Back to Journals » International Journal of Nanomedicine » Volume 21
Oral Microcapsules Encapsulating Endometrial Regenerative Cell-Derived Exosomes Promote Intestinal Epithelial Barrier Repair and Ameliorate Experimental Colitis
Authors Yang GM, Jiang HY, Ren SH ![]() , Xu YN, Wang HD, Shao B
, Xu YN, Wang HD, Shao B ![]() , Chen Q, Xiao YY, Sun CL, Liu X, Ren YF, Wang H
, Chen Q, Xiao YY, Sun CL, Liu X, Ren YF, Wang H ![]()
Received 29 May 2025
Accepted for publication 8 January 2026
Published 2 February 2026 Volume 2026:21 540230
DOI https://doi.org/10.2147/IJN.S540230
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Dong Wang
Guang-Mei Yang,1,2,* Hong-Yu Jiang,1,2,* Shao-Hua Ren,1– 3,* Yi-Ni Xu,1,2 Hong-Da Wang,1,2 Bo Shao,1,2 Qiang Chen,1,2 Yi-Yi Xiao,1,2 Cheng-Lu Sun,1,2 Xu Liu,1,2 Yu-Fan Ren,1,2 Hao Wang1,2,4
1Department of General Surgery, Tianjin Medical University General Hospital, Tianjin, People’s Republic of China; 2Tianjin General Surgery Institute, Tianjin, People’s Republic of China; 3Department of General Surgery, The Affiliated Hospital of Inner Mongolia Medical University, Hohhot, People’s Republic of China; 4Tianjin Key Laboratory of Precise Vascular Reconstruction and Organ Function Repair, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hao Wang, Department of General Surgery, Tianjin Medical University General Hospital, 154 Anshan Road, Heping District, Tianjin, 300052, People’s Republic of China, Tel +86-22-60362502, Email [email protected]; [email protected]
Background: Endometrial regenerative cell-derived exosomes (ERC-Exos) exhibit tissue repair and anti-inflammatory properties for ulcerative colitis (UC) treatment, but oral administration is limited by gastrointestinal degradation. This study first developed chitosan-sodium alginate microcapsules (CSE) for ERC-Exos delivery and explored the role of milk fat globule-EGF factor 8 (MFGE8).
Methods: MFGE8-knockdown ERC-Exos (MFGE8−/−-ERC-Exos) were constructed via lentiviral transfection. Colitis-targeted exosomes (CSE) were prepared, and their in vivo stability and targeting ability were verified. A dextran sulfate sodium (DSS)-induced murine colitis model was used to evaluate the effects of CSE. Immunofluorescence staining was utilized to assess colon DC infiltration; Flow cytometry was employed to detect CD4⁺ T cell proliferation and dendritic cell (DC) maturation. EdU/TUNEL staining and Western blotting were performed to determine intestinal epithelial cell proliferation and apoptosis, as well as the activation level of the PI3K/AKT/mTOR pathway.
Results: CSE achieved 89.2%± 1.5% encapsulation efficiency and 78% colon-targeted release, resisting gastric degradation. Oral CSE significantly alleviated DSS-induced weight loss (P< 0.001), colon shortening (P< 0.0001), and histopathological damage, while restoring intestinal barrier function (reduced permeability, P< 0.0001; upregulated tight junction proteins ZO-1/Occludin/Claudin-1). It also inhibited DC maturation (MLN: P< 0.0001; LP: P< 0.0001) and CD4⁺T cell proliferation (MLN: P< 0.001; LP: P< 0.01), and normalized cytokine profiles (reduced IL-6/IL-1β/TNF-α, P< 0.001; increased IL-10, P< 0.0001). MFGE8 knockdown significantly attenuated these effects. Mechanistically, MFGE8 mediated ERC-Exos uptake by intestinal epithelial cells via αvβ 5 integrin, activating PI3K/AKT/mTOR to inhibit apoptosis (P< 0.0001) and promote proliferation (P< 0.0001).
Conclusion: This study is the first to confirm that the CSE carrier achieves colonic targeted release, overcoming the limitations of oral exosome delivery. MFGE8 mediates the intestinal epithelial repair effect of ERC-Exos via the PI3K/AKT/mTOR pathway.
Keywords: oral administration, ulcerative colitis, endometrial regenerative cells, exosomes, milk fat globule-EGF factor 8, intestinal barrier
Introduction
Ulcerative colitis (UC) is a chronic intestinal inflammatory disorder.1 Its global incidence is increasing, especially in developing nations.2 Pathogenesis of UC involves genetic susceptibility, environmental factors, immune dysregulation, and intestinal barrier damage, leading to barrier dysfunction and clinical symptoms including hemorrhagic diarrhea, fever, and abdominal pain.3 Currently, the main treatments for UC include antibiotics, aminosalicylates, corticosteroids, and anti-tumor necrosis factor (TNF) inhibitors. However, these treatments are often associated with significant adverse effects, such as immune system dysfunction, gastrointestinal disorders, avascular necrosis, and hepatic dysfunction, which remain major challenges in clinical management.4 Therefore, the identification of safer and more effective therapeutic strategies for UC remains of utmost importance.
Recent investigations have increasingly focused on the therapeutic potential of mesenchymal stem cells (MSCs) in tissue regeneration and mitigation.5 Endometrial regenerative cells (ERCs) isolated from menstrual blood represent a novel type of MSCs that retains the fundamental properties of immunomodulation and tissue repair while exhibiting superior proliferative capacity compared to conventional MSCs.6 Furthermore, the collection of ERCs is safer, simpler, non-invasive, and more cost-effective than traditional MSCs sources such as adipose-derived or bone marrow-derived stem cells, with no significant ethical concerns.7 These attributes collectively enhance their value for clinical applications. A growing body of evidence suggests that the immunomodulatory effects of MSCs are largely attributable to their secretory exosomes.8 Exosomes are nanoscale extracellular vesicles ranging from 30 to 150 nm in size that are released by virtually all eukaryotic cells. They contain unique cargoes of nucleic acids, proteins, and lipids, which collectively modulate immune cell phenotype, function, and homing.9 Notably, exosomes derived from endometrial regenerative cells (ERC-Exos) demonstrate superior therapeutic efficacy and safety profiles,10 highlighting their significant potential for UC treatment.
