")
Back to Journals » Cancer Management and Research » Volume 13
Oral Tongue Cancer in a Patient with Fanconi Anemia: A Case Report and Literature Review
Authors Deng S, Ye W , Zhang S, Zhu G, Zhang P, Song Y , Duan F, Lang J , Lu S
Received 12 January 2021
Accepted for publication 17 March 2021
Published 12 April 2021 Volume 2021:13 Pages 3145—3154
DOI https://doi.org/10.2147/CMAR.S301582
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Siyao Deng,1 Wenjing Ye,1 Shichuan Zhang,2,3 Guiquan Zhu,4 Peng Zhang,2 Yanqiong Song,2 Fanglei Duan,3 Jinyi Lang,2,3 Shun Lu2,3
1School of Medicine, University of Electronic Science and Technology of China, Chengdu, People’s Republic of China; 2Department of Radiation Oncology, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Chengdu, People’s Republic of China; 3Radiation Oncology Key Laboratory of Sichuan Province, Chengdu, People’s Republic of China; 4State Key Laboratory of Oral Diseases, National Clinical Research Centre for Oral Diseases, Department of Head and Neck Oncology, West China Hospital of Stomatology, Sichuan University, Chengdu, People’s Republic of China
Correspondence: Shun Lu
Department of Radiation Oncology, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Renmin South Road 55#, Chengdu, 610041, People’s Republic of China
Tel +86 28 85420897
Fax +86 28 85420110
Email [email protected]
Purpose: Fanconi anemia (FA) is a rare genetic disorder characterized by congenital anomalies, progressive bone marrow failure and high susceptibility to solid tumors, especially head and neck squamous cell carcinoma (HNSCC). Management of FA patients with head and neck cancer is a challenge due to increased risk of surgery, poor tolerance of chemotherapy, and severe myelotoxicity of radiotherapy.
Patients and Methods: We present a case of a 33-year-old man with carcinoma of oral tongue (T1N2M0), who experienced prolonged and profound bone marrow failure as a consequence of concurrent cisplatin/radiation. The young patient who developed HNSCC without risk factors, the myelotoxicity after exposure to platinum-based agent cisplatin and the further evaluation of phenotypic characteristics raised suspicion of FA. Whole exome sequencing performed for the patient and parents ultimately established the diagnosis of FA.
Results: Genetic testing in 23 FANC genes revealed two novel heterozygous mutations, c.367C>T and c.3971_3972delCGinsTT in FANCA gene of the patient, which were inherited from his father and mother, respectively. Radiotherapy with reduced dose has successfully alleviated the symptoms of tumor invasion and progression, and the radiation-related side effects were acceptable. Unfortunately, the patient eventually died of locoregional disease progression.
Conclusion: This case highlights the importance of considering the diagnosis of FA in young patients who develop HNSCC in the absence of risk factors, thus permitting more effective oncological treatment strategies and improved outcomes. In conclusion, any decision on different modalities of management in such patients should be based on a balance between locoregional control and therapeutic toxicity.
Keywords: fanconi anemia, radiotherapy, cisplatin, head and neck squamous cell carcinoma, toxicity
Introduction
Fanconi anemia (FA) is a multisystem disorder characterized by congenital abnormalities, progressive bone marrow failure and pancytopenia, high susceptibility to acute myeloid leukemia and solid tumors.1 In FA patients, hematological abnormalities usually appear within the first decade of life, which eventually develop pancytopenia and even bone marrow failure.2 Congenital anomalies include skeletal malformations (short stature, microcephaly, or hypoplastic thumb), organ abnormalities (renal, ophthalmic, ear, cardiac, and genital), abnormal skin pigmentation like café au lait spots, and intellectual disability.3 These further prove that FA is a pleiotropic disease on the phenotype.
At least 23 genes have been implicated in FA that are responsible for the various complementation groups (A-C, D1, D2, E-G, I, J, L-W, Y). Mutations in FANCA are the most common, involved in 60–65% of reported cases.4 All mutations in FA genes are inherited in an autosomal recessive manner, with the exception of FANCB and RAD51, which are inherited in an X-linked manner and autosomal dominant manner, respectively.5 Products of these FA genes appear to interact in a common cellular pathway and participate in various aspects of DNA repair, particularly DNA interstrand crosslink repair, genomic stabilization, and regulation of downstream proteins.6,7 Eight of the FA proteins (FANCA/−B/−C/−E/−F/−G/−L/−M) are constitutively assembled in a multisubunit enzyme core complex to mediate the monoubiquitination of FANCD2 and FANCI after DNA damage, which is a central step in the FA pathway, as a response to cross-linking agents or reactive oxygen species.8–10 Any mutation or loss of the FA core complex components will result in failure of monoubiquitination. In addition, there is interaction with BRCA1/BRCA2 complexes and an array of proteins involved in cell cycle checkpoint regulation and DNA damage repair.11,12 Defects in any of these FA proteins result in genomic instability, defective DNA repair mechanisms and an increased risk of cancer.13
Although the risk of hematological abnormalities is higher in childhood, solid tumors are more common in adults.14 Interestingly, FA patients have a significantly higher risk of head and neck cancer, and most of them occur in the oral cavity, in which tongue carcinomas are the most common subsite.1 Compared with the general population, the risk of head and neck cancer in FA patients is increased by 500 to 700-fold, and the cumulative incidence of solid tumors was close to 20% by the age of 65 years.15–17 FA patients with hematopoietic dysfunction usually require supportive treatment such as blood transfusions, hematopoietic growth factor and androgen replacement therapy. Bone marrow transplantation is the preferred treatment option, but is associated with a high risk of the late development of FA-related head and neck cancer.18–20 The management of FA patients with head and neck cancer requires multidisciplinary coordination and is extremely challenging. First, potential pancytopenia results in increased risk of surgery and poor tolerance to nonsurgical treatment. Second, defective DNA repair mechanism and chromosomal fragility increase the sensitivity to cytotoxic chemotherapeutic agents such as cisplatin, typically used in head and neck cancer.21 Third, defective recognition of DNA damage and homologous recombination repair disorders may induce abnormal toxicity of radiotherapy.22
Here, we reported a new case of a young man with FA treated at our institution for the tongue squamous cell carcinoma, reviewed his clinical course and described the treatment-related toxicity. This study was in line with the Declaration of Helsinki and was approved by the Ethics Committee of Sichuan Cancer Hospital & Institute. The patient’s parents have provided the written informed consent for this article.
