative, 3-carboxypropyl-triphenylphosphine (CPTP), which enhances its retention on NPs. This approach enables effective mitochondrial targeting to mitigate rotenone-induced mitochondrial damage.58
Additionally, mitochondrial-targeting peptides can direct nanomedicines to mitochondria, which comprise either naturally occurring amino acid sequences (MTS) or synthetic peptides. MTS typically reside at the N-terminus of proteins and often contain hydrophobic and positively charged amino acids. MTS facilitates NPs entry into the mitochondrial matrix by interacting with translocase enzymes located in the outer and inner membrane complex.56,59 For instance, the Szeto-Schiller (SS) peptide combines MTS with an antioxidant motif, selectively accumulating in the IMM due to its affinity for mitochondrial lipids while also exhibiting antioxidant activity.53 Compared to MTS, mitochondrial localization signal peptides (MLSP) enable delivery of nanocarriers to more specialized mitochondrial compartments, facilitating targeted localization for precision medicine.60 Mitochondrial penetration peptides (MPP) are artificially designed permeable peptides possessing both cell membrane penetration capability and mitochondrial targeting specificity. MPP may target mitochondria through multiple mechanisms, such as inducing membrane depolarization, mimicking the evolutionary origin of antimicrobial peptides, and selectively binding to phosphatidylserine.61–63
The preparation of mitochondrial-targeted NP (MTN) is challenging, but they enhance mitochondrial targeting by improving lipid solubility, stability, and membrane permeability, thereby increasing the drug load, protein protection, and precise regulation of mitochondrial release. In summary, the design of MTN requires optimizing the characteristics of the NPs, such as size, charge, lipophilicity, biocompatibility, targeting ligands, and promoting intracellular escape, in order to successfully cross the biological barrier and deliver specific and highly efficient mitochondrial-protecting drugs.64
Mitochondria-Targeting Nano-Therapy Strategies
Therapeutic strategies leveraging MTN have demonstrated significant potential in treating PD, with mechanisms including alleviating oxidative stress, promoting antioxidant capacity, and enhancing mitophagy. The mitochondrial-protecting agents, nano-delivery systems, and key therapeutic outcomes reported in recent studies are systematically summarized in Table 2.
 |
Table 2 Examples of Mitochondria-Targeted Nanoparticles for Treatment of Parkinson’s Disease |
Antioxidation
Among MTNs, antioxidants are the most prevalent. Numerous neuroprotective agents with efficacy against oxidative stress are hindered from reaching pathological brain regions due to their inherent chemical properties, which limit their ability to cross the BBB and achieve targeted delivery. Thus, nanotechnology play a crucial role in enhancing drug delivery efficacy.79 NPs carrying natural antioxidant compounds has been extensively researched. For instance, naringenin (NAR) is a potent antioxidant with anti-apoptotic properties. Human serum albumin (HSA)-coated NAR nanocrystals (HSA-NAR-NCs) has been found maintained MMP and ATP production while regulating ROS levels, counteracting the neurotoxic effects of rotenone.65 Besides, the formulation of nanocrystals significantly enhancing solubility, dissolution rate, and stability of the agent, as HSA reduce immune responses and toxicity while prolonging circulation time.80 In another research, The RVG29 and RBC membrane-decorated curcumin nanocrystals effectively evaded reticuloendothelial system uptake, prolonged circulation and enhanced BBB penetration, Meanwhile, the nanomedicine restored dopamine level, inhibited αSyn aggregation, and reversed mitochondrial dysfunction in PD mice.20 Despite these preclinical successes, a critical translation gap. For instance, in a randomized controlled clinical trial, curcumin nanogels failed to significantly improve the quality of life and clinical symptoms of PD patients.81 This indicates that these preclinical trials with positive results still require further replication and validation. This single but important negative result underscores the frequent disconnect between promising preclinical data and clinical efficacy, possibly due to differences in disease chronicity, patient heterogeneity, or the inadequacy of toxin-based animal models to fully recapitulate human PD.
Additionally, emerging physical-assisted technologies offer new avenues for enhancing the efficacy of nanomedicines. Utilizing the photothermal conversion properties of special nanomaterials can enhance the targeting ability of antioxidants to the mitochondria within the brain. Under near infrared (NIR) irradiation, the photothermal effect produced by nanomaterials such as gold, Prussian blue, black phosphorus, etc. can cause local mild heating, temporarily expand the intercellular gaps of endothelial cells or activate the fluidity of the cell membrane, thereby increasing the permeability of the BBB.82,83 Researchers demonstrated that the zeolitic imidazolate framework 8-coated Prussian blue nanocomposite encapsulating quercetin (ZIF-8@PB-QCT) exhibits excellent NIR responsiveness. Guided by the photothermal effect, it penetrates BBB to reach sites of mitochondrial damage. In the mouse model of PD, ZIF-8@PB-QCT significantly elevated ATP levels, reduced oxidative stress, and reversed both dopaminergic neuronal damage and behavior deficits, without causing harm to normal tissues.75 Although photodynamic therapy has effectively enhanced the penetration and targeting properties of NPs, the optimal irradiation parameters, long-term safety and operability require systematic investigation.
Beyond natural or synthetic antioxidants, several reductive agent donors are being actively explored to enhance the release of endogenous antioxidants. For instance, H2S is an effective endogenous antioxidant. Reduced endogenous H2S levels may lead to sulfhydryl modification of Parkin protein and decreased enzymatic activity, thereby impairing its ability to recognize toxic proteins and mediating the formation of αSyn aggregates. Zhao et al designed a nanomotor-based H2S donor PCM (PEG-Cys-MPC). This H2S donor can cross the BBB and be catalytically formed into H2S in the mitochondria. Ultimately it eliminated ROS, alleviated neuroinflammation, and reduced αSyn aggregation.70 Furthermore, Wang et al encapsulated SNCA siRNA into PCM to reduce αSyn formation, while utilizing H2S generated during chemotaxis to reduce oxidative damage, achieving a multi-faceted therapeutic approach for PD.71
Notably, nanotherapeutics targeting mitochondrial energy metabolism have advanced to phase II clinical trials. CNM-Au8® is a faceted, clean-surfaced gold nanocrystal suspension designed to catalytically enhance energy metabolism in CNS cells, thereby supporting neuroprotection and remyelination. Its novel mechanism involves targeting the NAD⁺/NADH redox couple, which has been validated in multiple independent preclinical models. In a combined cohort of PD and multiple sclerosis (MS) patients, the average cerebral NAD⁺/NADH ratio increased by approximately 10.4% after over 12 weeks of CNM-Au8 treatment compared to baseline. However, likely due to the limited sample size, this improvement did not reach statistical significance when the disease cohorts were analyzed separately. Related long-term extension studies are currently underway.84
Nanozymes
Nanozymes are a class of nanomaterials exhibiting catalytic activity analogous to natural enzymes, capable of mimicking the catalytic functions of biological enzymes in complex biological environments.85 For instance, when the MoS2 structure is reduced from bulk to extremely thin monolayer quantum dot (QD) layers, it exhibited unique catalytic activity. Nanoscale MoS2 serves as an excellent antioxidant, eliminating various free radicals through nanozyme activity. This activity mimics intrinsic major cellular antioxidant enzymes.86 Studies indicate that TPP-MoS2 QDs reducing the pro-inflammatory transformation of microglia by alleviating oxidative stress.87
Many metallic nanozymes exhibit dual enzyme-like activities mimicking superoxide dismutase (SOD) and catalase. Octahedral palladium nanoparticles (Pd NPs) scavenge cytotoxic oxygen radicals, maintain MMP, and mitigate damage to lipids, proteins, and DNA under hyperthermia.88 The tri-element nanozyme (PtCuSe nanozyme) alleviates neuronal injury by scavenging cellular ROS and reduces behavioral and pathological symptoms in PD animal models.74 Furthermore, by binding to mitochondria-targeting peptides, nanozymes can more effectively rescue mitochondrial dysfunction. For instance, TPP-modified Cu(2-x)Se nanozymes (Cu(2-x)Se-TPP NPs) effectively scavenge ROS in microglial mitochondria, alleviating mitochondrial oxidative stress.89 To more efficiently exert the function of nanozymes, Liu et al integrated Fe-isolated single-atom nanozyme (Fe-ISAzyme) liposome with TPP to the microneedles patch, and adapt in situ delivery. The strategy significantly enhanced deep penetration into brain parenchyma compared to intravenous injection, particularly in key pathological sites like the SN and striatum. Besides, the nanozyme scavenged ROS and alleviated neuroinflammation at the lesion sites, effectively mitigating behavioral deficits and pathological symptoms in PD mice.73 While ligand-conjugated nanozymes like Cu(2-x)Se-TPP NPs show enhanced mitochondrial targeting, their synthetic complexity and potential for metal-ion leaching raise unresolved long-term safety questions. In contrast, “label-free” nanozymes like the Fe-ISAzyme microneedle patch demonstrate that strategic administration routes can partially compensate for a lack of molecular targeting ligands, achieving deep brain penetration by mechanical means.
Mitochondrial Autophagy
Dysregulation of the Parkin/PINK1 pathway in PD leads to impaired mitochondrial autophagy, preventing the clearance of damaged mitochondria and triggering neuronal dysfunction. Consequently, numerous studies target mitochondrial autophagy to rescue damaged neurons as a therapeutic strategy for PD.
Specifically modified nanocarriers delivering targeted drugs can clear damaged mitochondria by initiating the mitochondrial autophagy program, effectively restoring mitochondrial homeostasis, reducing the neurotoxicity caused by oxidative stress, and rescuing the motor dysfunction in animal models. For instance, Chen, Y. et al employed hyaluronic acid nanoparticles (HA-NPs) of varying molecular weights to protect mitochondria and repair mild, limited damage. For irreversible damage, NPs containing ubiquitin-specific protease 30 (USP30) siRNA were used to promote mitochondrial autophagy. Simultaneously, by incorporating PINK1 antibodies, these NPs selectively targeted irreversibly damaged mitochondria, preventing excessive clearance of healthy mitochondria.69 Similarly, Biswal, L. et al employed HSA nanocarrier to deliver melatonin to the brain. This nano-melatonin exhibited enhanced antioxidant and neuroprotective properties associated with promoting mitophagy by upregulating BMI1, thus counteracting rotundone-induced toxicity.67 Besides, Xia et al loaded lycopene nanodots into the cavity of recombinant human H-ferritin nanocages and attached TPP to the outer surface. H-ferritin nanocages were transported across the BBB by recognizing transferrin receptor-1, targeting neurons and mitochondria. The anti-ROS components protected mitochondrial function in vivo and in vitro and promoted PINK1/Parkin-mediated mitophagy, thereby facilitating the clearance of pathogenic αSyn and the survival of dopaminergic neurons.66 In addition, enhancing mitophagy can also effectively regulate glial cell-associated neuroinflammation. Zhang et al developed a nanomedicine based on stem cell-derived exosomes loaded with hydroxy-terminated phosphorus dendrimers and quercetin (QAE NPs), which exhibited favorable drug release characteristics and ideal cellular compatibility, capable of penetrating the nasal mucosa and accumulating in the brain. QAE NPs possess both anti-inflammatory and antioxidant properties, which induced mitophagy, scavenged ROS, and restored mitochondrial homeostasis, while promoting M2 microglia polarization and reducing neuroinflammation. This nanocomposite improves motor function, coordination, and alleviates depressive-like symptoms in PD mice.76
Collectively, these approaches highlight mitophagy modulation as a central and mechanistically coherent strategy for addressing mitochondrial dysfunction in PD; however, their relative efficacy and selectivity remain difficult to compare due to differences in carrier design and experimental models.
Targeting Lysosomes/Endosomes
Lysosomes/Endosomes in the Pathophysiology of PD
Lysosomes are key organelles responsible for degrading biomacromolecules. They eliminate dysfunctional extracellular or intracellular components—including prone-to-aggregate proteins and damaged organelles—via the endocytosis-lysosomal pathway or autophagy-lysosomal pathway (ALP).90 Lysosomes play vital roles in maintaining neuronal homeostasis, regulating synaptic signaling, promoting plasma membrane repair, and overseeing protein secretion.91–93 Currently, autophagy is known to comprise three primary pathways: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy. Macroautophagy, the most prevalent autophagy form, initiates with the engulfment of cytoplasmic contents into a double-membrane structure called an autophagosome, which ultimately fuses with lysosomes to degrade its contents. CMA does not require double-membrane vesicle formation. Instead, it promotes the unfolding and transport of CMA-targeted proteins to lysosomes through binding to KFERQ-like CMA target motifs, representing a selective form of autophagy. Microautophagy refers to the process where lysosomal or vacuolar membranes directly invaginate, engulf, and degrade cytoplasmic components. The mechanisms underlying microautophagy remain poorly understood.94 Given the irreplaceable nature and extensive branching volume of neurons, lysosomal function is crucial for maintaining normal neuronal function. Dysfunction in lysosomes may lead to abnormal protein accumulation and inadequate clearance of neurotoxic proteins, including αSyn, potentially causing dopaminergic neuronal dysfunction and subsequently resulting in PD.93
Numerous PD-associated genes have been implicated in lysosomal protein transport or lysosomal function, including LRRK2, GBA, ATP13A2, and TMEM175. LRRK2 possesses GTPase activity, and its mutations can cause autosomal dominant PD. LRRK2 is a dynamic protein capable of forming distinct complexes and regulating various functions across different subcellular locations, cell types, and conditions, including the autophagy-lysosome-endosome system.91 LRRK2 induces autophagy via the MAPK/ERK pathway and modulates autophagy through calcium-dependent pathways. LRRK2 mutations impair autophagosome maturation, disrupt LRRK2-v-ATPase interactions, and cause defective lysosomal acidification. Additionally, LRRK2 serves as a CMA substrate. LRRK2 mutations not only evade its own degradation but also inhibit the degradation of other CMA substrates, including αSyn.95 GBA encodes the lysosomal enzyme β-glucocerebrosidase (GCase). The estimated prevalence of GBA1 mutations among PD patients ranges from 5% to 25%. Postmortem PD tissues exhibit reduced GCase activity and protein levels. Within lysosomes, diminished GCase activity leads to sphingoglycolipid accumulation, disrupting lysosomal function and autophagy, thereby triggering αSyn accumulation. Furthermore, αSyn aggregates reduce GCase activity, forming a self-perpetuating cycle of lysosomal dysfunction and αSyn accumulation.95–97 The PD risk gene ATP13A2 encodes a lysosomal P5-type ATPase, considered as a pH regulator, as its mutations or deletions cause polyamine accumulation, elevated pH, and autophagosome/lysosome stacking. ATP13A2 mutations cause familial Parkinson’s syndrome with juvenile-onset.98 The endoplasmic reticulum/lysosomal proton channel TMEM175 is another key regulator of lysosomal pH and a risk factor for NDDs like PD and Lewy body dementia. Cells lacking TMEM175 exhibit reduced lysosomal hydrolase activity and pathological αSyn aggregation.99
The autophagy pathway plays a crucial role in maintaining appropriate levels of αSyn in neurons. Wild-type αSyn is primarily degraded via autophagy, but mainly through CMA.91 αSyn carries the sequence 95VKKDQ99, which is specifically recognized by HSP70 and promotes its translocation to the lysosomal surface, where it binds to LAMP2 for subsequent degradation. Alterations in αSyn due to mutations or post-translational modifications affect CMA-mediated turnover. Certain αSyn variants associated with familial PD cases cannot be efficiently degraded via CMA and further block CMA-dependent degradation of other substrates. Cathepsin D in lysosomes has been reported to directly participate in the degradation of both monomeric and aggregated α-syn. Blocking ALP with vancomycin A1 leads to the accumulation of α-syn aggregates in primary cortical neurons and non-neuronal cells.93
The acidic environment of lysosomes is critical for their normal hydrolytic function, with an optimal pH of 4.0–5.0.100 Lysosomal acidification is primarily maintained by a proton pump called the vacuolar-type H⁺-ATPase (V-ATPase) and multiple ion channels in the lysosomal membrane. Under pathological conditions, lysosomes with elevated pH either fail to fuse with autophagosomes or fuse to form poorly acidified autophagolysosomes, resulting in inefficient or absent degradation of these lysosomes during cellular turnover.101,102 In PD, impaired lysosomal acidification leads to mitochondrial dysfunction, reduced αSyn clearance, Lewy body pathology, and neurodegeneration. Notably, deficiency of TMEM175, which encodes a K⁺ channel, induces excessive lysosomal acidification rather than deacidification, yet similarly results in reduced cathepsin activity and impaired αSyn aggregation in vitro and in vivo tissues.99,103
Additionally, increased lysosomal membrane permeability and disrupted lysosomal calcium homeostasis have been implicated in PD.99 Various small-molecule compounds applied to PD models have been shown to promote clearance of abnormal protein aggregates in neurons and/or protect neurons from apoptosis by modulating ALP.104 Nanotechnology plays a crucial role in targeting drug delivery to the lysosomal pathway.