Owing to degradation by gastrointestinal digestive fluids, exosomes are typically administered intravenously, resulting in significantly lower targeting efficiency and drug bioavailability compared to oral administration.11 Nevertheless, existing oral exosome delivery systems typically suffer from low colonic release efficiency and suboptimal encapsulation performance.12 Specifically, the severe gastrointestinal microenvironment frequently compromises the structural integrity of delivery carriers. This results in exosomes being prematurely released in the upper gastrointestinal tract.13 Simultaneously, the deficient encapsulation performance of existing systems is predominantly demonstrated by a low loading capacity and poor compatibility between exosomes and carrier materials. These limitations result in significant, premature exosome leakage during preparation, storage, and gastrointestinal transit.14 Studies have demonstrated that chitosan (CS) and sodium alginate (SA) biopolymers can effectively encapsulate diverse therapeutic agents, including small-molecule drugs and bioactive compounds.15 CS have a protective effect on exosomes and facilitate their sustained release from SA hydrogels.16 Compared with existing liposomes17 and single sodium alginate carriers,18 the CSE carrier developed in this study demonstrates enhanced stability and colon - targeting ability in simulated gastrointestinal fluids. These hydrogels enables the encapsulation of biologics, making oral delivery of ERC-Exos feasible. Although oral ERC-Exos demonstrate promising therapeutic potential in alleviating UC, their oral delivery strategies and precise mechanisms of action remain unclear.
Milk fat globule-EGF factor 8 (MFGE8), a glycoprotein originally identified in breast and mammary epithelial cells is expressed in nearly all sources of MSCs.19 Structurally, MFGE8 contains two EGF-like domains, a proline/threonine-rich domain, and two factor VIII-homologous domains that enable interactions with various cell surface molecules including phosphatidylserine.20 Moreover, MFGE8 serves as a critical bridging molecule between αvβ3 and αvβ5 integrin receptors, thereby preventing the release of pro-inflammatory mediators from dying cells and contributing to the maintenance of immunological homeostasis.21 Critically, MFGE8 plays an indispensable role in maintaining intestinal barrier homeostasis and accelerating mucosal repair.22 Interestingly, during the initial exploratory phase of this study, we confirmed that MFGE8 is expressed in ERCs and ERC-Exos. We hypothesized that the anti-inflammatory and tissue repair potential of ERC-Exos may be attributed to the expression of MFGE8. Notably, the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway has been well-documented as a pivotal pathway regulating the survival of epithelial cells and tissue regeneration.23 Previous studies have demonstrated that the dysregulation of the PI3K/AKT pathway is closely associated with exacerbated intestinal barrier disruption and delayed mucosal healing in both dextran sulfate sodium (DSS)-induced murine colitis models and clinical patients with UC.24 Against this backdrop, the well-established role of the PI3K/AKT pathway in the maintenance of intestinal epithelial homeostasis and tissue repair lays a solid theoretical foundation for the mechanistic investigations conducted in the present study.
In this study, we engineered a CS-SA-encapsulated ERC-Exos nanocarrier (CSE) to effectively administer ERC-Exos and sought to further investigate the role of MFGE8 in the ERC-Exos-mediated amelioration of UC.
Materials and Methods
Preparations of ERCs and ERC-Exos
Human menstrual blood samples were collected from 20-to 30-year-old healthy female volunteers after obtaining informed consent. The study was approved by the Medical Ethics Committee of Tianjin Medical University General Hospital (IRB2024-YX-013-01) and conducted in compliance with the Declaration of Helsinki. ERCs were isolated via Ficoll density gradient centrifugation, cultured in DMEM/F12 with 10% FBS and 1% penicillin/streptomycin, and characterized at passage 3 for morphology and surface markers (CD44, CD73, CD79a, and HLA-DR) using microscopy and flow cytometry. For exosome collection, 60–70% confluent ERCs were washed with phosphate-buffered saline (PBS) and cultured in serum-free medium for 48 h, and the conditioned medium was sequentially centrifuged (300×g/10 min, 2000×g/20 min, 10,000×g/30 min) to remove debris, followed by ultracentrifugation (110,000×g/70 min) to isolate purified ERC-Exos. ERC-Exos were characterized by Western blotting, Coomassie brilliant blue staining.
Preparation of MFGE8 Knockdown ERCs (MFGE8−/−-ERCs) and MFGE8 Knockdown ERCs-Exos (MFGE8−/−-ERC-Exos)
To obtain MFGE8−/−-ERC-Exos, MFGE8 was knocked down in ERCs via lentiviral transfection (GeneChem, Shanghai, China) at an MOI of 25 in a biosafety cabinet. The target sequence for MFGE8 was 5′-GGGTACCATGTGGCCACAACT-3′. Subsequently, transduced ERCs were selected using 2 mg/mL puromycin (Solarbio, China). MFGE8−/−-ERCs were confirmed using fluorescence microscopy and Western blotting. MFGE8−/−-ERC-Exos were isolated using the aforementioned method and were characterized by Western blotting and ELISA. The exosome size distribution and morphology were assessed using nanoparticle tracking analysis (NTA) and transmission electron microscopy (TEM), respectively.
Preparation and Observation on the Intestinal Distribution of Oral Chitosan (CS)-Sodium Alginate (SA) Exosome Microcapsules (CSE)
For the preparation of CSE, 0.6 wt% CS and 2% w/v CaCl2 were dissolved in 1% acetic acid (pH 5 adjusted by NaOH), mixed with 1.4% w/w SA at 1:1 volume ratio, and incorporated with 0.1 mg/mL ERC-Exos to form exosome-loaded hydrogel.12 For the characterization of CSE, TEM was used to observe its morphology. CSE were labeled with fluorescein isothiocyanate (FITC) to mark the chitosan-alginate gel matrix, while exosomes derived from ERCs were labeled with PKH26. The results were observed using a fluorescence microscope. NTA was employed to determine the particle size of CSE, and the particle size changes were continuously monitored in PBS for 15 days to evaluate its stability. Zeta potential measurement was performed to assess the surface charge differences between CSE and ERC-Exos, and Western blotting was used to detect the protein markers of CSE. To visually observe gastrointestinal stability, capsules were prepared using CaCO3 particles instead of exosomes and exposed to simulated gastric fluid (SGF) and simulated colonic fluid (SCF). The bicinchoninic acid (BCA) protein assay was utilized to determine the encapsulation efficiency of CSE and its release efficiency in SGF and SCF. For gastrointestinal tract distribution evaluation, DIR-labeled ERC-Exos (0.6 mg protein/kg body weight) were embedded in chitosan-alginate hydrogel and orally gavaged to UC model mice.25 Mice were sacrificed at 2, 8, and 12 h post-administration to collect gastrointestinal tracts,26 which were imaged using an IVIS spectral imaging system to track the CSE distribution.