Patients and Methods
The Patient and Initial Treatment
A 33-year-old man without history of drinking and smoking was admitted to our hospital for examination of a lump on the right side of the tongue dorsum. Oral examination revealed a borderline ulcerative hyperplasia at the right side of tongue dorsum with white plaque covering it. No superficial lymph node enlargement was noted on neck examination. Computer tomography (CT) scan showed enhanced signals in the right tongue dorsal lesion and multiple bilateral cervical lymph nodes without any distant metastatic spread. Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) of the maxillofacial region revealed a 1.6 X 1.4 X 1.7 cm mass located at the right side of tongue dorsum, with significant cervical lymphadenopathy (Figure 1A). A pathological biopsy performed under local anesthesia demonstrated highly differentiated squamous cell carcinoma (Figure 2A and B). As a result, a diagnosis of T2N2M0 (stage III) squamous cell carcinoma of the oral tongue was made.
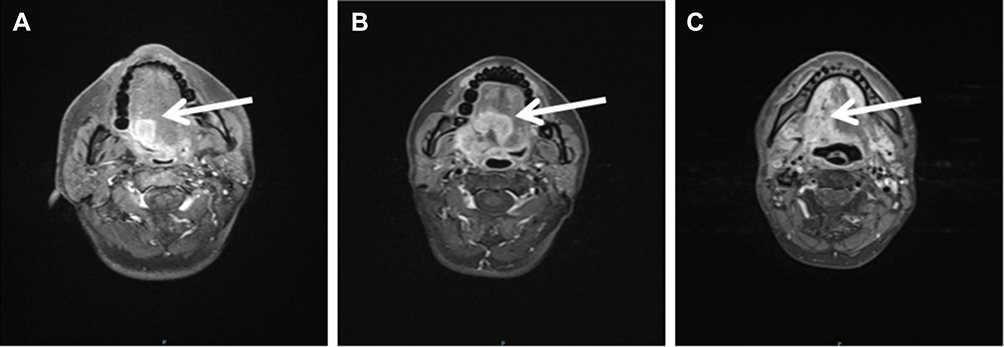 |
Figure 1 DCE-MRI of the maxillofacial region showed the mass located at the right side of tongue dorsum. (A) DCE-MRI before the treatment with concurrent cisplatin/radiation, the tumor size measured 1.6 X 1.4 X 1.7 cm. (B) DCE-MRI during the 8 months of bone marrow recovery therapy with cessation of concurrent cisplatin/radiation, the tumor size had increased to 3.7 X 3.0 X 3.1 cm. (C) DCE-MRI after completion of 21 fractions of reduced-dose radiotherapy, the tumor size had decreased to 3.2 X 2.3 X 2.8 cm. The white arrows indicated the location of the tumor. DCE-MRI, dynamic contrast-enhanced magnetic resonance imaging. |
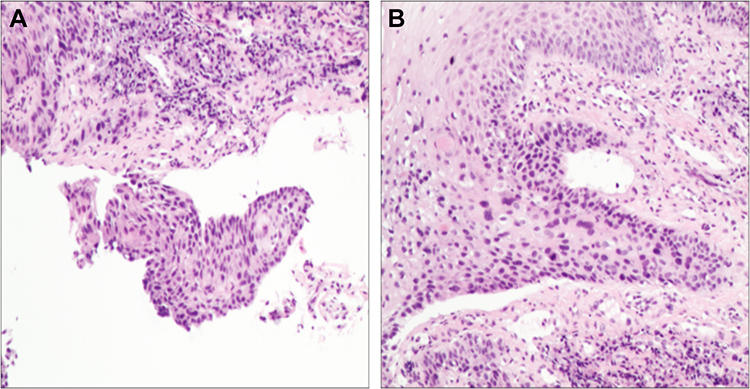 |
Figure 2 A pathological biopsy demonstrated highly differentiated squamous cell carcinoma. (A) Hematoxylin-eosin, original magnification 40X. (B) Hematoxylin-eosin, original magnification 200X. |
Initial hematological parameters showed features of mild pancytopenia, which ruled out aggressive surgical interventions. After receiving supportive care until the blood count returned to normal, the patient was treated with concurrent cisplatin/radiation, the standard of care for inoperable locally advanced HNSCC, according to the National Comprehensive Cancer Network (NCCN) guidelines. He received 13 days of single-dose cisplatin therapy (40 mg/m2, d1-d3) combined with 11 fractions of daily intensity-modulated radiation therapy (IMRT). The prescribed doses of each target area were as follows: 2.2Gy/f for the primary tumor determined by DCE-MRI (gross tumor volume, GTV), 2.2Gy/f for gross tumor volume of lymph nodes (GTVln), 2.0Gy/f for clinical target volume (CTV) which extended 1 cm from GTV and included the whole tongue, root of tongue, mouth floor and condylar tongue muscle, 1.8Gy/f for clinical target volume of lymph nodes (CTVln) which included I, II, III, IV and VA areas on both sides, 5 fractions/week. However, the treatment discontinued due to the pancytopenia (total leukocyte count: 2.02 × 109/L; hemoglobin count: 72 g/L; platelet count: 87 × 109/L). Additionally, he continued to have profound and persistent myelosuppression despite blood product support and daily administration of subcutaneous recombinant granulocyte-colony-stimulating factor.