Lysosomes/Endosomes-Targeting Nano-Delivery Strategies
Endocytosis is a crucial pathway for NPs to enter cells, representing a complex process strictly regulated by the plasma membrane. Primary endocytic mechanisms include clathrin-mediated endocytic mechanisms and endocytic pathways that proceed independently of clathrin.105 The vast majority of NPs enter the cytoplasm through endocytosis mediated by clathrin. The substances will pass through the early endosome - late endosome - lysosome transport pathway and are eventually degraded by hydrolases.106 These endocytic pathways have been extensively studied and are determined by the physicochemical properties of nanoparticles.107
Placing drug molecules in the lysosomal pathway may offer advantages, such as enhancing lysosomal acidification, replacing lysosomal enzymes, or promoting autophagy initiation. However, unmodified nanoparticles do not necessarily trigger endocytosis to enter this endocytic-lysosomal pathway. Therefore, researchers promote cellular endocytosis by biologically or chemically modifying nanoparticle surfaces. This includes cell-penetrating peptides, targeting ligands, cationic modifications and biomimetic modifications.108–111 Binding these natural or synthetic ligands to nanoparticles effectively promotes cell uptake, thereby enhancing the efficiency of drug delivery targeting the lysosomal pathway.13,106
In recent years, researchers have developed a novel approach to target protein degradation for clearing extracellular or intracellular harmful substances called lysosome-targeting chimeras (LYTACs) or autophagy-targeting chimeras (AUTACs). The chimera recognizes the target protein and links it to a lysosome-targeting receptor (LTR) on the cell membrane surface, initiating receptor-mediated endocytosis. The target protein is then transported to lysosomes for degradation, while the LTR is recycled back to the cell membrane via recycling endosomes.112,113 Combining LYTACs or AUTACs with nanodelivery significantly enhances the targeting efficiency. For example, Lu et al conjugating target protein and LTR ligands to gold nanoparticles. The large surface area of NPs allows binding of numerous ligands, greatly enhancing the efficiency of capturing pathological proteins and increasing the likelihood of lysosomal degradation.114 This strategy holds significant potential for PD to relieve pathological αSyn aggregation.
Lysosome/Endosome-Targeting Nano-Therapy Strategies
Designing nanostrategies targeting ALP can effectively improve lysosomal defects in PD, including impaired lysosomal acidification and promoting autophagy-mediated degradation of abnormal proteins. Table 3 summarizes recent applications of nanodelivery strategies targeting ALP in PD.
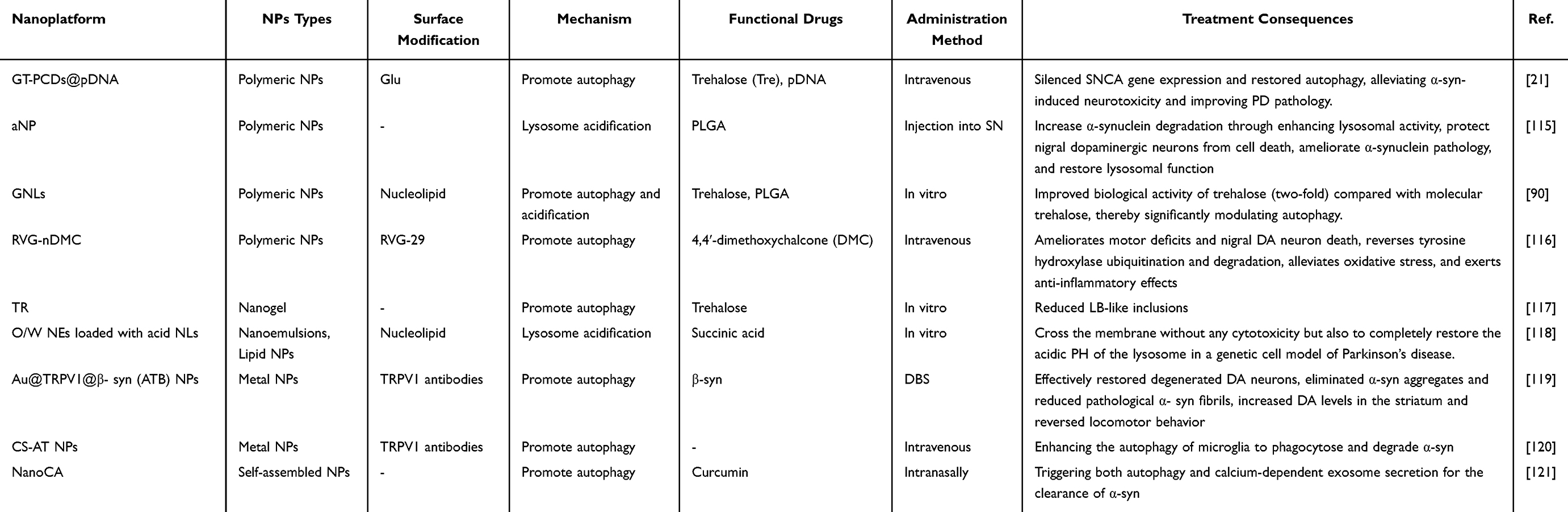 |
Table 3 Examples of Lysosomes-Targeted Nanoparticles for Treatment of Parkinson’s Disease |
Lysosomal Acidification
Extensive research indicates that lysosomal acidification is an effective strategy for restoring lysosomal function.76,122 Nanodelivery of lysosomal acidification drugs can significantly enhance lysosomal degradation capacity, eliminate pathological proteins, and slow disease progression.
Biocompatible poly(lactic-co-glycolic acid) (PLGA) are the most commonly used acidic nanomaterial. PLGA is completely biodegradable in vivo, ultimately metabolizing into carbon dioxide and water. It leaves no toxic residues and does not cause immune rejection reactions. For example, Arotcarena et al have found that PLGA-aNPs enhanced αSyn degradation by boosting lysosomal activity, thereby improving neurotoxin-induced dopaminergic neurodegeneration in mice.115 This study highlights the potential of acidification-based strategies to restore lysosomal function and promote pathogenic protein clearance, although their long-term effects on lysosomal homeostasis remain to be fully established. Besides, Brouillard, M. et al designed an innovative DNA derivative-based nanocarrier. Nucleolipids carry biocompatible organic acids as prodrugs, which were incorporated into oil-in-water nanomulsion to traverse biomembranes, followed by effective release of the biocompatible acidic components to restore functional pH in lysosomes of neuronal cells. The biosafety has also been validated.118 Compared with polymer-based systems, such prodrug-based designs may offer more controlled and localized pH modulation; however, their stability, release kinetics, and in vivo targeting efficiency require further validation.
Taken together, these approaches suggest that modulating lysosomal acidity is a promising strategy for restoring degradative function in PD. Nevertheless, achieving precise and sustained pH regulation without disrupting physiological lysosomal processes remains a key challenge for future development.
Ameliorate Autophagy
Modulating autophagy effectively clears abnormally accumulated proteins and senescent organelles within cells, improving intracellular protein homeostasis and associated pathological stress. Researchers therefore utilize nanoparticles to deliver natural or synthetic small-molecule drugs that modulate autophagy pathways, restoring impaired autophagy flux in disease states.
Trebose, a small natural autophagy enhancer molecule, is widely applied in NDDs to improve autophagy dysfunction. Cunha. et al designed a trehalose-based nanomedicine. They combined trehalose with an amphiphilic nucleolipid complex, which are incorporated in PLGA NPs or solid lipid NPs. It improved the uptake and internalization into neuronal cells associated with the trehalose-induced autophagy effect, and did not induce any cellular toxicity.90 Similarly, Mannose, a disaccharide capable of inducing autophagy, has been demonstrated to reduce αSyn aggregation in vivo. However, free mannose can be rapidly degraded by mannosidase, and its hydrophilic nature hinders cellular membrane penetration. Maruf. et al investigated a mannose-containing nanogel (TR) to promote its cellular uptake. TR inhibits αSyn aggregation formation and synergistically reduces Lewy body-like inclusions in primary hippocampal neurons by promoting aggregate clearance through mannitol-induced autophagy.117 Besides, Liu et al developed a curcumin analogue-based nanoscavenger (NanoCA) using a reprecipitated self-assembly process. This nanomaterial stimulates nuclear translocation of the transcription factor EB (TFEB), a key autophagy regulator, thereby triggering autophagy and calcium-dependent exosome secretion to clear α-syn. NanoCA protects neurons from MPP-induced neurotoxicity. Meanwhile, they designed a rapid-arousaling intranasal delivery system (RA-IDDS) for brain-targeted delivery, which provided potent neuroprotection and promoted clearance of α-syn within the midbrain in MPTP-induced PD mouse models.121 This strategy highlights the advantage of targeting upstream regulatory nodes such as TFEB to achieve broader activation of the autophagy–lysosome pathway; however, systemic modulation of such central regulators may also raise concerns regarding off-target effects and pathway overactivation.
In addition to natural medicinal compounds, existing drugs have been found to enhance autophagy. For instance, bionic microglial NPs carrying metformin could promote autophagy while reducing oxidative stress and neuroinflammation, which repaired dopaminergic neurons, cleared αSyn aggregates and improved motor function in PD mouse. Enhancing brain targeting of existing drugs like metformin through nanotechnology represents a promising direction for future nanomedicine design. Furthermore, repurposing this category of established drugs can help reduce research and development costs.
Furthermore, autophagy program could be initiated by leveraging the tunable physicochemical properties of nanoparticles and specific stimulus channels on the cell membrane surface. Transient Receptor Potential Vanilloid 1 (TRPV1), as a stimulus-responsive protein on the cell surface, can effectively sense thermal stimuli and regulate the flow of Ca2⁺, thereby activating neurons and precisely regulating neuronal activities. For example, Yuan et al designed photothermal Cu2- (x) Se-based nanoparticles targeting TRPV1 on microglia surfaces (CS-AT NPs). Under NIR-II laser irradiation, these NPs activate surface TRPV1 channels, inducing Ca2⁺ influx and activating ATG5 and Ca2⁺/CaMKK2/AMPK/mTOR signaling pathways to promote αSyn phagocytosis and degradation, significantly improved motor function in treated PD mice.120 Similarly, Wu, J. et al described a photothermal wireless DBS nanosystem (ATB NPs) composed of a gold nanocapsule shell conjugated with an anti-TRPV1 antibody. Under pulsed NIR irradiation, ATB NPs initiate autophagy to efficiently eliminate αSyn filaments in neurons. 119 These NIR-driven systems represent a sophisticated convergence of nanotechnology and neuromodulation. However, compared to chemically-driven autophagy enhancers like trehalose nanogels, they are technically more complex, require external hardware for clinical use, and their long-term safety profile within deep brain tissue is unexplored. The choice between a systemic, chemically-defined nanogel that passively enhances autophagy and an implant-free, externally-controlled photothermal system represents a fundamental design trade-off between clinical ease-of-use and spatiotemporal precision that the field has yet to formally evaluate.