Animals and Experimental Groups
The experimental animals used in this study were male BALB/c mice aged 6–8 weeks and weighing 22–25 g, purchased from an experimental animal center accredited by the China Food and Drug Administration (Beijing, China). All the animals were housed in a standard laboratory environment with adequate light, food, and water. The experimental protocol was approved by the Animal Ethics and Welfare Committee of Tianjin Medical University General Hospital (Approval No. IRB2024-DW-07, Tianjin, China). All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals: Eighth Edition and the Basel Declaration. Mice were randomly divided into four groups (n = 5 per group): control, untreated, CSE gavage, and MFGE8−/−-CSE gavage. Control mice received regular drinking water for 10 days, whereas Untreated, CSE, and MFGE8−/−-CSE groups were administered 3% DSS in water for 7 days. Additionally, CSE and MFGE8−/−-CSE groups received CSE orally (20 mg/kg body weight) or MFGE8−/−-CSE on days 2, 5, and 8, respectively.27 Sample size was calculated using G*Power 3.1 software (power = 0.8, α = 0.05), with 5 mice per group to ensure statistical reliability. Histological scoring was performed using a double-blind method, where scorers were unaware of sample grouping.
Assessment of Disease Activity and Colon Collection
The experimental mice were divided into groups as previously described and monitored daily for changes in body weight and the presence of mucopurulent bloody stools until day 10. On the terminal day, the excised colons were immediately measured for length documentation before collecting two 0.5 cm segments from the distal colon for subsequent histological examination. The cecum and entire colon were photographed under standardized conditions, followed by repeat length measurements to ensure data accuracy. All procedures were performed by investigators using calibrated instruments to maintain experimental consistency across samples.
Histopathological Analysis
Two distal colon segments (~0.5 cm) per mouse were fixed in 10% neutral buffered formalin for 48 h, dehydrated, paraffin-embedded, and sectioned into 3-µm slices. The sections were stained with hematoxylin and eosin (H&E). The severity of colonic epithelial damage and inflammatory cell infiltration were scored in a blinded manner. The scoring criteria for colonic epithelial injury were as follows: 0, nNormal; 1: Hyperplasia, loss of goblet cells, and irregular crypt foci; 2: Mild to moderate loss of crypt foci (10–50%); 3: Severe loss of crypt foci (50–90%); 4: Complete loss of crypt foci with intact epithelium; 5: Small to medium-sized ulcers (crypt width <10); 6: Large ulcers (crypt width ≥10). In addition, the mucosa, submucosa, and muscularis/serosa layers were scored separately as follows: 0: Normal; 1: Mild; 2: Moderate; 3: Severe. The total score ranged from 0 to 12, based on the combined assessment of epithelial damage and inflammatory infiltration.
Colonic Mucosal Ultrastructure and Intestinal Permeability Assessment
Transmission electron microscopy (TEM, Hitachi H-600, Japan) was employed to examine 70-nm ultrathin colon sections, with a particular focus on epithelial tight junctions and villous architecture. To evaluate intestinal permeability, mice were fasted for 6 hours and orally administered FITC-dextran (4 kDa; 0.6 mg/g body weight in 100 µL PBS) prior to sacrifice. Serum was collected from orbital blood and analyzed for fluorescein isothiocyanate (FITC)-dextran fluorescence (ex/em 488/520 nm) using an F-4500 spectrophotometer (Hitachi).
Alcian Blue Staining
To detect goblet cells and intestinal mucus in mouse colon tissues under different treatments, colon tissue sections were stained using an Alcian Blue Stain Kit (Solarbio, Beijing, China) according to the manufacturer’s protocol. Briefly, deparaffinized mouse colon tissues were treated with Alcian Blue acidification solution for 3 min, followed by staining with 1% Alcian Blue for 30 min. Sections were washed with water, counterstained with nuclear fast red for 5–10 min, dehydrated with ethanol, cleared with xylene, and mounted with neutral gum. Stained sections were examined under an optical microscope. For statistical analysis, the number of goblet cells was counted in 20 randomly selected, well-defined crypts from the colonic glands of each mice.
MUC2 Immunofluorescence Staining
Mice colon tissues were harvested, sectioned, and fixed in methanol as previously described. Immunostaining was performed using a rabbit anti-MUC2 antibody (Affinity Biosciences). The sections were then counterstained with DAPI (Sigma-Aldrich) at room temperature. Immunopositive signals were quantitatively assessed using fluorescence microscopy at 20× magnification. Images were analyzed using the ImageJ software.
Immunofluorescence Staining of Tunel and CD11c in vivo
Apoptotic cells in colon tissues were detected using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay according to the manufacturer’s protocol (Solarbio, Beijing). Additionally, immunofluorescence staining was performed to observe the infiltration of DCs in the colon. Paraffin-embedded colon sections were fixed with 4% paraformaldehyde for optimal preservation of tissue morphology. Following fixation, the sections were thoroughly washed with PBS. To facilitate reagent penetration, tissue permeabilization was performed using 0.2% Triton X-100 in PBS. The sections were then incubated with the TUNEL and CD11c reaction mixture in a humidified chamber for 1 h in the dark. The nuclei were stained with DAPI. Finally, sections were examined and photographed.