Phenotypic Features and Family History
To determine the cause of the patient’s continued pancytopenia, he underwent a bone marrow aspiration and biopsy which revealed a severely hypoplastic marrow with no granuloma, hematopoiesis, nor malignant cells. Given the development of HNSCC at a young age in the absence of risk factors, the development of profound and persistent bone marrow failure induced by the low dose of cisplatin, the possibility of a genetic DNA repair defect was proposed. Through a comprehensive evaluation of phenotypic characteristics, several physiological characteristics of FA were determined. The patient exhibited short stature and microcephaly. In addition, he also exhibited generalized hyperpigmentation of the skin and a thumb malformation of the right hand, accompanied by a few café au lait spots, particularly of the upper extremities (Figure 3A and B). The patient was born to unrelated parents and had a younger brother of normal stature. Besides, his parents displayed no phenotypic signs of hereditary syndromes.
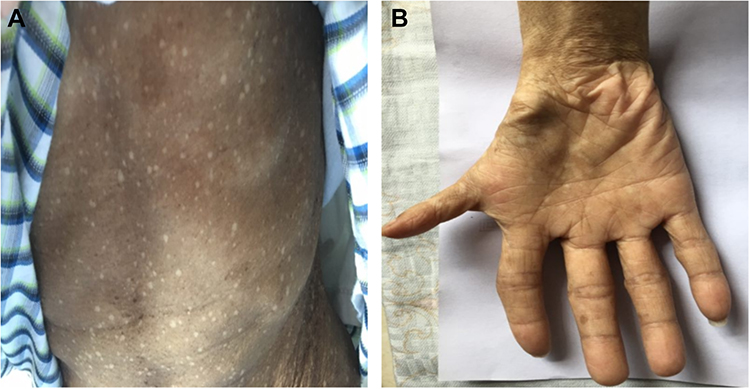 |
Figure 3 (A and B) Physical examination showed generalized hyperpigmentation of the skin and a thumb malformation of the right hand, accompanied by a few café au lait spots, particularly of the upper extremities. |
Genetic Testing
In consideration of his abnormal morphological features and unexplained hypoplastic marrow, the patient was advised for molecular genetic testing. After obtaining the informed consent of the family, peripheral blood was collected from proband and the parents for whole exome sequencing. Apart from the mutation analysis of the patient, targeted mutational analysis was performed on the parents to determine if they also had the mutations identified in the proband. As a result, the molecular study revealed compound heterozygous mutations in the FANCA gene of the proband. One mutation was a previously unreported heterozygous nonsense mutation, c.367C>T, which caused the protein encoded by FANCA gene to terminate prematurely at the 123rd amino acid. Another one was a novel heterozygous mutation, c.3971_3972delCGinsTT, caused by a combination of c.3971C>T and c.3972G>T linkage mutations (Figure 4A and B). The two heterozygous mutations were inherited from his father and mother, respectively (Figure 5). The variant was queried against the public version of the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/). Finally, clinical diagnosis of FA was made on the basis of clinical features, bone marrow biopsy and genetic testing analysis.