Targeting Endoplasmic Reticulum
ER in the Pathophysiology of PD
The endoplasmic reticulum (ER) is a multifunctional organelle primarily responsible for protein folding and post-translational modifications, biosynthesis of carbohydrates and lipids, signal transduction, and calcium homeostasis through storage and dynamic mobilization. It maintains close functional connections with other organelles such as mitochondria and lysosomes via mechanisms like Ca2⁺ signaling, playing a central role in sustaining cellular homeostasis.123 Protein quality control and cellular homeostasis depend on ER involvement. Genetic mutations, aging, oxidative stress, disrupted protein homeostasis, and other stimuli can induce protein misfolding and aggregation, leading to ER stress. To resolve ER stress, eukaryotic cells adapt by reducing protein synthesis rates, upregulating genes encoding chaperones and other proteins that prevent peptide aggregation, and degrading accumulated misfolded proteins—a process termed the “unfolded protein response” (UPR).124 The ER possesses three stress sensors: (1) inositol-requiring enzyme 1 (IRE1); (2) polyribose-6-phosphate-6-phosphate-dehydrogenase-2-like ER kinase (PERK); (3) Activated Transcription Factor 6 (ATF6). Each UPR sensor remains inactive or quiescent by binding to the ER chaperone BiP/grp78. They promptly relay information about ER disturbances to the cytoplasm and nucleus, thereby initiating subsequent adaptive responses.125 However, prolonged ER stress induces neuronal death, axonal degeneration, neuroinflammation, and synaptic dysfunction, leading to NDDs including Alzheimer’s disease (AD), PD, amyotrophic lateral sclerosis, viral diseases, and other disorders characterized by misfolded protein accumulation and aggregation.124
Past studies have confirmed the close association between PD pathology and activated ER stress. Thep-PERK and p-eIF2α, which served as markers of ER stress, have been detected in dopaminergic neurons within the SN of PD patients.126 MPTP can induce UPR by disrupting ER Ca2⁺ homeostasis through inhibiting calcium entry.127 Ablation of downstream ER stress signaling molecules protects dopaminergic neurons from 6-OHDA induced damage. Furthermore, common PD genetic factors, including Parkin and LRRK2, have also been involved in ER stress processes. For instance, under ER stress conditions, ATF4 binds to the Parkin promoter, leading to increased Parkin expression.128 Parkin has been shown to prevent UPR-induced cell death mediated by GPR37.129 The knockdown of LRRK2 can promote 6-OHDA or αSyn induced neuron death by eliminating the upregulation of BiP/grp78.130 These findings demonstrate the critical role of ER stress in promoting PD pathology. Additionally, extensive studies demonstrate that αSyn aggregates can activate the UPR signaling pathway, while inhibiting ER-Golgi vesicular transport accelerate overexpression of αSyn.131,132
Recently, the interaction and communication between mitochondria and ER have garnered significant attention.133 Mitochondria-associated ER membrane (MAM) maintains the structural and functional balance of the ER and mitochondria. Many important biological processes, such as calcium exchange, phospholipid exchange, mitochondrial fusion and fission, mitochondrial phagocytosis, oxidative stress and bioenergy production are precisely regulated by MAM. In the MPTP-induced PD mouse model, the morphological changes of MAM were blocked and its biological performance was disordered.134 Therapeutic strategies aimed at preserving MAM integrity hold promise for mitigating the pathological progression of PD.
ER-Targeting Nano-Delivery Strategies
Current ER-targeting drug delivery strategies primarily include lipid nanoparticles, small-molecule ligands, and ER-targeting sequence peptides. Identifying ER targets remains challenging due to the organelle’s complex spatial architecture and vast folding surface area. When designing ER-targeting strategies, identifying, constructing, and modifying structural sequences with selective affinity for the ER is crucial. In recent years, numerous compounds and small molecules targeting the UPR through diverse molecular mechanisms have been identified and tested. Attaching these to nanoparticles can promote their efficient accumulation within the ER, thereby enhancing drug delivery efficiency.135
Lipid nanoparticles possess inherent advantages for ER targeting due to their lipophilic nature, enabling fusion with the ER’s lipid membrane.136 Additionally, nanoparticle shape influences accumulation efficiency within the ER; for instance, rod-shaped nanorods exhibit higher concentration levels compared to spherical nanoparticles.137 However, unmodified liposomes exhibit poor specificity for ER targeting and typically require chemical group modifications.
Common small-molecule ligands for ER targeting include sulfonyl ligands, chloride ligands, and zwitterionic ligands, which achieve ER targeting by interacting with ATP-sensitive potassium channels, chloride channels, and positively charged phosphatidylcholine cytidine transferase (CCT) on the ER membrane surface, respectively.123 For example, studies have demonstrated that p-toluenesulfonyl-modified lipid nanoparticles (17AAG-ER-NP) can deliver drugs to disrupt ER function and inhibit tumor growth.138 Additionally, literature reports an ER-targeting probe (ER-Naph) bearing a sulfonylurea moiety that specifically detects glutathione (GSH) levels within the ER.139
ER-targeting peptides encompass both naturally occurring peptides and synthetic amino acid sequences. KDEL is a C-terminal signal peptide present in ER molecular chaperones, recognized by KDEL receptors on the ER membrane to facilitate chaperone retention within the ER. Additionally, during vesicular transport, KDEL mediates the retrograde transport of soluble proteins that escape from the Golgi apparatus back to the ER.140 To minimize endogenous biological activity, ER-targeted proteins and peptides are modified through truncation of sub-sequences, introduction of amino acid spacers, and residue substitutions, yielding novel ER localization signals such as the pardaxin (Par) sequence. Liu et al efficiently delivered antigens to the ER of dendritic cells by synthesizing ER-targeted vesicles containing the Par sequence and conjugating them to magnetic mesoporous silica particles, thereby promoting antigen presentation and enhancing immune responses.141 Similarly, Li et al designed a nanoparticle system modified with pardaxin peptides that induces ER stress via photodynamic/photothermal therapy, triggering immunogenic cell death and demonstrating enhanced antitumor efficacy.142 Furthermore, self-assembling nanoparticles such as enzyme-guided self-assembled (EISA) branched peptides and multifunctional peptide assemblies exert their effects by targeting enzymes or functional proteins on the ER surface.143
In recent years, ER-targeting strategies have demonstrated certain advantages and achieved some progress. However, how to effectively target the ER in non-cancerous contexts without compromising its structure or inducing ER stress remains an urgent challenge. This is further complicated by the intricate intracellular microenvironment, given that the ER is a structure extensively interconnected with multiple organelles and signaling pathways.
ER-Targeting Nano-Therapy Strategies
Recent studies indicate that nanoparticle-mediated delivery of small-molecule drugs to ameliorate ER stress in neurons represents a promising neuroprotective strategy. For example, Posadas et al demonstrated that engineered neutral phosphorous dendrimers reduced ER stress and UPR by modulating NMDA receptor-mediated Ca2⁺ influx. This intervention significantly attenuated excitotoxicity in primary cortical neurons, an effect that was also replicated in human brain organoid models.144 Mitchell et al employed a novel class of nanomaterials—transition metal dichalcogenide (TMD) nanoflowers (NFs)—and demonstrated that MoS2 and MoSe2 NFs suppress the α-Syn-induced UPR while promoting autophagic and exocytic clearance of αSyn. In Caenorhabditis elegans models treated with NFs, the burden of αSyn aggregates was significantly reduced, accompanied by a marked extension of lifespan.145 Similarly, studies have shown that nanocapsules carrying DA and catalase (CAT) effectively reduced neuroinflammation by down-regulating the expression of αSyn and ATF6 and alleviating ER stress.146 In addition, Chiang, M. C. et al showed, in an Alzheimer’s disease-related model (Aβ-treated human neural stem cells), that glutathione-conjugated gold nanoparticles (GSH-AuNPs) reversed ER stress gene expression and mitochondrial dysfunction. While this finding is not from a PD model, it provides indirect evidence that strategies ameliorating ER stress may be therapeutically relevant for PD and warrant further investigation in α-Syn-based systems.147
Furthermore, as ER serves as a Ca2⁺ storage site and a hub for multiple signaling pathways, many nanomedicines take advantage of this to regulate cellular physiology. For example, under NIR-II laser irradiation, mPDA-SeMn-IR nanoparticles effectively activate endogenously expressed IP3 receptors, inducing Ca2⁺ efflux from the ER. The enhanced Ca2⁺ signaling upregulates tyrosine hydroxylase (TH) expression and activity, triggering dopamine release and thereby enhancing dopaminergic function.148 Interestingly, Li et al engineered a “nanomassage” system based on PEGylated black phosphorus nanosheets (PEG-BPNS) with a lateral size of ~200 nm which can be administrated intranasally. By adhering to the cell membrane, it exerts external mechanical force on cells, promoting interactions among actin, mitochondria, and ER, thereby effectively enhancing cellular function. The nanosheets significantly improved motor performance in MPTP-induced PD mouse models.149 Notably, these nanodelivered therapeutics not only alleviate ER stress but also mitigate related pathological processes—including mitochondrial oxidative stress, impaired autophagy, and neuroinflammation, offers new insights for advancing nanomedicine in NDDs.
Although the nanomedicines targeting ER are limited, many existing drugs have been proven to have ER protection potential. They exert protective effects by alleviating ER stress or reducing misfolded proteins, such as small-molecule modulators,150 kinase inhibitors,151 miRNA,152 anti-prion drugs,153 antioxidants,154 natural compounds,155–158 and chemical chaperones.154 These drugs may potentially enhance their efficacy through integration with nanotechnology. Interestingly, moderate ER stress may favor cellular adaptation to exogenous stimuli and improve protein homeostasis. Some reports suggest that activating UPR transcription factors may improve pathological states in PD. In human and cellular studies, UPR-activating agents increased expression of UPR target genes in dopaminergic neurons and reduced neuronal death under MPTP-treated conditions.159 Therefore, how to effectively regulate the degree of ER modulation remains a question worth exploring.
Targeting Golgi Apparatus
GA in the Pathophysiology of PD
The Golgi apparatus (GA) is a vital organelle in eukaryotic cells. It plays a crucial role in the modification of proteins and lipids, the sorting and transport of cellular membranes, and the synthesis and repair of cell membranes. These functions ensure the proper functioning and localization of intracellular substances, thereby maintaining normal cellular physiological functions.160 Furthermore, it serves as an intracellular signaling platform and stress sensor, actively participating in regulating cell polarization, stress responses, directed migration, mitosis, metabolism, autophagy, apoptosis, DNA repair, inflammatory responses, and other cellular processes.161
Under normal conditions, the GA may experience transient and mild stress induced by oxidative stress, energy deficiency, or intracellular temperature fluctuations. To maintain cellular homeostasis, the GA primarily adjusts its structure and function by altering its volume, vesicle number, and protein modifications.160 When protein loads exceed the GA’s transport and modification capacity, ROS levels or glutathione (GSH) levels within the GA lumen rapidly increase or decrease, disrupting redox balance. This phenomenon, characterized by impaired GA function and disrupted redox balance, is termed GA stress.162 However, when subjected to sustained and irreparable pressure, GA stress may lead to structural fragmentation and abnormal protein accumulation by reducing protein synthesis and modification efficiency, impairing protein quality control, obstructing vesicular transport, and disrupting intracellular calcium homeostasis.160 As a pivotal signaling hub, GA stress engages multiple signaling pathways and physiological processes, including innate immune signaling, lysosomal autophagy, and cellular metabolism. Dysregulation of these intricate networks can subsequently trigger cell death and contribute to the onset and progression of diseases such as NDDs, autoimmune diseases, and cancer.163–167
Research indicates that GA fragmentation represents an early pathological event in PD. Literature suggests this fragmentation precedes neuronal cell death, disrupting intracellular protein synthesis and impairing axonal, dendritic, and synaptic transport.168 Early autopsy analyses of PD samples reveal highly fragmented GA in SN neurons.169 Some scholars propose that GA stress serves as the initial trigger for αSyn aggregation, Lewy body formation, and microtubule alterations. Changes in transport regulatory proteins and SNARE proteins may represent early disruptions in membrane transport within secretory pathways, leading to GA fragmentation.5,170 Additionally, multiple PD-associated genetic factors influence GA function. LRRK2 plays a crucial role in intracellular vesicular transport by phosphorylating Rab small GTPases. LRRK2 mutants disrupt GA integrity and vesicle transport. Furthermore, LRRK2 inactivation also leads to GA fragmentation via endocytosis and autophagy.171 Another PD-associated gene is PLA2G6. In fibroblasts from patients carrying this mutation, altered N- and O-linked glycosylation patterns occur, along with impaired ERGIC (ER-GA Intermediate Compartment) and GA function, potentially contributing to neurodegeneration.172 Animal models of PD demonstrate that αSyn can block membrane transport from the ER to the GA, leading to degeneration and loss of dopaminergic neurons.125 Collectively, these findings support GA stress as a critical component in the pathophysiology of PD.
GA-Targeting Nano-Delivery Strategies
To achieve effective targeting of the GA, researchers have identified and engineered a series of ligands and signaling peptides. Ligands accomplish active targeting by binding to receptors or enzymes on GA membranes or within its compartments, while peptide signals enable insertion, fusion, or localization within GA membranes and compartments to facilitate targeted cargo delivery.
Common small-molecule ligands targeting GA include chondroitin sulfate (CS), thioesters, and cysteine.173–175 CS is a typical sulfated glycosaminoglycan exhibiting excellent biocompatibility, biodegradability, non-immunogenicity, and low toxicity. In cancer, hydrophilic CS can self-assemble with hydrophobic drugs to target GA in liver cancer cells which overexpressing CD44. CS nanomicelles also demonstrate high affinity and effectively target GA in hepatic stellate cells.176,177 Nanopolymers modified with cell-penetrating peptides and CS have been demonstrated to effectively evade the endosomal degradation pathway while inhibiting ER/GA-mediated exocytosis, thereby enhancing the intracellular retention efficiency of loaded drugs.178 Research has revealed that peptide thioesters serve as substrates for GA-associated thioesterases. The resulting thiopeptides subsequently form dimers and accumulate within the GA. These peptide thioesters can enter cells via caveolae-mediated endocytosis and specifically accumulate in the GA, regardless of whether they are above or below the critical micelle concentration.174 Carbon quantum dots and silicon nanoparticles functionalized with L-cysteine residues have also been employed for targeting GA to achieve targeted imaging.179 These molecules, when conjugated to nanoparticles, demonstrate promising potential for targeted drug delivery to the GA.