Flow Cytometry Analysis
Flow cytometry was used to characterize ERCs surface markers and evaluate dendritic cell (DCs) maturation and CD4+ T cell proliferation in the Mesenteric Lymph Nodes (MLNs) and intestinal Lamina Propria (LP). Single-cell suspensions were pre-treated with Fc Blocking Buffer (eBioscience) for 10 min, followed by viability staining using Zombie NIR™ (BioLegend). Cells were then immunostained with a pre-optimized antibody panel, including anti-human markers (HLA-DR-PE, CD73-FITC, CD44-APC, and CD79a-PE) and anti-mouse antibodies (CD11c-PerCP-Cy5.5, CD80-PE, MHCII-FITC, CD3-FITC, CD4-PE, and KI67-APC) under light-protected conditions (4°C, 30 min). After thorough washing, the samples were analyzed for specific fluorescence signals.
Enzyme-Linked Immunosorbent Assay (ELISA)
To qualitatively and quantitatively assess inflammation and tissue damage in the colon, proinflammatory (TNF-α, IL-1β, and IL-6) and anti-inflammatory (IL-10) cytokines were measured in colon tissue homogenates using ELISA kits. All procedures were performed in strict accordance with the manufacturer’s instructions. Absorbance was measured at 450 nm and cytokine concentrations were determined based on standard curves generated for each assay.
CCK-8 Cell Viability Assay
To explore the therapeutic mechanism of ERC-Exos in UC, NCM460 human colonic epithelial cells were cultured in RPMI-1640/10% FBS with DSS (60 mg/L, 6 h),28 followed by 24-hour treatment with ERC-Exos (20 μg/mL),29 MFGE8−/−-ERC-Exos (equivalent dose), ERC-Exos, and cilengitide (1 mg/mL, αvβ5 integrin antagonist). Cell viability was assessed using the Cell Counting Kit-8 (CCK-8) reagent (Solarbio, Beijing, China) according to the manufacturer’s instructions. Briefly, NCM460 cells were seeded in 96-well plates at a density of 5 × 103cells per well. Subsequently, 10 µL of the CCK-8 reagent was added to each well and incubated for 1 h. The absorbance was measured at 450 nm using a multimode microplate reader to assess cell viability.
Uptake of Exosomes by NCM460
ERC-Exos from each experimental group were labeled with the red fluorescent dye PKH26 (Solarbio) following the manufacturer’s protocol. Labeled exosomes were co-incubated with NCM460 cells for 6 h. Next, the cells were fixed with 4% paraformaldehyde, and the cytoskeleton of NCM460 cells was stained with phalloidin (Sigma). Nuclei were counterstained with DAPI and the samples were imaged using fluorescence microscopy.
Immunofluorescence Staining of Tunel and EdU in vitro
To observe the proliferation and apoptosis of NCM460 cells in different experimental groups, fluorescence-based staining assays for 5-ethynyl-2′-deoxyuridine (EdU) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) were performed according to previously described methods. The results were observed and recorded using a fluorescence microscope.
Western Blotting
Protein lysates were extracted using RIPA buffer (Solarbio R0010) containing protease/phosphatase inhibitors (Solarbio P1261), separated by SDS-PAGE, and transferred to PVDF membranes (Millipore). After blocking with 5% BSA/TBST for 2 h, the membranes were probed with primary antibodies against CD9, CD63, CD81, Calnexin, MFGE8, ZO-1, Occludin, Claudin-1, p-PI3K, PI3K, p-AKT, AKT (Abcam), β-actin, p-MTOR, MTOR, BCL-2, BAX, Caspase-3 and GAPDH (ABclonal) followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Protein bands were detected by enhanced chemiluminescence (Thermo Fisher) and quantified using ImageJ software.
Statistical Analysis
All statistical analyses in this study were performed using GraphPad Prism version 8.0 (GraphPad Software). Results are presented as mean ± standard error of the mean (SEM). Differences between two groups were assessed using an unpaired two-tailed Student’s t-test, whereas differences among multiple groups were evaluated using a one-way ANOVA. Statistical significance is defined as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Characterization of ERCs and ERC-Exos
ERCs were extracted from menstrual blood and were highly positive for CD44 (99.9%) and CD73 (100%) but negative for CD79a (0.65%) and HLA-DR (0.62%) by flow cytometry analysis (Figure 1A). Light microscopy revealed that ERCs exhibited spindle-like morphology, forming tightly packed swirls, and Green fluorescent protein (GFP) serves as the endogenous reporter gene of the lentiviral vector for MFGE8 knockdown and was clearly visible under a fluorescence microscope. This GFP signal confirmed successful lentiviral transduction of ERCs, indirectly verifying effective MFGE8 knockdown. (Figure 1B). TEM imaging clearly showed that both groups of exosomes contained spherical vesicles with clear membrane structures (Figure 1C). Western blot (Figure 1D) confirmed that MFGE8 expression was significantly downregulated in both ERCs (P <0.05; Figure S1a) and ERC-Exos (P <0.01; Figure S1b). ELISA demonstrated MFGE8 concentration was 7.0±0.5 μg/mL in ERC-Exos but significantly reduced to 1.2±0.3 μg/mL in MFGE8−/−-ERC-Exos, with a 92.3%±2.1% knockdown efficiency (P < 0.0001; Figure 1E). To characterize the protein composition of ERC-Exos, Coomassie Brilliant Blue staining was performed (Figure 1F). ERC-Exos displayed a characteristic protein banding pattern, with bands spanning a molecular weight range of 25–140 kDa, indicative of the complex and intact protein cargo within the exosomes. NTA results showed that the diameter of the exosomes in both groups ranged from 30 to 150 nm (Figure 1G). ERC-Exos and MFGE8−/−-ERC-Exos were further characterized using Western blotting. The results of the two groups of exosomes confirmed the presence of exosomal markers CD9, CD63, and CD81, whereas Calnexin was undetectable in the isolated particles compared to the ERCs (Figure 1G).