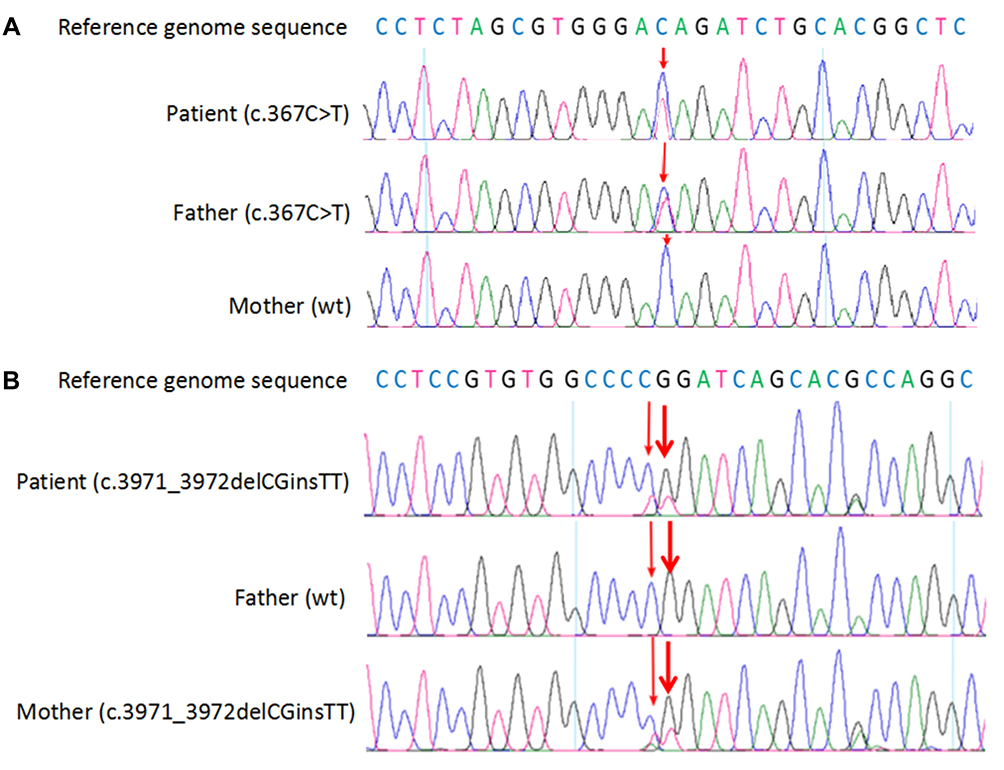 |
Figure 4 Fanconi anemia complementation group A gene sequence diagram of the patient. (A) c.367C>T was detected in the patient and his father. (B) c.3971_3972delCGinsTT was detected in the patient and his mother. Variants were indicated with arrows; wt, wild type. |
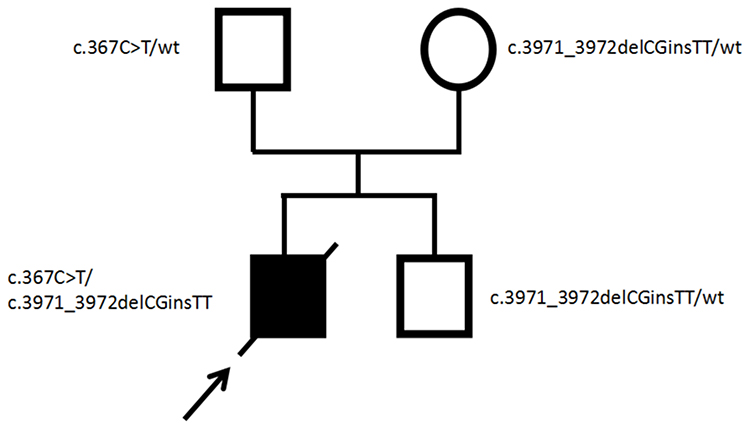 |
Figure 5 Pedigree of the family. Filled and open symbols denoted affected and healthy individuals, respectively. An arrow indicated the index patient; diagonal line indicated deceased status. The mutation status was shown next to each symbol; wt, wild type. |
Results
In consideration of the pancytopenia and substantially increased risk of toxicity with chemotherapy, especially DNA cross-linking agents like cisplatin, thus immediate discontinuation of cisplatin was determined. However, during the 8 months of bone marrow recovery therapy with cessation of concurrent cisplatin/radiation, DCE-MRI showed the tumor had increased in size to 3.7 X 3.0 X 3.1 cm (Figure 1B). Given his aggressive disease, the risk of locoregional failure and symptomatic progression was considered to outweigh the risk of treatment-related adverse event. After multidisciplinary diagnosis and treatment with experts of hematology, radiation oncology and oncology internal medicine, we modified the IMRT plan to reduce radiation toxicity. The revised doses of each target area were as follows: 2.0Gy*30f for GTV, 2.0Gy*30f for cross tumor volume of left lymph nodes (GTVlnL), 2.0Gy*30f for cross tumor volume of right lymph nodes (GTVlnR), 1.8Gy*30f for CTV. After improvement of blood cells with the intensive support care, the patient completed 21 fractions of IMRT and the tumor size had decreased to 3.2 X 2.3 X 2.8 cm (Figure 1C). Since there was an 8-month interruption between the two radiotherapy, we treated the patient with re-irradiation for the second process radiotherapy and planned 60 Gy for this course. However, canker sores occurred when the patient received 36 Gy radiation, and it aggravated with the increase of dose. The severity of mucositis was evaluated as grade III according to Radiation Therapy Oncology Group (RTOG V4.3) when the patient received 42 Gy radiation. Given the decreased lesion size and relief of tumor invasion, he was considered not suitable to finish the remaining doses of radiotherapy in order to reduce the radiation-related toxicity. Unfortunately, he deteriorated progressively after discharge and died from disease progression after 5 months of follow-up.