On the other hand, identifying signal sequences capable of efficient GA localization provides favorable conditions for GA-targeted nano-delivery. Analysis of certain cytoplasmic domains of glycoproteins present in GA transport network revealed a series of GA-localizing sequences. For example, Alexandra P et al identified a 37-amino acid alternative open reading frame (altORF) within the mRNA of the centromere protein CENP-R. The peptide specifically localizes to the GA surface.180 Cheng, G. et al discovered a targeting domain within the matrix protein GM130 that localizes to the GA.181 Besides, SXYQRL and RS1-reg peptides promote GA localization by initiating retrograde transport from endosomes to the GA. They have been effectively utilized to selectively deliver therapeutics to target sites within the GA network.177,182 Concurrently, Li et al designed transformable peptide C6RVRRF4KY that can self-assemble into nanoparticles and convert into destructive fibers only under the action of enzymes specifically expressed in the GA of cancer cells, and which incorporate cysteine residues, enhancing GA targeting.183 Furthermore, caveolin-mediated endocytosis is linked to cargo transport within the GA. When drugs and their delivery systems utilize vesicular structures, they can bypass lysosomes to reach ER/GA. This allows them to avoid significant loss due to degradation in lysosomes and enables specific targeting of the ER/GA to achieve their therapeutic mechanisms.162 Since the ER and GA are closely linked cellular structures along the protein synthesis and secretion pathway, transport between them typically lacks specificity. Therefore, when designing ER- or GA-targeted systems, the system should exploit differences between the ER and GA to exert drug efficacy immediately upon reaching the target organelle.
GA-Targeting Nano-Therapy Strategies
Although the role of the GA in regulating cellular homeostasis cannot be overlooked, the exploratory application of improving GA function in PD and even other neurodegenerative diseases remains considerably limited. This scarcity may stem from the unclear mechanisms of the GA in neurological disorders, coupled with the lack of understanding regarding how to leverage the GA for neuroprotective effects. Therefore, we primarily discuss several proof-of-concept studies that are still in the exploratory stage, with the aim of providing a theoretical basis for subsequent targeted nanotherapies. In PD, modulating disease-related genes such as LRRK2, PLA2G6, and VPS35 can indirectly alleviate GA stress. The Rab GTPases family, acting as LRRK2 substrates, plays a role in vesicle transport within the GA. Inhibiting LRRK2 mutations alleviates GA stress by promoting Rab29-mediated recruitment of LRRK2 to the GA, subsequently phosphorylating Rab8A to maintain normal vesicular transport.184 Similarly, suppressing PLA2G6 mutations reduces GA stress, rescues abnormal glycosylation, and mitigates protein aggregation.172 Furthermore, VPS35 mutations are associated with hereditary PD and indirectly linked to GA stress. Inhibiting VPS35 mutations promotes cathepsin transport, reduces α-syn accumulation, and ensures post-transport functions of the GA.185 Additionally, GA-associated proteins such as STX5, Rab1A, and GRASP65 are also considered potential intervention targets.160 These targets may potentially be precisely targeted through nanotechnology in the future.
However, it must be stressed that the application of GA-targeting NPs in NDDs, and particularly in PD, remains almost entirely unexplored territory. The strategies discussed above, such as CS-based prodrug NPs for cancer or Golgi disruption for immunotherapy, are currently at the proof-of-concept stage in oncology.186 Their translation to a neuroprotective context in PD is highly speculative and requires a fundamental shift from cytotoxic Golgi disruption to restoring Golgi structural integrity and function. A critical future step is thus to elucidate how to therapeutically promote Golgi recovery in PD models, rather than simply adapting destructive cancer-targeting strategies.
Targeting the Nucleus
Nucleus in the Pathophysiology of PD
The nucleus serves as the primary repository for genetic material DNA. By precisely regulating processes such as DNA replication, transcription, and RNA processing, it directly determines cellular fate and function, acting as the principal organelle governing reproduction, growth, metabolism, and the cell cycle.187–189 The nucleus maintains its integrity through structures such as the nuclear envelope, nuclear pore complexes (NPCs), and nuclear cytoskeleton.190 Recent discoveries indicate that nucleophagy selectively removes nuclear components to maintain genomic stability, while abnormalities in nuclear morphology or structure significantly impair cellular activities.191 The nucleus also serves as a hub for inter-organelle communication, coordinating organelle functions and responses to developmental or environmental stimuli through retrograde signaling pathways. NPC-mediated nuclear-cytoplasmic transport is critical for maintaining neuronal function.192
Dysfunction of the NPCs is a common feature in NDDs, including ALS, AD, and PD.193–195 The NPC governs macromolecular exchange between the nucleus and cytoplasm; its disruption leads to abnormal cytoplasmic accumulation and nuclear depletion of RNA-binding proteins like TDP-43 and FUS, as well as transcription factors.196 In PD, abnormal nuclear localization of multiple transcription factors has been observed. For instance, both total and phosphorylated cyclic AMP response element-binding protein (CREB) and phosphorylated extracellular signal-regulated kinase (ERK) accumulate in the cytoplasm of dopamine neurons exposed to 6-OHDA and in neurons from PD patients.195 Furthermore, abnormal αSyn blocks NCT-mediated transport of macromolecules in the nucleus by interacting with Ran GTPase.197
Numerous studies indicate that PD-associated gene mutations disrupt NE architecture, while abnormal processing of the nuclear lamin network destabilizes the nuclear skeleton and accelerates neuronal aging. Triple mutations at the A53T and SNCA loci exacerbate nuclear senescence and compromise NE integrity in stem cell-derived DA neurons. LRRK2 gene mutations accelerate genomic instability by impairing nuclear structural integrity.197,198
Oxidative stressors increase nuclear translocation of αSyn in DA neurons both in vitro and in vivo.199 Nuclearly localized αSyn promotes neurotoxicity by altering transcription, histone modifications, and inducing DNA damage.200,201 It also exacerbates mitochondrial dysfunction and disrupts nuclear-mitochondrial communication through transcriptional regulation of the mitochondrial regulator PGC1-α.202 Currently, multiple epigenetic abnormalities—such as dysregulated histone methylation, promoter methylation of PARK2 and SNCA genes, and DNA methyltransferase (DNMT1) deficiency.203 Furthermore, specific defects in DNA repair can affect the dopaminergic system and are related to the pathology of human PD.204 The above evidence indicates that nuclear dysfunction plays an important role in the occurrence and development of the disease.
Collectively, disturbances in the nucleus of dopaminergic neurons correlate with PD-associated neurodegeneration. These disturbances involve structural damage to the nuclear envelope and mislocalization of key transcription factors, with oxidative stress, αSyn, and pathogenic mutations potentially exacerbating nuclear injury.
Nucleus-Targeting Nano-Delivery Strategies
The nuclear envelope possesses a double-layered structure that provides highly protective and selective containment for nuclear genetic material. This protective architecture presents a major barrier to nucleolar targeting. After entering cells, nanoparticles must overcome multiple barriers before reaching the nucleus, including capture by endosomes and lysosomes, complex transport pathways within the cytoplasmic sol, and the highly selective nuclear membrane.205,206 These obstacles significantly limit the therapeutic efficacy of drugs targeting nuclear components. Therefore, designing and preparing nanomaterials capable of entering the nucleus is crucial for enhancing the effectiveness of these therapies.
For nuclear targeting, nanoparticle size is a primary consideration. Studies indicate small particles can enter the nucleus, while larger particles remain confined to the cytoplasm.207 However, excessively small nanoparticles compromise drug loading, necessitating additional targeting modifications. During transport from the cytoplasm to the nucleus, cargo undergoes a precise recognition and transport process where nuclear import proteins play a vital role. Nuclear localization signals (NLS) are recognized by import proteins, allowing NLS-tagged cargo to form complexes with these proteins and enter the nucleus via NPC.208 Consequently, conjugating NLS to various functionalized nanoparticles has proven effective for targeting the nucleus, demonstrating significant value across multiple biomedical fields including drug delivery, gene therapy, and diagnostic imaging.209 The TAT peptide, an 11-amino acid sequence from the HIV-1 transcription activator protein, is one of the most commonly used and versatile NLS peptides for guiding nanoparticles to the nucleus. This peptide has been employed to couple with various types of nanoparticles to achieve intracellular drug delivery.210–212 Additionally, nuclear localization sequences from other sources—such as those derived from simian virus SV40, bipartite nucleoplasmin, and adenovirus—have been demonstrated to efficiently deliver nanoparticles of varying sizes to the nucleus.13 For instance, Li et al designed a nuclear-targeted siRNA delivery system by modifying gold nanoparticles with NLS peptides and siRNA. The NLS peptides translocate the nanoparticle carrier into the nucleus, enabling sustained gene silencing and effectively inhibiting cancer cell proliferation.213 This diversity highlights the potential of NLS-modified nanoparticles as a powerful strategy for enhancing nuclear drug delivery.
Nucleus-Targeting Nano-Therapy Strategies
Nucleic acid-based therapeutics have been extensively studied for their ability to regulate functional protein expression. However, due to their simple structure, they are highly susceptible to peripheral degradation. Consequently, nanotechnology holds significant potential for enhancing the targeting efficiency and therapeutic efficacy of nucleic acids. siRNA can specifically silence disease-related genes through the RNA interference (RNAi) mechanism, offering an effective therapeutic strategy by interfering with αSyn expression. Nano-delivery effectively shield siRNA from serum enzyme degradation, prolonging its half-life in vivo. For example, Feja et al developed a nanoparticle-based intranasal siRNA delivery method. Tyrosine-modified polyethyleneimines (PEIs) or polypropyleneimine dendrimers (PPIs) were complexed with siRNA targeting the SNCA gene (siSNCA) and conjugated with liposomes. In Thy1-aSyn mice, the NPs exhibited extensive distribution throughout the brain, significantly reducing brain aSyn protein and SNCA mRNA levels while demonstrating favorable safety profiles.214 Furthermore, certain systems enable co-delivery of siRNA with other therapeutic agents. Katamesh et al developed an intranasal albumin-coated liposome system for co-delivering the MAO-B inhibitor selegiline (SEL) and αSyn-targeted siRNA (C-LipSel-siSNCA2). In PD rats, C-LipSel-siSNCA2 mitigated dopamine degradation and αSyn aggregation, reduced MAO-B levels, and improved motor and non-motor functions.215 Although siRNA has shown promising results in animal studies, its clinical advancement has been slow, with no nucleic acid therapeutics yet entering clinical trials for PD treatment.
Plasmid DNA (pDNA) can serve as a vector to introduce target genes into the cell nucleus, enabling replication and expression independent of the host chromosome. It is commonly used for overexpressing endogenous genes or correcting non-functional genes to treat genetic disorders.216 In the context of PD, pDNA-based nanodelivery offers a direct approach to restoring deficient pathways; however, its therapeutic potential is closely tied to efficient nuclear delivery and sustained transgene expression, which remain key technical challenges. Recent studies have demonstrated localized GBA1 gene therapy using pDNA-loaded nanoparticles. For example, PEGylated poly(β-amino ester) nanoparticles (PEG-PBAE NPs) were designed to reduce adhesion to the brain extracellular matrix and improve physiological stability. When administered via convection-enhanced delivery, these particles achieved broad distribution within the brain. In a α-Syn preformed fibril (PFF)-induced PD mouse model, they enhanced human GBA1 transgene expression in the SN, reduced α-Syn aggregation, preserved dopaminergic neurons, and alleviated neuroinflammation.217 This approach highlights the advantage of combining optimized carrier design with physical delivery strategies to overcome tissue-level barriers, although its clinical applicability may be limited by the invasiveness and complexity of administration. In addition to carrier design, delivery strategies play a critical role in improving therapeutic outcomes. Combining focused ultrasound (FUS) with intranasal administration has been shown to enhance nanoparticle penetration into deep brain regions. Aly et al used this approach to facilitate microglial transfection and increase plasmid nanoparticle delivery to brain parenchyma, resulting in sustained expression of the neurotrophic factor GDNF in PD-affected regions.218
Beyond pDNA-based systems, other nucleic acid therapeutics are also being explored. For instance, delivering mRNA encoding immunoregulatory factors via LNPs can activate immune tolerance pathways, thereby achieving indirect neuroprotection in the CNS. Studies indicate that LNPs carrying granulocyte-macrophage colony-stimulating factor (GM-CSF) mRNA induce peripheral Treg expansion in PD animal models, reducing reactive microgliosis and thereby protecting dopaminergic neurons in the SN and striatum.219
Additionally, gene editing technologies such as CRISPR/Cas9 may offer the potential to fundamentally correct defective genes at the genetic level. However, the underlying genetic mechanisms of PD remain poorly understood, and significant challenges persist regarding precision, off-target effects, and the long-term implications of genetic modification. While precise nanodelivery systems could potentially mitigate some of these issues, rigorous ethical review and extensive preliminary trials remain essential prerequisites for their application.
Overall, nucleic acid–based nanotherapeutics provide a versatile platform for targeting multiple pathogenic mechanisms in PD, including gene deficiency, protein aggregation, and neuroinflammation. However, achieving efficient and cell-specific nuclear delivery, maintaining controlled gene expression, and balancing efficacy with safety remain critical challenges for clinical translation.
Conclusions and Perspective
Parkinson’s disease remains a formidable therapeutic challenge, as current treatments primarily provide symptomatic relief without halting the underlying neurodegenerative process. This review systematically evaluates the potential of organelle-targeted nanotherapeutics—directed toward mitochondria, lysosomes, the endoplasmic reticulum, the Golgi apparatus, and the nucleus—to intervene at the core of PD pathogenesis by restoring subcellular homeostasis. The convergence of nanotechnology and organelle biology represents a promising paradigm for precision medicine in PD. However, a critical and balanced appraisal of the field reveals a markedly uneven landscape of translational maturity, which must be explicitly recognized to more effectively inform and guide future research efforts.
Cumulatively, current preclinical and clinical evidence demonstrates that mitochondria-targeted therapeutic strategies stand out as the most well-established class for PD intervention. Among these, mitochondria-targeted antioxidants and nanozymes have been thoroughly validated and extensively characterized in a wide range of animal disease models, with the lead candidate CNM-Au8 already advancing to Phase II clinical investigation. By comparison, lysosome- and nucleus-targeted therapeutic approaches, while holding considerable therapeutic potential, remain largely limited to preclinical research and face substantial technical challenges and high regulatory barriers to subsequent clinical translation. In sharp contrast, strategies targeting the ER and Golgi apparatus remain far less mature in the PD research field; most available supporting evidence is merely indirect, extrapolated from non-PD models rather than dedicated PD experimental systems. Such marked disparities in research maturity, preclinical validity, and translational progress across subcellular-targeted modalities underscore the urgent need for well-designed, direct head-to-head comparative studies under unified, standardized experimental conditions.