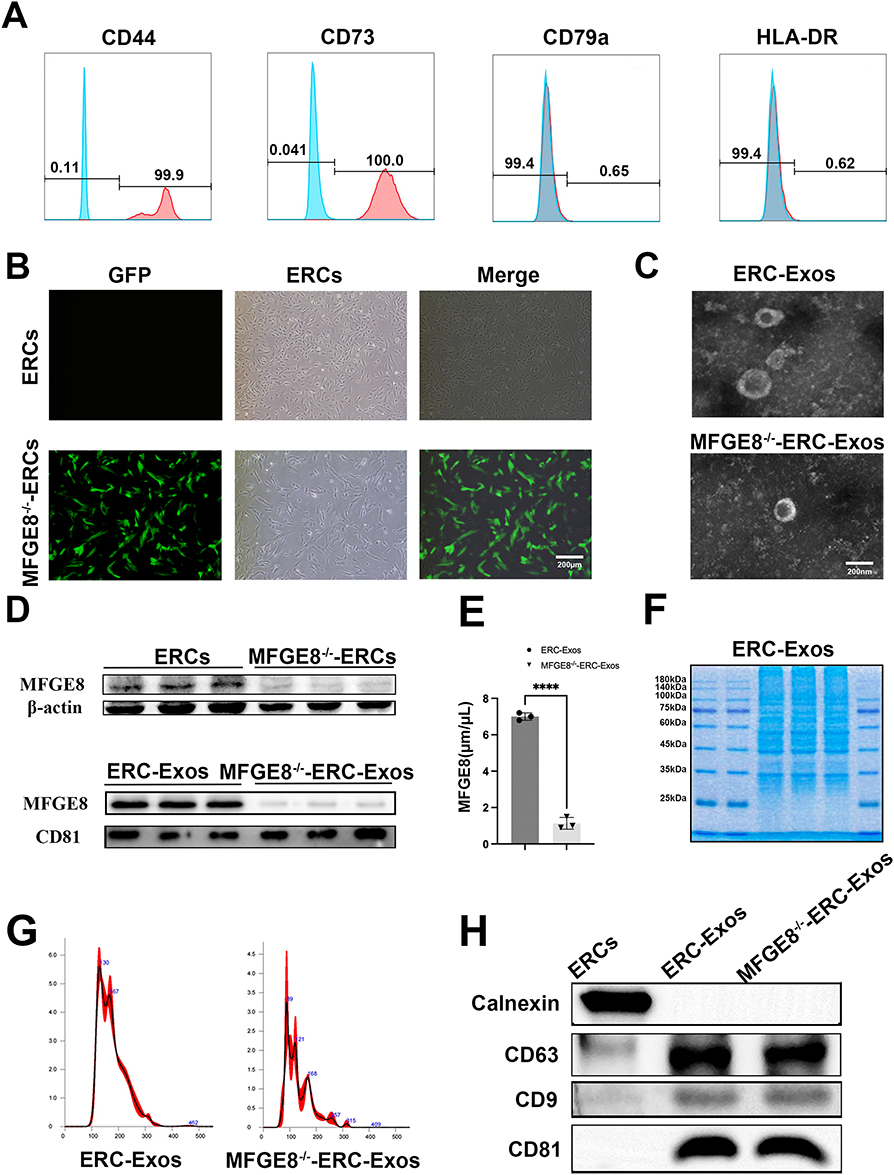 |
Figure 1 ERCs and ERC-Exos Characterization. (A) Flow cytometric analysis of ERC surface markers, demonstrating positive expression of CD44 and CD73, and negative expression of CD79a and HLA-DR. (B) Fluorescence microscopy images showing GFP staining (green fluorescence) in ERCs and MFGE8−/−-ERCs. (ERCs lack GFP signal, MFGE8−/−-ERCs exhibit robust GFP fluorescence) Scale bar = 200 μm. (C) Transmission electron microscopy (TEM) images of ERC-Exos and MFGE8−/−-ERC-Exos Scale bar = 200 nm. (D) Western blotting analysis of MFGE8 expression in ERCs and MFGE8−/−-ERCs. (E) ELISA quantification of MFGE8 protein levels in ERC-Exos and MFGE8−/−-ERC-Exos (****P < 0.0001). (F) SDS-PAGE protein profile of ERC-Exos, illustrating the characteristic protein composition of exosomes. (G) Nanoparticle tracking analysis (NTA) of the size distribution of ERC-Exos and MFGE8−/−-ERC-Exos. (H) Western blotting analysis of exosomal markers (CD9, CD63, CD81, and calnexin) in ERCs, ERC-Exos, and MFGE8−/−-ERC-Exos. Data were analyzed using an unpaired two-tailed Student’s t-test; *P < 0.05; **P < 0.01. |
Preparation and Characterization of CSE
To prevent gastric degradation of ERC-Exos, CS and sodium SA self-assembled into CSE nanomembranes (Figure 2A). Transmission electron microscopy (TEM) showed CSE as spherical microcapsules, with encapsulated ERC-Exos (30–150 nm) clearly visible in the matrix, confirming successful encapsulation (Figure 2B). Fluorescence staining verified complete ERC-Exos (red) encapsulation in the biodegradable CS (green) membrane (Figure 2C). Western blotting detected exosomal markers CD63 and CD9 in CSE (Figure 2D). NTA characterized CSE as monodisperse with a mean diameter of 169 nm (Figure 2E). Zeta potential analysis revealed ERC-Exos had a negative zeta potential, while CSE was positive (Figure 2F). NTA assessed CSE stability in PBS over 15 days, showing particle size stably maintained at ~166–169.8 nm (Figure 2G).