Discussion
FA is a disease in which underlying human genome instability plays an important role. With a rare incidence of one to five cases per million persons, heterozygous carrier frequency of FA is one in 300 persons.23 In certain populations, such as Ashkenazi Jews and Spanish Romani, the carrier frequency is believed to be as high as 1/100.24,25 Cases of FA are usually diagnosed through recognition of congenital and hematological abnormalities, with a median age of diagnosis at 4.8–7.5 years;26,27 therefore, it is rarely considered for diagnosis in adults.
FA patients are at high risk for the development of hematological malignancies and various solid tumors especially HNSCC. Onset of tumors in patients with FA is as early as 16 to 31 and shows a more aggressive behavior. Interestingly, 22% of FA patients who develop solid tumors are diagnosed with FA only after discovery of their cancer.28 In this case, the diagnosis of FA was atypical, presenting in an adult, and made only after unexpected toxicity of DNA cross-linking agents in an initially assumed sporadic malignancy. Therefore, FA should be considered in early-onset malignancies, especially if a typical FA-related malignancy is diagnosed. In addition, severe hematological abnormalities of the patient did not appear until initiation of systemic tumor therapy. In some FA patients, genomic duplication is reversed to wild-type alleles in hematopoietic tissues, which can induce spontaneous correction of hematological alterations. In non-hematopoietic tissue, the biallelic inactivation stills remains, and thus the patient harbors mosaicism for the mutated FA gene.29 This phenomenon may explain a milder hematological phenotype for this patient with FA. Notably, FA is a heterogeneous disease and 30% of FA patients can present without any congenital abnormalities.3,30 Thus, lack of physical manifestations does not preclude a diagnosis of FA.
Standard treatments for non-FA HNSCC recommended by NCCN guidelines include surgery, radiotherapy and chemotherapeutic agents such as cisplatin. However, FA patients diagnosed with HNSCC have limited options for treatment because of low tolerance to adverse effects from highly toxic radiation and chemotherapy, leaving surgery as the preferred primary therapy.31,32 Most commonly, radical surgical excision with negative margins is required. Extensive radical surgery is sometimes needed to remove advanced cancer, and microvascular free flap reconstruction may be required.15 Previous studies have shown that FA patient can tolerate surgeries with free flap reconstruction without significant morbidity or mortality; therefore, such large complex surgeries should be carefully considered before developing a treatment plan.33,34 Moreover, there are potentially increased perioperative risks. Surgeons must be aware of potential need for blood or platelet transfusion both preoperatively and postoperatively, and the increased susceptibility to postoperative complications such as hematomas, poor wound healing, infections and aspiration pneumonia.15 In a study of HNSCC associated with FA, postoperative complications occurred in 7 of 30 patients (23%), but most patients tolerated surgeries well and did not develop severe bone marrow failure during the postoperative hospitalization.35 Although the surgical removal of tumor is currently the optimal option, this is usually not curative for FA patients who are diagnosed with secondary tumors or highly aggressive tumors that are difficult to remove. In addition, even after successful surgeries with clear margins, rapid local recurrence is common in FA patients. In our case, given the pancytopenia of his disease, it was not felt that he was an appropriate candidate for surgical resection of the tumor. Despite the improvement of blood counts with supportive care, the hematological parameters remained unstable Considering the great intraoperative risk, after discussion with the patient and his family, the decision was made for concurrent radio-chemotherapy to reduce the tumor size. In general, appropriately aggressive oncologic surgery should be considered in FA patients, and special considerations regarding inadequate management with surgical resection alone for locally advanced disease should also be a part of the normal therapy evaluation of FA patients.
Chemotherapy is one treatment option of most patients presenting with recurrent/metastatic HNSCC. It also can be used as a radiation sensitizer. Cisplatin is a DNA cross-linking agent recommended to be used in patients with oral cancer. However, people with FA have a substantially increased risk of toxicity to this kind of agent. It has been reported that one patient developed high-grade mucositis, cytopenia, tracheal stenosis, radiation pneumonitis, recurrent pneumonia, persistent myelosuppression and hemorrhage after receiving radiotherapy with cisplatin, bleomycin and methotrexate.35 In the present patient, the cisplatin chemotherapeutic regimen was given before the diagnosis of FA. Severe and persistent bone marrow failure was observed and the treatment plan was interrupted, which most likely resulted from the chemotherapy. Although concurrent radio-chemotherapy is recommended to treat inoperable oral cancer patients, patients with FA may experience potentially fatal bone marrow toxicity from concurrent radio-chemotherapy; therefore, cytotoxic chemotherapy is discouraged for these patients. Most importantly, timely identification of underlying FA would lead to a different therapeutic approach with a potentially better outcome.