Nanomedicines have introduced a paradigm shift in the biomedical field. However, fundamental barriers that impede or delay their clinical translation remain, including delivery efficiency, safety, reproducibility, storage stability, and scalable manufacturing prior to clinical application.220 These challenges have been extensively discussed in previous studies, therefore, here we focus specifically on the outstanding issues that remain to be addressed for organelle-targeted nanotherapeutics in PD. First, the disease model gap remains a major limitation. A large proportion of current studies rely on simplified in vitro systems that primarily capture cellular phenotypes, such as oxidative stress or α-synuclein aggregation, without accounting for key physiological barriers, including BBB, systemic circulation, and nanoparticle clearance mechanisms. As a result, these models may overestimate intracellular delivery efficiency and therapeutic efficacy. The widespread use of acute toxin-based models (MPTP, 6-OHDA, rotenone) provides proof-of-concept but does not reflect the chronic and progressive nature of human PD or the interaction between aging, genetic susceptibility, and environmental factors. Validation in non-human primate models with higher construct validity is essential for assessing disease-modifying effects, including α-Syn PFF models and genetic models (eg, LRRK2, GBA1). Second, there is a clear lack of rigorous subcellular targeting validation. Many studies infer organelle specificity based on ligand design (eg, TPP, NLS), but direct in vivo evidence in relevant brain cell types remains limited. Future studies should adopt quantitative and standardized methods, such as transmission electron microscopy with immunogold labeling,221 subcellular fractionation combined with mass spectrometry,222 and super-resolution imaging in tissue sections,223 to allow more reliable and comparable evaluation. Third, a significant gap in long-term safety data persists. While acute toxicity is increasingly reported, chronic exposure studies remain scarce. Key issues include immunogenicity, unintended interference with organelle function, nanoparticle accumulation and clearance, and material-specific risks.224 Given the need for long-term treatment in PD, systematic evaluation under repeated dosing conditions is critical. Fourth, manufacturing and quality control challenges remain underaddressed.220 The batch-to-batch reproducibility directly impact biodistribution and efficacy, and parameters such as particle size, surface charge, ligand density, and drug loading require tight control.225
In summary, organelle-targeted nanotherapeutics represent a promising but still developing strategy for PD treatment. The gap between preclinical findings and clinical application remains substantial. Progress will depend less on expanding the range of nanoplatforms and more on addressing key translational challenges, including model relevance, targeting validation, long-term safety, and manufacturing standardization. Establishing clear and comparable benchmarks will be essential for advancing the most robust candidates toward clinical testing.
Although integration into a precision medicine framework remains a long-term goal, achieving this will first require systematic resolution of the translational challenges outlined above.
Data Sharing Statement
No datasets were generated or analyzed during the current study.
Acknowledgments
This work was supported by grants 82471456 from the National Natural Science Foundation of China (to YX), and grants 82371270 from the National Natural Science Foundation of China (to YX).
Author Contributions
MaoYu Liu is the first author. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants 82471456 from the National Natural Science Foundation of China (to YX), and grants 82371270 from the National Natural Science Foundation of China (to YX).
Disclosure
The authors declare no competing interests.
References
1. Ben-Shlomo Y, Darweesh S, Llibre-Guerra J, et al. The epidemiology of Parkinson’s disease. Lancet. 2024;403:283–30. doi:10.1016/s0140-6736(23)01419-8
2. Zhu J, Cui Y, Zhang J, et al. Temporal trends in the prevalence of Parkinson’s disease from 1980 to 2023: a systematic review and meta-analysis. Lancet Healthy Longev. 2024;5:e464–e479. doi:10.1016/s2666-7568(24)00094-1
3. Tolosa E, Garrido A, Scholz SW, Poewe W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021;20:385–397. doi:10.1016/s1474-4422(21)00030-2
4. Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912. doi:10.1016/s0140-6736(14)61393-3
5. Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernández MA, et al. Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front Neurosci. 2019;13:1399. doi:10.3389/fnins.2019.01399
6. Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Progr Neurobiol. 2013;106-107:17–32. doi:10.1016/j.pneurobio.2013.04.004
7. Nguyen M, Wong YC, Ysselstein D, Severino A, Krainc D. Synaptic, mitochondrial, and lysosomal dysfunction in parkinson’s disease. Trends Neurosci. 2019;42:140–149. doi:10.1016/j.tins.2018.11.001
8. Costa CAD, Manaa WE, Duplan E, Checler F. The endoplasmic reticulum stress/unfolded protein response and their contributions to parkinson’s disease physiopathology. Cells. 2020;9:2495. doi:10.3390/cells9112495
9. Heidari A, Yazdanpanah N, Rezaei N. The role of Toll-like receptors and neuroinflammation in Parkinson’s disease. J Neuroinflamm. 2022;19:135. doi:10.1186/s12974-022-02496-w
10. Armstrong MJ, Okun MS. Diagnosis and treatment of parkinson disease: a review. JAMA. 2020;323:548–560. doi:10.1001/jama.2019.22360
11. Singh D. Organelle targeted drug delivery: key challenges, recent advancements and therapeutic implications. Endocr Metab Immune Disord Drug Targets. 2024;24:1480–1487. doi:10.2174/0118715303282573240112104035
12. Ma X, Gong N, Zhong L, Sun J, Liang XJ. Future of nanotherapeutics: targeting the cellular sub-organelles. Biomaterials. 2016;97:10–21. doi:10.1016/j.biomaterials.2016.04.026
13. Soukar J, Peppas NA, Gaharwar AK. Organelle-targeting nanoparticles. Adv Sci. 2025;12:e2411720. doi:10.1002/advs.202411720
14. Guo ZH, Khattak S, Rauf MA, et al. Role of nanomedicine-based therapeutics in the treatment of CNS disorders. Molecules. 2023;28. doi:10.3390/molecules28031283
15. Huang W, Ye X, Huang Z, et al. Nanozymes in Parkinson’s disease: strategic approaches, clinical considerations, and challenges. J Mat Chem B. 2025;13:7919–7933. doi:10.1039/d5tb00295h
16. Kim S, Kim HD. Multifunctional nanomaterial-mediated tumor therapeutics: enhancing efficacy and specificity. Int J Pharm. 2025;682:125982. doi:10.1016/j.ijpharm.2025.125982
17. Sun M, Sen Gupta A. Vascular nanomedicine: current status, opportunities, and challenges. Semin Thromb Hemost. 2020;46:524–544. doi:10.1055/s-0039-1692395
18. Chen L, Li Q. Nanomaterials in the diagnosis and treatment of gastrointestinal tumors: new clinical choices and treatment strategies. Mater Today Bio. 2025;32:101782. doi:10.1016/j.mtbio.2025.101782
19. Monge-Fuentes V, Biolchi mayer A, Lima MR, et al. Dopamine-loaded nanoparticle systems circumvent the blood-brain barrier restoring motor function in mouse model for Parkinson’s Disease. Sci Rep. 2021;11:15185. doi:10.1038/s41598-021-94175-8
20. Liu Y, Luo J, Liu Y, et al. Brain-targeted biomimetic nanodecoys with neuroprotective effects for precise therapy of Parkinson’s disease. ACS Cent Sci. 2022;8:1336–1349. doi:10.1021/acscentsci.2c00741
21. Zhai L, Gao Y, Yang H, et al. A ROS-Responsive nanoparticle for nuclear gene delivery and autophagy restoration in Parkinson’s disease therapy. Biomaterials. 2025;321:123345. doi:10.1016/j.biomaterials.2025.123345
22. Li L, Sun C, Cai S, et al. Biomimetic nanoparticles enhance recovery of movement disorders in parkinson’s disease by improving microglial mitochondrial homeostasis and suppressing neuroinflammation. ACS Appl Mater Interfaces. 2025;17:23536–23552. doi:10.1021/acsami.4c22181
23. Kaur J, Thakran A, Naqvi S. Recent advances in cell membrane-based biomimetic delivery systems for Parkinson’s disease: perspectives and challenges. Asian J Pharm Sci. 2025;20:101060. doi:10.1016/j.ajps.2025.101060
24. Yang YP, Nicol CJB, Chiang MC. A review of the neuroprotective properties of exosomes derived from stem cells and exosome-coated nanoparticles for treating neurodegenerative diseases and stroke. Int J Mol Sci. 2025;26. 10.3390/ijms26083915.
25. Zheng Q, Liu H, Zhang H, et al. Ameliorating mitochondrial dysfunction of neurons by biomimetic targeting nanoparticles mediated mitochondrial biogenesis to boost the therapy of parkinson’s disease. Adv Sci. 2023;10:e2300758. doi:10.1002/advs.202300758
26. Li H, Fan Y, Shen Y, et al. Acid-activated TAT peptide-modified biomimetic boron nitride nanoparticles for enhanced targeted codelivery of doxorubicin and indocyanine green: a synergistic cancer photothermal and chemotherapeutic approach. ACS Appl Mater Interfaces. 2024;16:25101–25112. doi:10.1021/acsami.4c01622
27. Liu Y, Xia X, Zheng M, Shi B. Bio-nano toolbox for precision alzheimer’s disease gene therapy. Adv Mater. 2024;36:e2314354. doi:10.1002/adma.202314354
28. Wang H. Rapid deep brain chemogenetics: Minimally invasive, genetically targeted deep brain stimulation is achieved using ultrasound-activated nanocrystals. Science. 2025;389:469. doi:10.1126/science.aea0852
29. Ahmad J, Haider N, Khan MA, et al. Novel therapeutic interventions for combating Parkinson’s disease and prospects of nose-to-brain drug delivery. Biochem Pharmacol. 2022;195:114849. doi:10.1016/j.bcp.2021.114849
30. van Vliet EF, Knol MJ, Schiffelers RM, Caiazzo M, Fens M. Levodopa-loaded nanoparticles for the treatment of Parkinson’s disease. J Control Release. 2023;360:212–224. doi:10.1016/j.jconrel.2023.06.026
31. Khatri DK, Preeti K, Tonape S, et al. Nanotechnological advances for nose to brain delivery of therapeutics to improve the parkinson therapy. Curr Neuropharmacol. 2023;21:493–516. doi:10.2174/1570159x20666220507022701
32. Torres-Ortega PV, Saludas L, Hanafy AS, Garbayo E, Blanco-Prieto MJ. Micro- and nanotechnology approaches to improve Parkinson’s disease therapy. J Control Release. 2019;295:201–213. doi:10.1016/j.jconrel.2018.12.036
33. Chen Z, Chen L, Lyu TD, et al. Targeted mitochondrial nanomaterials in biomedicine: Advances in therapeutic strategies and imaging modalities. Acta Biomater. 2024;186:1–29. doi:10.1016/j.actbio.2024.08.008
34. Jamwal S, Blackburn JK, Elsworth JD. PPARgamma/PGC1alpha signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol Ther. 2021;219:107705. doi:10.1016/j.pharmthera.2020.107705
35. Zong Y, Li H, Liao P, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9:124. doi:10.1038/s41392-024-01839-8
36. Klemmensen MM, Borrowman SH, Pearce C, Pyles B, Chandra B. Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics. 2024;21:e00292. doi:10.1016/j.neurot.2023.10.002
37. Chen F, Xu K, Han Y, et al. Mitochondrial dysfunction in pancreatic acinar cells: mechanisms and therapeutic strategies in acute pancreatitis. Front Immunol. 2024;15:1503087. doi:10.3389/fimmu.2024.1503087
38. Xie Y, Liu X, Xie D, et al. Voltage-dependent anion channel 1 mediates mitochondrial fission and glucose metabolic reprogramming in response to ionizing radiation. Sci Total Environ. 2024;946:174246. doi:10.1016/j.scitotenv.2024.174246
39. Zhang Y, Ma P, Wang S, Chen S, Deng H. Restoring calcium crosstalk between ER and mitochondria promotes intestinal stem cell rejuvenation through autophagy in aged Drosophila. Nat Commun. 2025;16:4909. doi:10.1038/s41467-025-60196-4
40. Liu Z, Shan S, Kang K, et al. Mitochondrial transfer of alpha-synuclein mediates carbon disulfide-induced mitochondrial dysfunction and neurotoxicity. Ecotoxicol Environ Saf. 2024;281:116613. doi:10.1016/j.ecoenv.2024.116613
41. Liao J, He W, Li L, et al. Mitochondria in brain diseases: bridging structural-mechanistic insights into precision-targeted therapies. Cell Biomat. 2025;1(100016). doi:10.1016/j.celbio.2025.100016
42. Zambrano K, Barba D, Castillo K, et al. Fighting Parkinson’s disease: the return of the mitochondria. Mitochondrion. 2022;64:34–44. doi:10.1016/j.mito.2022.02.003
43. Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139(Suppl 1):216–231. doi:10.1111/jnc.13731
44. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19:170–178. doi:10.1016/s1474-4422(19)30287-x
45. Borsche M, Pereira SL, Klein C, Grünewald A. Mitochondria and Parkinson’s disease: clinical, molecular, and translational aspects. J Parkinsons Dis. 2021;11:45–60. doi:10.3233/jpd-201981
46. Jiao H, Jiang D, Hu X, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021;184:2896–2910.e2813. doi:10.1016/j.cell.2021.04.027
47. Eldeeb MA, Thomas RA, Ragheb MA, Fallahi A, Fon EA. Mitochondrial quality control in health and in Parkinson’s disease. Physiol Rev. 2022;102:1721–1755. doi:10.1152/physrev.00041.2021
48. Ge P, Dawson VL, Dawson TM. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol Neurodegener. 2020;15:20. doi:10.1186/s13024-020-00367-7
49. Wang W, Thomas ER, Xiao R, et al. Targeting mitochondria-regulated ferroptosis: a new frontier in Parkinson’s disease therapy. Neuropharmacology. 2025;274:110439. doi:10.1016/j.neuropharm.2025.110439
50. Huang E, Qu D, Huang T, et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat Commun. 2017;8:1399. doi:10.1038/s41467-017-01435-1
51. Matuz-Mares D, González-Andrade M, Araiza-Villanueva MG, Vilchis-Landeros MM, Vázquez-Meza H. Mitochondrial calcium: effects of its imbalance in disease. Antioxidants. 2022;11. 10.3390/antiox11050801.