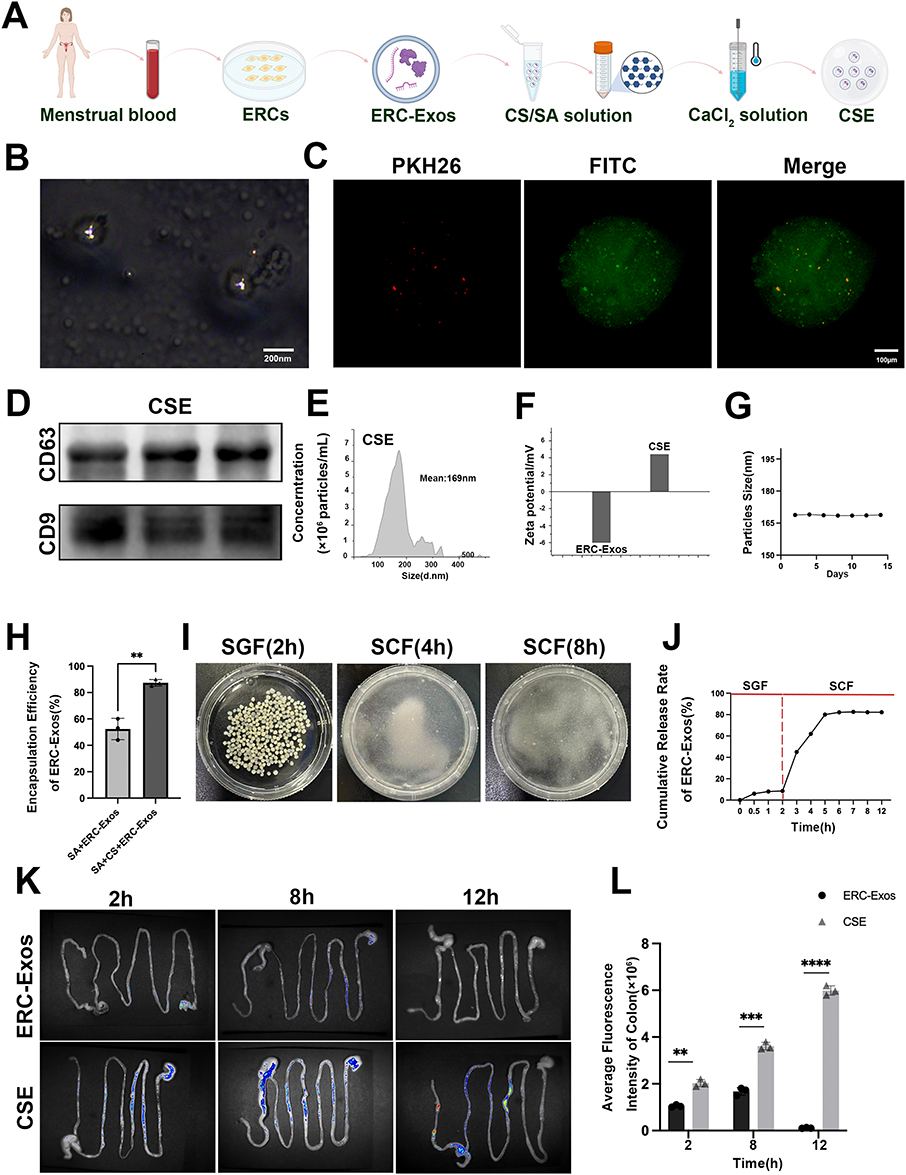 |
Figure 2 Preparation and characterization of CSE. (A) Schematic workflow for CSE preparation. Menstrual blood is used to isolate and culture ERCs, from which ERC-Exos are purified. ERC-Exos are then encapsulated into chitosan/sodium alginate (CS/SA) matrix via ionic crosslinking with CaCl2 solution to form CSE. (B) Transmission electron microscopy (TEM) image of CSE. Scale bar = 200 nm. (C) Fluorescence microscopy images of CSE stained with PKH26 (red, for ERC-Exos labeling) and FITC (green, for CS/SA matrix labeling). Scale bar = 100 μm. (D) Western blotting analysis of exosomal markers CD63 and CD9 in CSE (E) Nanoparticle tracking analysis (NTA) of CSE. (F) Zeta potential analysis of ERC-Exos and CSE, characterizing their surface charge properties. (G) Stability assay of CSE particle size over 15 days. (H) Encapsulation efficiency of ERC-Exos in sodium alginate (SA-ERC-Exos) versus chitosan/sodium alginate (SA-CS-ERC-Exos) formulations. (**P < 0.01). (I) Morphological observation of CSE after incubation in simulated gastric fluid (SGF, 2 h), simulated colonic fluid (SCF, 4 h), and SCF (8 h), respectively. (J) Cumulative release kinetics of ERC-Exos from CSE in sGF and sCF within 12 h. (K) In vivo tracking of ERC-Exos at 2 h, 8 h, and 12 h after oral administration of CSE in mice. (L) Quantification of average fluorescence intensity in the colon at different time points. (**P < 0.01, ***P< 0.001, ****P < 0.0001). |
Encapsulation Efficiency and Gastrointestinal Stability of CSE
To assess the encapsulation efficiency of ERC-Exos in SA and SA+CS carriers, the BCA quantification assay was employed. As presented in Figure 2H, the encapsulation efficiency of SA-ERC-Exos was approximately 55%, whereas that of SA+CS-ERC-Exos was significantly elevated to around 85%, with a statistically significant difference noted (P < 0.01). These findings indicate that the integration of chitosan into the SA-based carrier substantially enhances the encapsulation capacity for ERC-Exos. CSE morphology was observed after incubation in SGF (2 h) and SCF (4 h, 8 h, Figure 2I).30 CSE remained intact in SGF but gradually degraded in SCF (more obvious at 2 h), indicating gastric stability and colonic-responsive degradation for site-specific targeting. Release profile showed negligible ERC-Exos release in SGF (2 h), while SCF triggered rapid, sustained release (>80% within 12 h, Figure 2J), confirming pH-responsive protection in the stomach and controlled release in the colon. In vivo fluorescence imaging at 2 h, 8 h, and 12 h (Figure 2K) showed CSE had more prominent intestinal retention than ERC-Exos at 2 h, stronger and wider colonic fluorescence at 8 h, and sustained detectability at 12 h. Quantitative colonic fluorescence intensity (Figure 2L) showed CSE significantly outperformed ERC-Exos at all time points (2 h: P < 0.01, 8 h: P < 0.001, 12 h: P < 0.0001), indicating CSE reduced ERC-Exos degradation and enhanced colonic absorption.