However, FA patients with HNSCC tend to have a poor prognosis, which is associated with the aggressive disease presentation and the limitations of multimodality therapy due to potential bone marrow failure.36 Given the bone marrow abnormalities and low tolerance to standard therapies, treatment is more challenging in these patients. Radiotherapy has been served as a definitive treatment in many FA patients with HNSCC given the significant but acceptable toxicity. For instance, 70 Gy was recommended for the definitive initial treatment in a previous study.37 However, these patients have a significantly increased complication rate compared with the general HNSCC population, sometimes patients with FA even cannot complete full course of the treatment.38 Based on the International Fanconi Anemia Registry (IFAR) series, radiation-related side effects including anemia, thrombocytopenia, myelosuppression, skin ulceration and stenosis of the trachea were observed in the 8 FA patients treated with 40–61Gy radiotherapy.15 Previous studies have reported that doses of radiotherapy ranged from 3.2 to 80 Gy and the acute hematologic toxicity was individualized. Severe radiation-related toxicity could occur in a low dose such as 8 Gy in some FA patient, while the higher dose was acceptable in others.39–41 In this case, the patient was initially treated with 24.2 Gy IMRT in 11 fractions. After 8 months interruption of treatment, standard chemotherapy agent was not considered after diagnosis of FA in the patient due to the risk of unacceptable myelosuppression and irreversible aplastic anemia. IMRT of 42Gy/21f was then treated to the progressed tumor. From the DCE-MRI evaluation, radiotherapy has successfully alleviated the symptoms of tumor invasion and progression, and the radiation-related side effects were acceptable Combined with the above evidence, a precise dose of radiotherapy with careful toxicity assessment is needed for each patient to enhance the treatment, since other therapeutic options are not available because of the high risk.
For FA patients with HNSCC, longer duration treatment courses with lower dose per fraction of radiotherapy may maximize the therapeutic effect and reduce complications. Furthermore, in view of the adaptability of dose and target volume, application of the technique allowing the best sparing of healthy tissues, such as IMRT with image-guided radiotherapy and intensity-modulated proton therapy, may be a safer option.42 Overall, any decision on different modalities of management in such patients should be based on a balance between locoregional control and therapeutic toxicity.
Tremendous strides have been made recently for cancer treatment in non-FA cancers. Currently, drugs approved by Food and Drug Administration (FDA) for the treatment of HNSCC in the general population include conventional cytotoxic agents and antibodies such as cetuximab and gefitinib, which target the epidermal growth factor receptor (EGFR), and nivolumab and pembrolizumab, which target the immune checkpoint protein. Cetuximab is so far the only targeted therapy known to demonstrate an overall survival (OS) benefit in patients with recurrent and/or metastatic HNSCC, whether in combination with radiotherapy or chemotherapy.43,44 A previous case report provided detailed evidence that the patient treated with postoperative radiotherapy with concurrent cetuximab.36 Although the patient developed grade 3 dermatitis and mucositis during treatment, he was able to finish the prescribed 70.2 Gy without interruption. After an initial dose of 400 mg/m2 of cetuximab, he developed neutropenia, prompting a subsequent dose reduction to 200 mg/m2. Another study reported on three patients who received targeted chemotherapy with cetuximab in addition to adjuvant or neoadjuvant radiotherapy. Of these three patients treated with cetuximab, only one patient experienced complications.35 These evidences above suggest that adjuvant radiation with concurrent cetuximab is feasible with careful monitoring.
Currently, several large Phase III clinical trials have evaluated the efficacy of tyrosine kinase inhibitors (TKIs) in patients with advanced HNSCC, such as gefitinib and afatinib (alone or in combination with docetaxel). As a result, in a recurrent and/or metastatic setting, gefitinib and afatinib failed to improve OS when compared with standard treatments.45–47 However, a previous study reported that a FA patient with advanced tongue cancer was treated with gefitinib 250 mg/day for palliation. Two months later, the lesion size decreased (from 2.6 X 3.6 cm to 1.5 X 1.3 cm) and no skin rash or diarrhea developed during the treatment.48 In addition, Montanuy et al recently present a complete preclinical evidence that gefitinib and afatinib both inhibited tumor growth in xenograft experiments in immunodeficient mice using two fanconi anemia patient-derived HNSCC and displayed manageable side effects.49 Taken together, gefitinib appears to be a safe and effective therapeutic modality without severe toxicity and may be applicable to FA patients with advanced HNSCC, but more research needs to be done in the future to validate its potential use.
Immunotherapy for HNSCC such as the application of immune checkpoint inhibitors (ICIs), is considered as a nonnegligible treatment in recent years, focusing on mobilizing and strengthening the anti-tumor ability of human immune system. However, the immunotherapeutic potential of FA patients with HNSCC is currently unknown. Factors to be considered include an understanding of the mutational burden in HNSCC associated with FA, the role of bone marrow transplantation, the presence of acute and chronic graft-versus-host disease (GvHD) and the adverse effects of ICIs.50 In our case, the patient was newly diagnosed with tongue cancer. Nevertheless, based on recent results of clinical trials, ICIs are recommended to treat recurrent or metastatic HNSCC patients.51,52 Since other treatment options were limited by low tolerance to adverse effects, ICIs were recommended for this patient, but the patient refused because of economic reasons.