52. Gambardella S, Limanaqi F, Ferese R, et al. ccf-mtDNA as a potential link between the brain and immune system in neuro-immunological disorders. Front Immunol. 2019;10:1064. doi:10.3389/fimmu.2019.01064
53. Li Y, Li XM, Wei LS, Ye JF. Advancements in mitochondrial-targeted nanotherapeutics: overcoming biological obstacles and optimizing drug delivery. Front Immunol. 2024;15:1451989. doi:10.3389/fimmu.2024.1451989
54. Han Y, Chu X, Cui L, et al. Neuronal mitochondria-targeted therapy for Alzheimer’s disease by systemic delivery of resveratrol using dual-modified novel biomimetic nanosystems. Drug Delivery. 2020;27:502–518. doi:10.1080/10717544.2020.1745328
55. Liu Y, Zhao D, Yang F, et al. In situ self-assembled phytopolyphenol-coordinated intelligent nanotherapeutics for multipronged management of ferroptosis-driven alzheimer’s disease. ACS Nano. 2024;18:7890–7906. doi:10.1021/acsnano.3c09286
56. Zielonka J, Joseph J, Sikora A, et al. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev. 2017;117:10043–10120. doi:10.1021/acs.chemrev.7b00042
57. Yuan X, Bi Y, Liu J, et al. In vitro evaluation of mitochondrial-targeted andrographolide nanoparticles against 4T1 breast cancer cells. Int J Nanomed. 2025;20:15395–15414. doi:10.2147/ijn.S525568
58. Schlichtmann BW, Kalyanaraman B, Schlichtmann RL, et al. Functionalized polyanhydride nanoparticles for improved treatment of mitochondrial dysfunction. J Biomed Mater Res Part B. 2022;110:450–459. doi:10.1002/jbm.b.34922
59. Cilleros-Holgado P, Gómez-Fernández D, Piñero-Pérez R, et al. Mitochondrial quality control via mitochondrial unfolded protein response (mtUPR) in ageing and neurodegenerative diseases. Biomolecules. 2023;13:1789. doi:10.3390/biom13121789
60. Chin MY, Espinosa JA, Pohan G, Markossian S, Arkin MR. Reimagining dots and dashes: visualizing structure and function of organelles for high-content imaging analysis. Cell Chem Biol. 2021;28:320–337. doi:10.1016/j.chembiol.2021.01.016
61. Pan F, Li Y, Ding Y, et al. Anticancer effect of rationally designed alpha-helical amphiphilic peptides. Colloids Surf B. 2022;220:112841. doi:10.1016/j.colsurfb.2022.112841
62. Caspari OD, Garrido C, Law CO, et al. Converting antimicrobial into targeting peptides reveals key features governing protein import into mitochondria and chloroplasts. Plant Commun. 2023;4:100555. doi:10.1016/j.xplc.2023.100555
63. Shin G, Hyun S, Kim D, et al. Cyclohexylalanine-containing alpha-helical amphipathic peptide targets cardiolipin, rescuing mitochondrial dysfunction in kidney injury. J Med Chem. 2024;67:3385–3399. doi:10.1021/acs.jmedchem.3c01578
64. Nahar N, Sohag MSU. Advancements in mitochondrial-targeted antioxidants: organelle-specific drug delivery for disease management. Adv Redox Res. 2025;17(100142). doi:10.1016/j.arres.2025.100142
65. Giradkar V, Mhaske A, Shukla R. Naringenin nanocrystals mitigate rotenone neurotoxicity in SH-SY5Y cell line by modulating mitophagy and oxidative stress. AAPS Pharm Sci Tech. 2024;25:227. doi:10.1208/s12249-024-02936-1
66. Xia X, Li H, Xu X, Zhao G, Du M. Facilitating pro-survival mitophagy for alleviating parkinson’s disease via sequence-targeted lycopene nanodots. ACS Nano. 2023;17:17979–17995. doi:10.1021/acsnano.3c04308
67. Biswal L, Sardoiwala MN, Kushwaha AC, Mukherjee S, Karmakar S. Melatonin-loaded nanoparticles augment mitophagy to retard parkinson’s disease. ACS Appl Mater Interfaces. 2024;16:8417–8429. doi:10.1021/acsami.3c17092
68. Biswal L, Sahu VK, Sardoiwala MN, Karmakar S, Choudhury SR. Antibody conjugated targeted nanotherapy epigenetically inhibits calpain-mediated mitochondrial dysfunction to attenuate Parkinson’s disease. Carbohydr Polym. 2024;346:122575. doi:10.1016/j.carbpol.2024.122575
69. Chen Y, Zhang B, Yu L, et al. A novel nanoparticle system targeting damaged mitochondria for the treatment of Parkinson’s disease. Biomat Adv. 2022;138:212876. doi:10.1016/j.bioadv.2022.212876
70. Zhao Z, Chen L, Yang C, et al. Nanomotor-based H(2)S donor with mitochondrial targeting function for treatment of Parkinson’s disease. Bioact Mater. 2024;31:578–589. doi:10.1016/j.bioactmat.2023.09.001
71. Wang W, Zhao Z, Zhang Z, et al. Delivery of small interfering RNA by hydrogen sulfide-releasing nanomotor for the treatment of Parkinson’s disease. J Control Release. 2025;377:648–660. doi:10.1016/j.jconrel.2024.11.069
72. Pham KY, Khanal S, Bohara G, et al. HDAC6 inhibitor-loaded brain-targeted nanocarrier-mediated neuroprotection in methamphetamine-driven Parkinson’s disease. Redox Biol. 2025;79:103457. doi:10.1016/j.redox.2024.103457
73. Liu Y, Liu Y, Shi P, et al. Single-atom nanozyme liposome-integrated microneedles for in situ drug delivery and anti-inflammatory therapy in Parkinson’s disease. J Nanobiotechnol. 2024;22(1):643. doi:10.1186/s12951-024-02924-4
74. Xu H, Ding X, Li L, et al. Tri-element nanozyme PtCuSe as an ingenious cascade catalytic machine for the amelioration of Parkinson’s disease-like symptoms. Front Bioeng Biotechnol. 2023;11:1208693. doi:10.3389/fbioe.2023.1208693
75. Liu Y, Hong H, Xue J, et al. Near-infrared radiation-assisted drug delivery nanoplatform to realize blood-brain barrier crossing and protection for parkinsonian therapy. ACS Appl Mater Interfaces. 2021;13:37746–37760. doi:10.1021/acsami.1c12675
76. Zhang L, Zhan M, Sun H, et al. Mesenchymal stem-cell-derived exosomes loaded with phosphorus dendrimers and quercetin treat parkinson’s disease by modulating inflammatory immune microenvironment. ACS Appl Mater Interfaces. 2025;17:32013–32027. doi:10.1021/acsami.5c05809
77. Li B, Bai Y, Yion C, et al. Single-atom nanocatalytic therapy for suppression of neuroinflammation by inducing autophagy of abnormal mitochondria. ACS Nano. 2023;17:7511–7529. doi:10.1021/acsnano.2c12614
78. Zhang S, Li M, Li Y, et al. Mitochondria-targeted nanovesicles for ursodeoxycholic acid delivery to combat neurodegeneration by ameliorating mitochondrial dysfunction. J Nanobiotechnol. 2025;23:202. doi:10.1186/s12951-025-03258-5
79. Nguyen-Thi PT, Ho TT, Nguyen TT, Vo GV. Nanotechnology-based drug delivery for alzheimer’s and parkinson’s diseases. Current Drug Delivery. 2024;21:917–931. doi:10.2174/1567201820666230707113405
80. Sun Z, Fu H, Zhang R, et al. Advances in chemically modified HSA as a multifunctional carrier for transforming cancer therapy regimens. Int J Biol Macromol. 2025;305:141373. doi:10.1016/j.ijbiomac.2025.141373
81. Ghodsi H, Rahimi HR, Aghili SM, Saberi A, Shoeibi A. Evaluation of curcumin as add-on therapy in patients with Parkinson’s disease: a pilot randomized, triple-blind, placebo-controlled trial. Clin Neurol Neurosurg. 2022;218:107300. doi:10.1016/j.clineuro.2022.107300
82. Zhang P, Bai H, Yao Z, et al. Tumor microenvironment responsive chitosan-coated W-doped MoO(x) biodegradable composite nanomaterials for photothermal/chemodynamic synergistic therapy. Int J Biol Macromol. 2024;276:133583. doi:10.1016/j.ijbiomac.2024.133583
83. Ding Q, Shang J, Yang L, et al. Enhanced anti-tumor efficacy of berberine-loaded mesoporous polydopamine nanoparticles for synergistic chemotherapy and photothermal therapy. Int J Pharm. 2025;670:125151. doi:10.1016/j.ijpharm.2024.125151
84. Ren J, Dewey RB, Rynders A, et al. Evidence of brain target engagement in Parkinson’s disease and multiple sclerosis by the investigational nanomedicine, CNM-Au8, in the REPAIR Phase 2 clinical trials. J Nanobiotechnol. 2023;21:478. doi:10.1186/s12951-023-02236-z
85. Zhang Y, Yu W, Zhang L, Li P. Nanozyme-based visual diagnosis and therapeutics for myocardial infarction: the application and strategy. J Adv Res. 2025;70:187–201. doi:10.1016/j.jare.2024.04.019
86. Alomari OA, Qusti S, Balgoon M, et al. Modified TPP-MoS(2) QD blend as a bio-functional model for normalizing microglial dysfunction in alzheimer’s disease. Neurol Int. 2023;15:954–966. doi:10.3390/neurolint15030061
87. Ren C, Li D, Zhou Q, Hu X. Mitochondria-targeted TPP-MoS(2) with dual enzyme activity provides efficient neuroprotection through M1/M2 microglial polarization in an Alzheimer’s disease model. Biomaterials. 2020;232:119752. doi:10.1016/j.biomaterials.2019.119752
88. Jia Z, Yuan X, Wei J-A, et al. A functionalized octahedral palladium nanozyme as a radical scavenger for ameliorating alzheimer’s disease. ACS Appl Mater Interfaces. 2021;13:49602–49613. doi:10.1021/acsami.1c06687
89. Wang L, Yuan X, Cai Q, et al. Mitochondria-targeting Cu(2-x)Se-TPP with dual enzyme activity alleviates Alzheimer’s disease by modulating oxidative stress. Colloids Surf B. 2025;245:114244. doi:10.1016/j.colsurfb.2024.114244
90. Cunha A, Gaubert A, Verget J, et al. Trehalose-based nucleolipids as nanocarriers for autophagy modulation: an in vitro study. Pharmaceutics. 2022;14:857. doi:10.3390/pharmaceutics14040857
91. Navarro-Romero A, Montpeyo M, Martinez-Vicente M. The emerging role of the lysosome in parkinson’s disease. Cells. 2020;9:2399. doi:10.3390/cells9112399
92. Galper J, Dean NJ, Pickford R, et al. Lipid pathway dysfunction is prevalent in patients with Parkinson’s disease. Brain. 2022;145:3472–3487. doi:10.1093/brain/awac176
93. Dai L, Liu M, Ke W, et al. Lysosomal dysfunction in alpha-synuclein pathology: molecular mechanisms and therapeutic strategies. Cell Mol Life Sci. 2024;81:382. doi:10.1007/s00018-024-05419-5
94. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi:10.1016/j.cell.2011.10.026
95. Pang SY, Lo RCN, Ho PW-L, et al. LRRK2, GBA and their interaction in the regulation of autophagy: implications on therapeutics in Parkinson’s disease. Transl Neurodegener. 2022;11:5. doi:10.1186/s40035-022-00281-6
96. Rubilar JC, Outeiro TF, Klein AD. The lysosomal beta-glucocerebrosidase strikes mitochondria: implications for Parkinson’s therapeutics. Brain. 2024;147:2610–2620. doi:10.1093/brain/awae070
97. Wildburger NC, Hartke AS, Schidlitzki A, Richter F. Current evidence for a bidirectional loop between the lysosome and alpha-synuclein proteoforms. Front Cell Develop Biol. 2020;8:598446. doi:10.3389/fcell.2020.598446
98. Hopfner F, Mueller SH, Szymczak S, et al. Rare variants in specific lysosomal genes are associated with parkinson’s disease. Mov Disord. 2020;35:1245–1248. doi:10.1002/mds.28037
99. Nixon RA, Rubinsztein DC. Mechanisms of autophagy-lysosome dysfunction in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2024;25:926–946. doi:10.1038/s41580-024-00757-5
100. Lo CH, Zeng J. Defective lysosomal acidification: a new prognostic marker and therapeutic target for neurodegenerative diseases. Transl Neurodegener. 2023;12:29. doi:10.1186/s40035-023-00362-0
101. Mindell JA. Lysosomal acidification mechanisms. Ann Rev Physiol. 2012;74:69–86. doi:10.1146/annurev-physiol-012110-142317