Oral Administration of MFGE8-Expressing CSE Alleviates Colitis
To evaluate the therapeutic potential of orally administered CSE, a DSS-induced colitis model was established using BALB/c mice that were gavaged with CSE or MFGE8−/−-CSE. Compared to the untreated group, CSE treatment significantly reduced bloody stools (Figure 3A), attenuated colon shortening (P <0.0001, Figure 3B and C), improved weight maintenance (P <0.001, Figure 3D), and lowered disease activity index scores (Figure 3E). Histological analysis indicated that CSE protected against DSS-induced damage by preserving the goblet cells and reducing crypt loss, ulceration, and inflammation (Figure 3F). Notably, these therapeutic effects were largely abolished in the MFGE8−/−-CSE group, highlighting the critical role of MFGE8 in CSE-mediated protection against colitis.
 |
Figure 3 Oral administration of MFGE8-expressing CSE alleviates ulcerative colitis. (A) Representative photographs of BALB/c mice from each experimental group on day 10 after DSS induction. (B and C) Macroscopic images of colons from each group and quantitative analysis of colon length on day 10. (D) Body weight changes of mice in each group over a 10-day period. (E) Quantitative assessment of colonic tissue damage using histopathology scores (n = 5). (F) Representative hematoxylin and eosin (H&E)-stained sections of colonic tissues from each group. Red arrows indicate epithelial detachment, blue arrows denote inflammatory cell infiltration, and black boxes highlight ulcerative regions. Scale bar = 100μm. Data were analyzed by one-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. |
MFGE8 Is a Key Factor Involved in Orally Administered CSE That Repairs the Intestinal Barrier
Destruction of the intestinal barrier is critical for colitis pathogenesis. To investigate whether CSE and MFGE8 expressed in CSE reversed the intestinal barrier injury, we assessed the integrity and function of the intestinal barrier in each group. TEM (Figure 4A) showed DSS-induced damage to microvilli and tight junctions (TJs) were repaired by CSE, with less improvement in the MFGE8−/−-CSE group. TUNEL staining (Figure 4B and C) demonstrated that the apoptosis of intestinal epithelial cells in the untreated group was higher than that in the control group (P < 0.0001), and this increase was reduced in the CSE group (P < 0.0001). However, the therapeutic effect of CSE was partially reversed (P < 0.01) by MFGE8 knockdown. Intestinal permeability assays (Figure 4D) confirmed that CSE reduced DSS-induced leakage (P < 0.0001), with diminished repair in the MFGE8−/−-CSE groups (P < 0.05). Western blotting revealed that DSS significantly downregulated the expression of the tight junction proteins ZO-1, occludin, and Claudin-1; Figure 4E–H). This effect was reversed by CSE treatment but attenuated in the MFGE8−/−- CSE group.
 |
Figure 4 Oral administration of CSE repairs the intestinal barrier: MFGE8 as a key factor. (A) TEM images showing the ultrastructure of colonic epithelial cells. Scale bar = 10 μm. Red arrows indicate microvilli, and blue arrows denote tight junctions between adjacent epithelial cells. (B) Tunel staining for assessing apoptosis of intestinal epithelial cells in each group. (green, Tunel marker) and DAPI (blue, nuclear staining) Scale bar = 10 μm. (C) Quantitative analysis of apoptotic cells in colon tissues. (D) Intestinal permeability was evaluated by measuring serum FITC-dextran levels (n = 5). (E) Western blotting analysis of ZO-1, Occludin, and Claudin-1 expression in colon tissues, normalized to GAPDH. (F-H) Quantitative analysis of ZO-1, Occludin, Claudin-1 expression in intestinal epithelial cells normalized to GAPDH (n = 3). Data were analyzed using an unpaired two-tailed Student’s t-test; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 00001. |
MFGE8 Knockdown Attenuates CSE-Mediated Restoration of the Intestinal Mucus Barrier
To evaluate intestinal mucus barrier damage, we performed Alcian Blue staining (Figure 5A). The results indicated that the untreated group had severely depleted goblet cells (blue) compared with the control group (P < 0.0001, Figure 5B). Conversely, the therapeutic effects of CSE were significantly reversed in the MFGE8−/−-CSE group. The number of goblet cells in the CSE group was 7-fold higher than that in the untreated (P < 0.001), and 5-fold higher than that in the MFGE8−/−-CSE (P < 0.001). Immunofluorescence for MUC2 (red) (Figure 5C) revealed that the significant reduction in fluorescence in the untreated group (P < 0.001, Figure 5D) was rescued by CSE (P < 0.01), but the rescue effect was blunted by MFGE8 knockdown (P < 0.05). Collectively, oral CSE repairs colitis-damaged intestinal barriers and MFGE8 is indispensable for this protective effect.
 |
Figure 5 MFGE8 knockdown attenuates CSE-mediated restoration of the intestinal mucus barrier. (A) Representative Alcian blue-stained micrographs showing goblet cells (blue). Scale bar = 100 μm (B) Quantitative analysis of goblet cells in colon tissues. (C) MUC2 immunofluorescence staining (red, muc2 marker) and DAPI (blue, nuclear staining) in colonic tissues. Scale bar = 100 μm. (D) Statistical analysis of the mean fluorescence intensity of MUC2. (E) Immunofluorescence staining of CD11c (green, dendritic cell marker) and DAPI (blue, nuclear staining) in colonic tissues. Scale bar = 100 μm. (F) Integral optical density quantification of CD11c staining. Data were analyzed using one-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. |
MFGE8 Deficiency Compromises the Ability of CSE to Inhibit DC Maturation in vivo
To investigate the infiltration of DCs in intestinal tissues, immunofluorescence staining and quantitative analysis were performed (Figure 5E). CD11c (green) was detected to identify DCs, and DAPI (blue) was used for nuclear staining. In the Control group, CD11c fluorescence was barely detectable The Untreated group showed prominent green fluorescence of CD11c. The CSE group exhibited a significant reduction in CD11c fluorescence intensity, while the MFGE8−/−-CSE group displayed a rebound in CD11c fluorescence. For quantitative analysis (Figure 5F), the integral optical density of CD11c was measured. Compared with the Control group, the Untreated group showed a significantly increased CD11c expression (P < 0.0001). The CD11c expression in the CSE group was significantly lower than that in the Untreated group (P < 0.001), whereas the MFGE8−/−-CSE group exhibited a significantly higher CD11c expression compared to the CSE group (P < 0.001). To elucidate the roles of CSE and MFGE8 in modulating maturation of DCs in vivo. We analyzed the proportions of MHC II+ and CD80+ DCs in the MLN and intestinal LP using flow cytometry (Figure 6A and B). Flow cytometric analysis revealed that the maturation of DCs in the MLN (Figure 6C and D) and LP (Figure 6E and F) of colitis mice was significantly inhibited by CSE treatment compared with that in the untreated group (MLN-MHCII: P < 0.0001, LP-MHCII: P < 0.0001) (MLN-CD80: P < 0.05, LP-CD80: P < 0.05). However, this effect was significantly attenuated in the MFGE8−/−-CSE-treated group (MLN-MHCII: P < 0.0001, LP-MHCII: P < 0.001) (MLN-CD80: P < 0.05, LP-CD80: P < 0.05). Collectively, these results indicated that MFGE8 is essential for CSE to inhibit DCs maturation and infiltration in vivo.