Conclusion
In conclusion, the patient with a classically FA-associated early-onset cancer in this case can serve as a reminder to consider the diagnosis of FA in young patients who develop HNSCC in the absence of risk factors. Clinicians involved in the care of such patients should conduct an in-depth investigation of the patients’ family history and accurate genetic testing, in order to timely diagnosis with FA to permit more effective oncological treatment strategies and improved outcomes. Moreover, for the treatment of inoperable FA patients with local HNSCC, chemotherapy is not suggested due to the fatal myelotoxicity, while reduced dose of radiotherapy on the tumor may be associated with a better survival.
Abbreviations
FA, fanconi anemia; HNSCC, head and neck squamous cell carcinoma; CT, computer tomography; DCE-MRI, dynamic contrast-enhanced magnetic resonance imaging; NCCN, National Comprehensive Cancer Network; IMRT, intensity modulated radiation therapy; GTV, gross tumor volume; GTVln, gross tumor volume of lymph nodes; CTV, clinical target volume; CTVln, clinical target volume of lymph nodes; GTVlnL, gross tumor volume of left lymph nodes; GTVlnR, gross tumor volume of right lymph nodes; RTOG, Radiation Therapy Oncology Group; IFAR, International Fanconi Anemia Registry; FDA, Food and Drug Administration; EGFR, epidermal growth factor receptor; OS, overall survival; TKIs, tyrosine kinase inhibitors; ICIs, immune checkpoint inhibitors; GvHD, graft-versus-host disease.
Ethics Approval and Informed Consent
This study was in line with the Declaration of Helsinki and was approved by the Ethics Committee of Sichuan Cancer Hospital & Institute.
Consent for Publication
The patient’s parents have provided the written informed consent for this article.
Acknowledgments
We thank all of our colleagues who have contributed to this study.
Author Contributions
Genetic testing: DS, YW and DF; Writing-original draft: DS and YW; Management of the patient: ZS, ZG, ZP and SY; Resources and Funding acquisition: LJ and LS; Writing-review & editing: DS and LS. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Department of Science and Technology of Sichuan Province [20YYJC3815, 20GJHZ0088].
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bhandari J, Thada PK, Puckett Y. Fanconi anemia. In: StatPearls. Treasure Island (FL): StatPearls Publishing LLC; 2020.
2. Furlong E, Carter T. Aplastic anaemia: current concepts in diagnosis and management. J Paediatr Child Health. 2020;56(7):1023–1028.
3. Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood. 2003;101(4):1249–1256.
4. Solomon PJ, Margaret P, Rajendran R, et al. A case report and literature review of Fanconi Anemia (FA) diagnosed by genetic testing. Ital J Pediatr. 2015;41:38.
5. Meetei AR, Levitus M, Xue Y, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004;36(11):1219–1224.
6. Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17(6):337–349.
7. Bhattacharjee S, Nandi S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun Signal. 2017;15(1):41.
8. Cheung RS, Taniguchi T. Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int J Hematol. 2017;106(3):335–344.
9. Sims AE, Spiteri E, Sims RJ
10. Smogorzewska A, Matsuoka S, Vinciguerra P, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129(2):289–301.
11. D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362(20):1909–1919.
12. Medhurst AL, El Laghmani H, Steltenpool Jet al. Evidence for subcomplexes in the Fanconi anemia pathway. Blood. 2006;108(6):2072–2080.
13. Sumpter R
14. Scheckenbach K, Wagenmann M, Freund M, Schipper J, Hanenberg H. Squamous cell carcinomas of the head and neck in Fanconi anemia: risk, prevention, therapy, and the need for guidelines. Klin Padiatr. 2012;224(3):132–138.
15. Kutler DI, Auerbach AD, Satagopan J, et al. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg. 2003;129(1):106–112.
16. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica. 2018;103(1):30–39.
17. Rosenberg PS, Alter BP, Ebell W. Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica. 2008;93(4):511–517.
18. Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol. 2014;27(3–4):214–221.
19. Salum FG, Martins GB, de Figueiredo MA, Cherubini K, Yurgel LS, Torres-Pereira C. Squamous cell carcinoma of the tongue after bone marrow transplantation in a patient with Fanconi anemia. Braz Dent J. 2006;17(2):161–165.
20. Guardiola P, Socié G, Li X, et al. Acute graft-versus-host disease in patients with Fanconi anemia or acquired aplastic anemia undergoing bone marrow transplantation from HLA-identical sibling donors: risk factors and influence on outcome. Blood. 2004;103(1):73–77.
21. Spanier G, Pohl F, Giese T, Meier JK, Koelbl O, Reichert TE. Fatal course of tonsillar squamous cell carcinoma associated with Fanconi anaemia: a mini review. J Cranio-Maxillo Facial Surg. 2012;40(6):510–515.
22. Francies FZ, Wainwright R, Poole J, et al. Diagnosis of Fanconi Anaemia by ionising radiation- or mitomycin C-induced micronuclei. DNA Repair (Amst). 2018;61:17–24.
23. Rosenberg PS, Tamary H, Alter BP. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am J Med Genet A. 2011;155a(8):1877–1883.
24. Kutler DI, Auerbach AD. Fanconi anemia in Ashkenazi Jews. Fam Cancer. 2004;3(3–4):241–248.
25. Callén E, Casado JA, Tischkowitz MD, et al. A common founder mutation in FANCA underlies the world’s highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood. 2005;105(5):1946–1949.
26. Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003;101(3):822–826.
27. Risitano AM, Marotta S, Calzone R, Grimaldi F, Zatterale A. Twenty years of the Italian Fanconi Anemia Registry: where we stand and what remains to be learned. Haematologica. 2016;101(3):319–327.
28. Alter BP, Greene MH, Velazquez I, Rosenberg PS. Cancer in Fanconi anemia. Blood. 2003;101(5):2072.
29. Asur RS, Kimble DC, Lach FP, et al. Somatic mosaicism of an intragenic FANCB duplication in both fibroblast and peripheral blood cells observed in a Fanconi anemia patient leads to milder phenotype. Mol Genetics Genomic Med. 2018;6(1):77–91.
30. Fiesco-Roa MO, Giri N, McReynolds LJ, Best AF, Alter BP. Genotype-phenotype associations in Fanconi anemia: a literature review. Blood Rev. 2019;37:100589.
31. Best DL, Spresser W, Shivers P, Edwards SP, Ward BB. Squamous cell carcinoma of the tongue in young patients: a case series and literature review. J Oral Maxillofacial Surg. 2020.
32. Nolan M, Courtney R, Sexton P, Barry T, McCann PJ. Aggressive recurrence of oral squamous cell carcinoma in a patient with Fanconi’s Anaemia (FA). Ir Med J. 2017;110(3):533.
33. Chao JW, Cohen BD, Rohde CH, Kutler DI, Spector JA. Free fibular flap reconstruction of the mandible in a patient with Fanconi anemia. Plast Reconstr Surg. 2010;125(2):61e–63e.
34. Kaplan KA, Reiffel AJ, Kutler DI, Rohde CH, Spector JA. Sequential second free flap for head and neck reconstruction in a patient with fanconi anemia and metachronous squamous cell carcinoma. Plast Reconstr Surg. 2011;128(1):18e–19e.
35. Kutler DI, Patel KR, Auerbach AD, et al. Natural history and management of Fanconi anemia patients with head and neck cancer: a 10-year follow-up. The Laryngoscope. 2016;126(4):870–879.
36. Wong WM, Parvathaneni U, Jewell PD, et al. Squamous cell carcinoma of the oral tongue in a patient with Fanconi anemia treated with radiotherapy and concurrent cetuximab: a case report and review of the literature. Head Neck. 2013;35(10):E292–298.
37. Marur S, Forastiere AA. Head and neck squamous cell carcinoma: update on epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2016;91(3):386–396.
38. Birkeland AC, Auerbach AD, Sanborn E, et al. Postoperative clinical radiosensitivity in patients with fanconi anemia and head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2011;137(9):930–934.
39. Alter BP. Radiosensitivity in Fanconi’s anemia patients. Radiother Oncol. 2002;62(3):345–347.
40. Bremer M, Schindler D, Gross M, Dork T, Morlot S, Karstens JH. Fanconi’s anemia and clinical radiosensitivity report on two adult patients with locally advanced solid tumors treated by radiotherapy. Strahlenther Onkol. 2003;179(11):748–753.
41. Budrukkar A, Shahid T, Murthy V, et al. Squamous cell carcinoma of base of tongue in a patient with Fanconi’s anemia treated with radiation therapy: case report and review of literature. Head Neck. 2010;32(10):1422–1427.
42. Bernard ME, Kim H, Berhane H, et al. GS-nitroxide (JP4-039)-mediated radioprotection of human Fanconi anemia cell lines. Radiat Res. 2011;176(5):603–612.
43. Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–1127.
44. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567–578.
45. Argiris A, Ghebremichael M, Gilbert J, et al. Phase III randomized, placebo-controlled trial of docetaxel with or without gefitinib in recurrent or metastatic head and neck cancer: an eastern cooperative oncology group trial. J clin oncol. 2013;31(11):1405–1414.
46. Stewart JS, Cohen EE, Licitra L, et al. Phase III study of gefitinib compared with intravenous methotrexate for recurrent squamous cell carcinoma of the head and neck [corrected]. J clin oncol. 2009;27(11):1864–1871.
47. Machiels JP, Haddad RI, Fayette J, et al. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): an open-label, randomised Phase 3 trial. Lancet Oncol. 2015;16(5):583–594.
48. Jung HS, Byun GW, Lee KE, et al. Gefitinib trial in a fanconi’s anemia patient with multiple squamous cell carcinomas and hepatocellular carcinoma. Cancer Res Treatment. 2005;37(6):370–373.
49. Montanuy H, Martínez-Barriocanal Á, Antonio casado J, et al. Gefitinib and afatinib show potential efficacy for fanconi anemia-related head and neck cancer. Clin Cancer Res. 2020;26(12):3044–3057.
50. Ayas M, Siddiqui K, Al-Jefri A, et al. Successful outcome in patients with Fanconi Anemia undergoing T cell-replete mismatched related donor hematopoietic cell transplantation using reduced-dose cyclophosphamide post-transplantation. Biol Blood Marrow Transplant. 2019;25(11):2217–2221.
51. Ferris RL, Haddad R, Even C, et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open-label phase III study. Ann Oncol. 2020;31(7):942–950.
52. Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17(7):956–965.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.