102. Quick JD, Silva C, Wong JH, et al. Lysosomal acidification dysfunction in microglia: an emerging pathogenic mechanism of neuroinflammation and neurodegeneration. J Neuroinflamm. 2023;20:185. doi:10.1186/s12974-023-02866-y
103. Hu M, Li P, Wang C, et al. Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell. 2022;185:2292–2308.e2220. doi:10.1016/j.cell.2022.05.021
104. Jiao F, Meng L, Du K, Li X. The autophagy-lysosome pathway: a potential target in the chemical and gene therapeutic strategies for Parkinson’s disease. Neural Regen Res. 2025;20:139–158. doi:10.4103/nrr.Nrr-d-23-01195
105. Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi:10.1146/annurev.biochem.78.081307.110540
106. Sharma A, Vaghasiya K, Ray E, Verma RK. Lysosomal targeting strategies for design and delivery of bioactive for therapeutic interventions. J Drug Targeting. 2018;26:208–221. doi:10.1080/1061186x.2017.1374390
107. Salatin S, Maleki Dizaj S, Yari Khosroushahi A. Effect of the surface modification, size, and shape on cellular uptake of nanoparticles. Cell Biol Int. 2015;39:881–890. doi:10.1002/cbin.10459
108. Jiang L, Liu J, Chen S, et al. Cyclic cell-penetrating peptide-engineered ceria nanoparticles for non-invasive alleviation of ultraviolet radiation-induced cataract. J Nanobiotechnol. 2025;23(1):337. doi:10.1186/s12951-025-03402-1
109. Bai M, Cui N, Liao Y, et al. Astrocytes and microglia-targeted Danshensu liposomes enhance the therapeutic effects on cerebral ischemia-reperfusion injury. J Control Release. 2023;364:473–489. doi:10.1016/j.jconrel.2023.11.002
110. Dong L, Lou W, Xu C, Wang J. Naringenin cationic lipid-modified nanoparticles mitigate MASLD progression by modulating lipid homeostasis and gut microbiota. J Nanobiotechnol. 2025;23:168. doi:10.1186/s12951-025-03228-x
111. Cao Z, Liu X, Zhang W, et al. Biomimetic macrophage membrane-camouflaged nanoparticles induce ferroptosis by promoting mitochondrial damage in glioblastoma. ACS Nano. 2023;17(23):23746–23760. doi:10.1021/acsnano.3c07555
112. Chen Y, Liu F, Pal S, Hu Q. Proteolysis-targeting drug delivery system (ProDDS): integrating targeted protein degradation concepts into formulation design. Chem Soc Rev. 2024;53:9582–9608. doi:10.1039/d4cs00411f
113. Lin J, Jin J, Shen Y, et al. Emerging protein degradation strategies: expanding the scope to extracellular and membrane proteins. Theranostics. 2021;11(17):8337–8349. doi:10.7150/thno.62686
114. Lu W, Chen J, Guo Z, et al. Targeted degradation of ABCG2 for reversing multidrug resistance by hypervalent bispecific gold nanoparticle-anchored aptamer chimeras. Chem Commun. 2023;59(21):3118–3121. doi:10.1039/d3cc00168g
115. Arotcarena ML, Soria FN, Cunha A, et al. Acidic nanoparticles protect against alpha-synuclein-induced neurodegeneration through the restoration of lysosomal function. Aging Cell. 2022;21:e13584. doi:10.1111/acel.13584
116. Zhang W, Chen H, Ding L, et al. Trojan horse delivery of 4,4’-dimethoxychalcone for parkinsonian neuroprotection. Adv Sci. 2021;8:2004555. doi:10.1002/advs.202004555
117. Maruf A, Gerasymchuk D, Hlushchuk I, et al. Trehalose-releasing nanogels reduce α-synuclein-induced Lewy body-like inclusions in primary mouse hippocampal neurons. J Mat Chem B. 2025;13:5845–5857. doi:10.1039/d4tb02704c
118. Brouillard M, Barthelemy P, Dehay B, Crauste-Manciet S, Desvergnes V. Nucleolipid acid-based nanocarriers restore neuronal lysosomal acidification defects. Front Chem. 2021;9:736554. doi:10.3389/fchem.2021.736554
119. Wu J, Cui X, Bao L, et al. A nanoparticle-based wireless deep brain stimulation system that reverses Parkinson’s disease. Sci Adv. 2025;11:eado4927. doi:10.1126/sciadv.ado4927
120. Yuan J, Liu H, Zhang H, et al. Controlled activation of TRPV1 channels on microglia to boost their autophagy for clearance of alpha-synuclein and enhance therapy of parkinson’s disease. Adv Mater. 2022;34:e2108435. doi:10.1002/adma.202108435
121. Liu J, Liu C, Zhang J, et al. A self-assembled α-synuclein nanoscavenger for parkinson’s disease. ACS Nano. 2020;14:1533–1549. doi:10.1021/acsnano.9b06453
122. Zeng J, Acin-Perez R, Assali EA, et al. Restoration of lysosomal acidification rescues autophagy and metabolic dysfunction in non-alcoholic fatty liver disease. Nat Commun. 2023;14(1):2573. doi:10.1038/s41467-023-38165-6
123. Shi Y, Wang S, Wu J, Jin X, You J. Pharmaceutical strategies for endoplasmic reticulum-targeting and their prospects of application. J Control Release. 2021;329:337–352. doi:10.1016/j.jconrel.2020.11.054
124. Ghemrawi R, Khair M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int J Mol Sci. 2020;21:6127. doi:10.3390/ijms21176127
125. Colla E. Linking the endoplasmic reticulum to parkinson’s disease and alpha-synucleinopathy. Front Neurosci. 2019;13:560. doi:10.3389/fnins.2019.00560
126. Yang J, Kim KS, Iyirhiaro GO, et al. DJ-1 modulates the unfolded protein response and cell death via upregulation of ATF4 following ER stress. Cell Death Dis. 2019;10:135. doi:10.1038/s41419-019-1354-2
127. Calì T, Ottolini D, Brini M. Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. BioFactors. 2011;37:228–240. doi:10.1002/biof.159
128. Bouman L, Schlierf A, Lutz AK, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011;18(5):769–782. doi:10.1038/cdd.2010.142
129. Marazziti D, Di Pietro C, Golini E, et al. Induction of macroautophagy by overexpression of the Parkinson’s disease-associated GPR37 receptor. FASEB J. 2009;23:1978–1987. doi:10.1096/fj.08-121210
130. Yuan Y, Cao P, Smith MA, et al. Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PLoS One. 2011;6(8):e22354. doi:10.1371/journal.pone.0022354
131. Thayanidhi N, Helm JR, Nycz DC, et al. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell. 2010;21:1850–1863. doi:10.1091/mbc.e09-09-0801
132. Kim DY, Shin JY, Lee JE, et al. A selective ER-phagy exerts neuroprotective effects via modulation of α-synuclein clearance in parkinsonian models. Proc Natl Acad Sci USA. 2023;120:e2221929120. doi:10.1073/pnas.2221929120
133. Yang Y, Chen M, Ding L, et al. Mitochondria-associated endoplasmic reticulum membranes and calcium ion exchange: a novel direction for aging and neurodegenerative diseases. Neural Regen Res. 2025. doi:10.4103/nrr.Nrr-d-25-00857
134. Liu J, Liu Y, Gao C, et al. The ultrastructural and proteomic analysis of mitochondria-associated endoplasmic reticulum membrane in the midbrain of a Parkinson’s disease mouse model. Aging Cell. 2025;24:e14436. doi:10.1111/acel.14436
135. Girigoswami K, Pallavi P, Girigoswami A. Intricate subcellular journey of nanoparticles to the enigmatic domains of endoplasmic reticulum. Drug Delivery. 2023;30:2284684. doi:10.1080/10717544.2023.2284684
136. Shi Y, Luo Z, You J. Subcellular delivery of lipid nanoparticles to endoplasmic reticulum and mitochondria. WIREs Nanomed Nanobiotechnol. 2022;14. 10.1002/wnan.1803.
137. Zhang W, Yu M, Xi Z, et al. Cancer cell membrane-camouflaged nanorods with endoplasmic reticulum targeting for improved antitumor therapy. ACS Appl Mater Interfaces. 2019;11:46614–46625. doi:10.1021/acsami.9b18388
138. Ghosh C, Nandi A, Basu S. Lipid nanoparticle-mediated induction of endoplasmic reticulum stress in cancer cells. ACS Appl Bio Mater. 2019;2:3992–4001. doi:10.1021/acsabm.9b00532
139. Wi Y, Le HT, Verwilst P, et al. Modulating the GSH/Trx selectivity of a fluorogenic disulfide-based thiol sensor to reveal diminished GSH levels under ER stress. Chem Commun. 2018;54:8897–8900. doi:10.1039/c8cc04846k
140. Cela I, Dufrusine B, Rossi C, et al. KDEL receptors: pathophysiological functions, therapeutic options, and biotechnological opportunities. Biomedicines. 2022;10:1234. doi:10.3390/biomedicines10061234
141. Liu M, Zhao Y, Shi Z, Zink JI, Yu Q. Virus-like magnetic mesoporous silica particles as a universal vaccination platform against pathogenic infections. ACS Nano. 2023;17:6899–6911. doi:10.1021/acsnano.3c00644
142. Li W, Yang J, Luo L, et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat Commun. 2019;10(1):3349. doi:10.1038/s41467-019-11269-8
143. Kim BJ, Fang Y, He H, Xu B. Trypsin-instructed self-assembly on endoplasmic reticulum for selectively inhibiting cancer cells: dedicated to Professor George M. Whitesides on the occasion of his 80th birthday. Adv Healthcare Mater. 2021;10:e2000416. doi:10.1002/adhm.202000416
144. Posadas I, Romero-Castillo L, Ronca R-A, et al. Engineered neutral phosphorous dendrimers protect mouse cortical neurons and brain organoids from excitotoxic death. Int J Mol Sci. 2022;23(8):4391. doi:10.3390/ijms23084391
145. Mitchell CL, Matveyenka M, Brown HC, et al. Neuroprotective parkinson’s disease therapeutic: transition metal dichalcogenide nanoflower treatments alleviate pathological cell stress. bioRxiv. 2025. doi:10.1101/2025.09.29.679305
146. Liu Z, Xiang S, Chen B, et al. Parkinson disease -targeted nanocapsules for synergistic treatment: combining dopamine replacement and neuroinflammation mitigation. Adv Sci. 2024;11(46):e2404717. doi:10.1002/advs.202404717
147. Chiang MC, Nicol CJB. GSH-AuNP anti-oxidative stress, ER stress and mitochondrial dysfunction in amyloid-beta peptide-treated human neural stem cells. Free Radic Biol Med. 2022;187:185–201. doi:10.1016/j.freeradbiomed.2022.05.025
148. Zhou C, Wu F, He L, et al. An implant-free nanosystem enabling synergistic oxidative damage mitigation and deep brain stimulation for alleviating parkinsonian symptoms. ACS nano. 2025;19(29):26715–26734. doi:10.1021/acsnano.5c06227
149. Li T, Huang L, Guo C, et al. Massage-mimicking nanosheets mechanically reorganize inter-organelle contacts to restore mitochondrial functions in parkinson’s disease. Adv Sci. 2025;12:e2413376. doi:10.1002/advs.202413376
150. Ke M, Chong C-M, Zeng H, et al. Azoramide protects iPSC-derived dopaminergic neurons with PLA2G6 D331Y mutation through restoring ER function and CREB signaling. Cell Death Dis. 2020;11:130. doi:10.1038/s41419-020-2312-8
151. Siwecka N, Galita G, Granek Z, et al. IRE1/JNK is the leading UPR pathway in 6-OHDA-induced degeneration of differentiated SH-SY5Y cells. Int J Mol Sci. 2024;25:7679. doi:10.3390/ijms25147679
152. Krammes L, Hart M, Rheinheimer S, et al. Induction of the endoplasmic-reticulum-stress response: microRNA-34a targeting of the IRE1α-branch. Cells. 2020;9:1442. doi:10.3390/cells9061442
153. Gentile L, Nasir A, Simon L, et al. Anti-prion drugs reduce endoplasmic reticulum stress and protect human dopaminergic neurons from death. Biomed Pharmacother. 2025;193:118758. doi:10.1016/j.biopha.2025.118758
154. Kim S, Kim DK, Jeong S, Lee J. The common cellular events in the neurodegenerative diseases and the associated role of endoplasmic reticulum stress. Int J Mol Sci. 2022;23. 10.3390/ijms23115894.
155. Ahmadi A, Hayes AW, Karimi G. Resveratrol and endoplasmic reticulum stress: a review of the potential protective mechanisms of the polyphenol. Phytother Res. 2021;35:5564–5583. doi:10.1002/ptr.7192
156. Zhou HY, Sun YY, Chang P, Huang HC. Curcumin inhibits cell damage and apoptosis caused by thapsigargin-induced endoplasmic reticulum stress involving the recovery of mitochondrial function mediated by Mitofusin-2. Neurotox Res. 2022;40:449–460. doi:10.1007/s12640-022-00481-y
157. He Y, Ruganzu JB, Lin C, et al. Tanshinone IIA ameliorates cognitive deficits by inhibiting endoplasmic reticulum stress-induced apoptosis in APP/PS1 transgenic mice. Neurochem Int. 2020;133(104610):104610. doi:10.1016/j.neuint.2019.104610
158. Lin Z, Xie R, Zhong C, et al. Recent progress (2015–2020) in the investigation of the pharmacological effects and mechanisms of ginsenoside Rb1, a main active ingredient in Panax ginseng Meyer. J Ginseng Res. 2022;46(1):39–53. doi:10.1016/j.jgr.2021.07.008
159. Zeeshan HM, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci. 2016;17:327. doi:10.3390/ijms17030327
160. Liu M, Duan Y, Dong J, et al. Early signs of neurodegenerative diseases: possible mechanisms and targets for Golgi stress. Biomed Pharmacother. 2024;175:116646. doi:10.1016/j.biopha.2024.116646
161. Zhang M, Xu N, Xu W, Ling G, Zhang P. Potential therapies and diagnosis based on Golgi-targeted nano drug delivery systems. Pharmacol Res. 2022;175:105861. doi:10.1016/j.phrs.2021.105861
162. Cho H, Huh KM, Shim MS, et al. Selective delivery of imaging probes and therapeutics to the endoplasmic reticulum or Golgi apparatus: current strategies and beyond. Adv Drug Delivery Rev. 2024;212:115386. doi:10.1016/j.addr.2024.115386
163. Tao Y, Yang Y, Zhou R, Gong T. Golgi apparatus: an emerging platform for innate immunity. Trends Cell Biol. 2020;30:467–477. doi:10.1016/j.tcb.2020.02.008
164. Chang HY, Yang WY. Golgi quality control and autophagy. IUBMB Life. 2022;74:361–370. doi:10.1002/iub.2611
165. Hu Z, Zeng L, Huang Z, Zhang J, Li T. The study of Golgi apparatus in Alzheimer’s disease. Neurochem Res. 2007;32:1265–1277. doi:10.1007/s11064-007-9302-4
166. Mărunţelu I, Constantinescu AE, Covache-Busuioc RA, Constantinescu I. The Golgi apparatus: a key player in innate immunity. Int J Mol Sci. 2024;25:4120. doi:10.3390/ijms25074120
167. Liu J, Huang Y, Li T, et al. The role of the Golgi apparatus in disease (Review). IntJ Mol Med. 2021;47(4). doi:10.3892/ijmm.2021.4871
168. Nakagomi S, Barsoum MJ, Bossy-Wetzel E, et al. A Golgi fragmentation pathway in neurodegeneration. Neurobiol Dis. 2008;29(2):221–231. doi:10.1016/j.nbd.2007.08.015
169. Fujita Y, Ohama E, Takatama M, Al-Sarraj S, Okamoto K. Fragmentation of Golgi apparatus of nigral neurons with alpha-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 2006;112:261–265. doi:10.1007/s00401-006-0114-4
170. Martínez-Menárguez J, Tomás M, Martínez-Martínez N, Martínez-Alonso E. Golgi fragmentation in neurodegenerative diseases: is there a common cause? Cells. 2019;8(7):748. doi:10.3390/cells8070748
171. Mohan AG, Calenic B, Ghiurau NA, et al. The Golgi apparatus: a voyage through time, structure, function and implication in neurodegenerative disorders. Cells. 2023;12(15):1972. doi:10.3390/cells12151972
172. Davids M, Kane MS, He M, et al. Disruption of Golgi morphology and altered protein glycosylation in PLA2G6-associated neurodegeneration. J Med Genet. 2016;53(3):180–189. doi:10.1136/jmedgenet-2015-103338
173. Li H, Zhang P, Luo J, et al. Chondroitin sulfate-linked prodrug nanoparticles target the golgi apparatus for cancer metastasis treatment. ACS Nano. 2019;13:9386–9396. doi:10.1021/acsnano.9b04166
174. Tan W, Zhang Q, Quiñones-Frías MC, et al. Enzyme-responsive peptide thioesters for targeting golgi apparatus. J Am Chem Soc. 2022;144:6709–6713. doi:10.1021/jacs.2c02238
175. Li RS, Gao PF, Zhang HZ, et al. Chiral nanoprobes for targeting and long-term imaging of the Golgi apparatus. Chem Sci. 2017;8(10):6829–6835. doi:10.1039/c7sc01316g
176. Luo J, Zhang P, Zhao T, et al. Golgi apparatus-targeted chondroitin-modified nanomicelles suppress hepatic stellate cell activation for the management of liver fibrosis. ACS Nano. 2019;13(4):3910–3923. doi:10.1021/acsnano.8b06924
177. Wang SH, Xu XL, Chen W. How do organelle-targeting nanotherapeutics treat inflammatory diseases? A comprehensive review of the literature. Int J Nanomed. 2025;20:7133–7152. doi:10.2147/ijn.S516260
178. Wang Y, Yin S, He D, et al. Dual strategies based on golgi apparatus/endoplasmic reticulum targeting and anchoring for high-efficiency siRNA delivery and tumor RNAi therapy. ACS Nano. 2025;19:3791–3806. doi:10.1021/acsnano.4c14778
179. Wei YY, Chen L, Zhang X, et al. Orange-emissive carbon quantum dots for ligand-directed Golgi apparatus-targeting and in vivo imaging. Biomater Sci. 2022;10:4345–4355. doi:10.1039/d2bm00429a
180. Navarro AP, Cheeseman IM. Identification of a Golgi-localized peptide reveals a minimal Golgi-targeting motif. Mol Biol Cell. 2022;33:ar110. doi:10.1091/mbc.E22-03-0091
181. Cheng G, Chang J, Gong H, Zhou W. A distinct Golgi-targeting mechanism of dGM130 in Drosophila neurons. Front Mol Neurosci. 2023;16:1206219. doi:10.3389/fnmol.2023.1206219
182. Deng C, Zhao X, Chen Y, et al. Engineered platelet microparticle-membrane camouflaged nanoparticles for targeting the golgi apparatus of synovial fibroblasts to attenuate rheumatoid arthritis. ACS Nano. 2022;16(11):18430–18447. doi:10.1021/acsnano.2c06584
183. Li RS, Liu J, Shi H, et al. Transformable helical self-assembly for cancerous golgi apparatus disruption. Nano Lett. 2021;21:8455–8465. doi:10.1021/acs.nanolett.1c03112
184. Madero-Pérez J, Fernández B, Lara Ordóñez AJ, et al. RAB7L1-mediated relocalization of LRRK2 to the golgi complex causes centrosomal deficits via RAB8A. Front Mol Neurosci. 2018;11:417. doi:10.3389/fnmol.2018.00417
185. Rahman AA, Morrison BE. Contributions of VPS35 mutations to Parkinson’s disease. Neuroscience. 2019;401:1–10. doi:10.1016/j.neuroscience.2019.01.006
186. Li H, Deng C, Tan Y, et al. Chondroitin sulfate-based prodrug nanoparticles enhance photodynamic immunotherapy via Golgi apparatus targeting. Acta Biomater. 2022;146:357–369. doi:10.1016/j.actbio.2022.05.014
187. Li Y, Ge S, Liu J, et al. Nuclear structure, size regulation, and role in cell migration. Cells. 2024;13(24):2130. doi:10.3390/cells13242130
188. Kalukula Y, Stephens AD, Lammerding J, Gabriele S. Mechanics and functional consequences of nuclear deformations. Nat Rev Mol Cell Biol. 2022;23:583–602. doi:10.1038/s41580-022-00480-z
189. Shan L, Li P, Yu H, Chen LL. Emerging roles of nuclear bodies in genome spatial organization. Trends Cell Biol. 2024;34:595–605. doi:10.1016/j.tcb.2023.10.012
190. Cantwell H, Dey G. Nuclear size and shape control. Semin Cell Dev Biol. 2022;130:90–97. doi:10.1016/j.semcdb.2021.10.013
191. Ji F, Dai E, Kang R, et al. Mammalian nucleophagy: process and function. Autophagy. 2025;21(7):1396–1412. doi:10.1080/15548627.2025.2455158
192. Calderon RH, Strand Å. How retrograde signaling is intertwined with the evolution of photosynthetic eukaryotes. Curr Opin Plant Biol. 2021;63:102093. doi:10.1016/j.pbi.2021.102093
193. Coyne AN, Baskerville V, Zaepfel BL, et al. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci Trans Med. 2021;13(604). doi:10.1126/scitranslmed.abe1923
194. Eftekharzadeh B, Daigle JG, Kapinos LE, et al. Tau protein disrupts nucleocytoplasmic transport in alzheimer’s disease. Neuron. 2018;99:925–940.e927. doi:10.1016/j.neuron.2018.07.039
195. Riaz Z, Richardson GS, Jin H, et al. Nuclear pore and nucleocytoplasmic transport impairment in oxidative stress-induced neurodegeneration: relevance to molecular mechanisms in Pathogenesis of Parkinson’s and other related neurodegenerative diseases. Mol Neurodegener. 2024;19:87. doi:10.1186/s13024-024-00774-0
196. Bitetto G, Di Fonzo A. Nucleo–cytoplasmic transport defects and protein aggregates in neurodegeneration. Transl Neurodegen. 2020;9:25. doi:10.1186/s40035-020-00205-2
197. Chen V, Moncalvo M, Tringali D, et al. The mechanistic role of alpha-synuclein in the nucleus: impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Human Mol Genet. 2020;29:3107–3121. doi:10.1093/hmg/ddaa183
198. Chen X, Xie C, Tian W, et al. Parkinson’s disease-related Leucine-rich repeat kinase 2 modulates nuclear morphology and genomic stability in striatal projection neurons during aging. Mol Neurodegener. 2020;15:12. doi:10.1186/s13024-020-00360-0
199. Musgrove RE, Helwig M, Bae E-J, et al. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular α-synuclein transfer. J Clin Invest. 2019;129:3738–3753. doi:10.1172/jci127330
200. Pan Y, Zong Q, Li G, et al. Nuclear localization of alpha-synuclein affects the cognitive and motor behavior of mice by inducing DNA damage and abnormal cell cycle of hippocampal neurons. Front Mol Neurosci. 2022;15:1015881. doi:10.3389/fnmol.2022.1015881
201. Sugeno N, Hasegawa T. Unraveling the complex interplay between alpha-synuclein and epigenetic modification. Int J Mol Sci. 2023;24:6645. doi:10.3390/ijms24076645
202. Murali Mahadevan H, Hashemiaghdam A, Ashrafi G, Harbauer AB. Mitochondria in neuronal health: from energy metabolism to parkinson’s disease. Adv Biol. 2021;5:e2100663. doi:10.1002/adbi.202100663
203. Labbé C, Lorenzo-Betancor O, Ross OA. Epigenetic regulation in Parkinson’s disease. Acta Neuropathol. 2016;132:515–530. doi:10.1007/s00401-016-1590-9
204. Sepe S, Milanese C, Gabriels S, et al. Inefficient DNA repair is an aging-related modifier of parkinson’s disease. Cell Rep. 2016;15:1866–1875. doi:10.1016/j.celrep.2016.04.071
205. Remaut K, Oorschot V, Braeckmans K, Klumperman J, De Smedt SC. Lysosomal capturing of cytoplasmic injected nanoparticles by autophagy: an additional barrier to non viral gene delivery. J Control Release. 2014;195:29–36. doi:10.1016/j.jconrel.2014.08.002
206. Li Z, Zou J, Chen X. In response to precision medicine: current subcellular targeting strategies for cancer therapy. Adv Mater. 2023;35:e2209529. doi:10.1002/adma.202209529
207. Tammam SN, Azzazy HM, Breitinger HG, Lamprecht A. Chitosan nanoparticles for nuclear targeting: the effect of nanoparticle size and nuclear localization sequence density. Mol Pharmaceut. 2015;12:4277–4289. doi:10.1021/acs.molpharmaceut.5b00478
208. Goswami R, Gupta A, Bednova O, et al. Nuclear localization signal-tagged systems: relevant nuclear import principles in the context of current therapeutic design. Chem Soc Rev. 2024;53:204–226. doi:10.1039/d1cs00269d
209. Nie Y, Fu G, Leng Y. Nuclear delivery of nanoparticle-based drug delivery systems by nuclear localization signals. Cells. 2023;12:1637. doi:10.3390/cells12121637
210. Krpetić Z, Saleemi S, Prior IA, et al. Negotiation of intracellular membrane barriers by TAT-modified gold nanoparticles. ACS Nano. 2011;5:5195–5201. doi:10.1021/nn201369k
211. Rao KS, Reddy MK, Horning JL, Labhasetwar V. TAT-conjugated nanoparticles for the CNS delivery of anti-HIV drugs. Biomaterials. 2008;29:4429–4438. doi:10.1016/j.biomaterials.2008.08.004
212. Moku G, Layek B, Trautman L, et al. Improving payload capacity and anti-tumor efficacy of mesenchymal stem cells using TAT peptide functionalized polymeric nanoparticles. Cancers. 2019;11:491. doi:10.3390/cancers11040491
213. Drescher D, Büchner T, Schrade P, et al. Influence of nuclear localization sequences on the intracellular fate of gold nanoparticles. ACS Nano. 2021;15:14838–14849. doi:10.1021/acsnano.1c04925
214. Feja M, Drath I, Weiß S, et al. Nose-to-brain siRNA delivery by PEI/PPI-based nanoparticles reduces α-synuclein expression in a Parkinson’s disease mouse model. Mol Ther Nucleic Acids. 2025;36:102671. doi:10.1016/j.omtn.2025.102671
215. Katamesh AA, Abdel-Bar HM, Break MKB, et al. Tailored intranasal albumin caged selegiline-α synuclein siRNA liposome with improved efficiency in parkinson’s model. Pharmaceutics. 2025;17:243. doi:10.3390/pharmaceutics17020243
216. Bellefroid C, Lechanteur A, Evrard B, Piel G. Lipid gene nanocarriers for the treatment of skin diseases: current state-of-the-art. Eur J Pharm Biopharm. 2019;137:95–111. doi:10.1016/j.ejpb.2019.02.012
217. Kwatra M, Kwak G, Li H, Suk JS, Ko HS. Polymeric nanoparticle-mediated GBA1 gene therapy is neuroprotective in a preclinical model of Parkinson’s disease. Drug Delivery Transl Res. 2025;16:894–910. doi:10.1007/s13346-025-01944-3
218. Aly AE, Sun T, Zhang Y, et al. Focused ultrasound enhances transgene expression of intranasal hGDNF DNA nanoparticles in the sonicated brain regions. J Control Release. 2023;358:498–509. doi:10.1016/j.jconrel.2023.04.041
219. Moosavi SG, Rahiman N, Jaafari MR, Arabi L. Lipid nanoparticle (LNP) mediated mRNA delivery in neurodegenerative diseases. J Control Release. 2025;381:113641. doi:10.1016/j.jconrel.2025.113641
220. Joyce P, Allen CJ, Alonso MJ, et al. A translational framework to DELIVER nanomedicines to the clinic. Nature Nanotechnol. 2024;19:1597–1611. doi:10.1038/s41565-024-01754-7
221. Ermini F, Low VF, Song JJ, et al. Ultrastructural localization of Porphyromonas gingivalis gingipains in the substantia nigra of Parkinson’s disease brains. NPJ Parkinson’s Dis. 2024;10:90. doi:10.1038/s41531-024-00705-2
222. Kretz R, Walter L, Raab N, et al. Spatial proteomics reveals differences in the cellular architecture of antibody-producing CHO and plasma cell-derived cells. Mol Cell Proteomics. 2022;21:100278. doi:10.1016/j.mcpro.2022.100278
223. Villegas-Hernández LE, Dubey V, Nystad M, et al. Chip-based multimodal super-resolution microscopy for histological investigations of cryopreserved tissue sections. Light Sci Appl. 2022;11:43. doi:10.1038/s41377-022-00731-w
224. Rafati N, Zarepour A, Bigham A, et al. Nanosystems for targeted drug Delivery: innovations and challenges in overcoming the Blood-Brain barrier for neurodegenerative disease and cancer therapy. Int J Pharm. 2024;666:124800. doi:10.1016/j.ijpharm.2024.124800
225. De Jong WH, Geertsma RE, Borchard G. Regulatory safety evaluation of nanomedical products: key issues to refine. Drug Delivery Transl Res. 2022;12:2042–2047. doi:10.1007/s13346-022-01208-4
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
How Nanotherapeutic Platforms Play a Key Role in Glioma? A Comprehensive Review of Literature

Yang Y, Cheng N, Luo Q, Shao N, Ma X, Chen J, Luo L, Xiao Z
International Journal of Nanomedicine 2023, 18:3663-3694
Published Date: 3 July 2023
Multiomic Profiling and Neuroprotective Bioactivity of Salvia Hairy Root-Derived Extracellular Vesicles in a Cellular Model of Parkinson’s Disease

Vestuto V, Conte M, Vietri M, Mensitieri F, Santoro V, Di Muro A, Alfieri M, Moros M, Miranda MR, Amante C, Delli Carri M, Campiglia P, Dal Piaz F, Del Gaudio P, De Tommasi N, Leone A, Moltedo O, Pepe G, Cappetta E, Ambrosone A
International Journal of Nanomedicine 2024, 19:9373-9393
Published Date: 11 September 2024
How Do Organelle-Targeting Nanotherapeutics Treat Inflammatory Diseases? A Comprehensive Review of the Literature
Wang SH, Xu XL, Chen W
International Journal of Nanomedicine 2025, 20:7133-7152
Published Date: 3 June 2025