 |
Figure 6 Effects of CSE on immune cell modulation and inflammatory cytokine levels in mice with colitis. (A) Flow cytometry plots of mature dendritic cells (DCs) in the mesenteric lymph nodes (MLN). (B) Flow cytometry plots of mature DCs in the lamina propria (LP). (C and D) Quantitative analysis of dendritic cells (DCs) in the mesenteric lymph nodes (MLN). (E and F) Quantitative analysis of dendritic cells (DCs) in the lamina propria (LP). (G and H) Flow cytometry plots and quantitative analysis of CD4+ T cells proliferation (Ki67+) in the MLN. (I and J) Flow cytometry plots and quantitative analysis of CD4+ T cells proliferation (Ki67+) in the LP. (K–N) Concentrations of pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α) and anti-inflammatory cytokine (IL-10) in colonic tissues were measured by ELISA (n = 5). Data were analyzed using one-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. |
MFGE8 Knockdown Reverses the Impact of CSE on CD4+ T Cell Activation in vivo
We assessed the proliferation of CD4⁺ T cells in the MLN (Figure 6G and H) and LP (Figure 6I and J) of each experimental group. The results indicated that CSE treatment significantly suppressed the activation of CD4⁺ T cells compared with that in the untreated group. This was evidenced by a notable decrease in the mean fluorescence intensity (MFI) of Ki67 (MLN, P < 0.001; LP, P < 0.01). However, in the MFGE8−/−-CSE group, this suppressive effect was attenuated compared with that in the CSE group (MLN: P < 0.01; LP: P < 0.05). These findings suggest that MFGE8-expressing CSE effectively curtails the overactivation of CD4⁺ T cells within the inflamed intestinal microenvironment, thereby facilitating colitis repair.
MFGE8 Mediates the Therapeutic Effect of CSE in Modulating Pro-Inflammatory and Anti-Inflammatory Cytokine Profiles in the Colitis Model
Cytokine profiles were analyzed by ELISA to evaluate the intestinal inflammation levels in each group (Figure 6K–N). These profiles include those of pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α) and anti-inflammatory cytokines (IL-10). ELISA analysis of the intestinal cytokine profiles revealed significantly elevated pro-inflammatory cytokines (IL-6, P < 0.0001; IL-1β, P < 0.0001; TNF-α, P < 0.0001) and reduced anti-inflammatory IL-10 (P < 0.0001) in the untreated group compared to the controls. However, CSE treatment effectively normalized these changes (IL-6: P < 0.001; IL-1β: P < 0.001; TNF-α: P < 0.0001; and IL-10: P < 0.0001). MFGE8 knockdown substantially attenuated these therapeutic effects (IL-6: P < 0.05; IL-1β: P < 0.05; TNF-α: P < 0.05; IL-10: P < 0.001). These results demonstrate that CSE microcapsules exert potent anti-inflammatory effects, and that MFGE8 expressed in ERC-Exos plays a critical role in this process.
MFGE8 Enhances the Uptake of ERC-Exos by Intestinal Epithelial Cells in vitro
To further elucidate the reparative mechanisms of MFGE8 expression in ERC-Exos, we investigated intestinal epithelial cell phagocytosis by ERC-Exos (Figure 7A). Fluorescence microscopy analysis of PKH26-labeled ERC-Exos (red) and phalloidin-stained DSS-pretreated NCM460 cells (green) demonstrated that MFGE8 knockdown significantly attenuated the phagocytosis of ERC-Exos by intestinal epithelial cells. (P < 0.0001, Figure 7B). Furthermore, pharmacological inhibition of the αvβ5 integrin receptor impaired the phagocytosis of ERC-Exos by DSS-pretreated intestinal epithelial cells, paralleling the effects observed with MFGE8 knockdown (P < 0.0001). In conclusion, MFGE8 mediates ERC-Exos phagocytosis by intestinal epithelial cells under inflammatory conditions and this process is dependent on the αvβ5 integrin receptor.
 |
Figure 7 MFGE8 suppresses apoptosis and promotes proliferation in damaged intestinal epithelial cells. (A) Immunofluorescence staining of intestinal epithelial cells with Phalloidin (green, actin), PKH26 (red, ERC-Exos labeling), and DAPI (blue, nuclei) in ERC-Exos, MFGE8−/−-ERC-Exos, and Cilengitide groups. Merged (Merge) images illustrate the cellular uptake of exosomes. Scale bar = 100 μm. (B) Quantification of phagocytosis rate, in ERC-Exos, MFGE8−/−-ERC-Exos, and Cilengitide groups. (C) TUNEL staining (green, apoptotic cells) and DAPI (blue, nuclei) in colonic tissues from Control, Untreated, ERC-Exos, MFGE8−/−-ERC-Exos, and Cilengitide groups. Merged (Merge) images depict apoptotic cell distribution. Scale bar = 100 μm. (D) Quantitative of Tunel analysis shown in the statistical graphs. (E) EdU staining (green, proliferating cells) and DAPI (blue, nuclei) in colonic tissues from Control, Untreated, ERC-Exos, MFGE8−/−-ERC-Exos, and Cilengitide groups. Merged (Merge) images show cell proliferation. Scale bar = 100 μm. (F) Quantitative analysis of EdU shown in the statistical graphs. (G) Cell viability of NCM460 cells in each group was determined using the CCK-8 assay. (H) Western blotting analysis of PI3K/AKT/mTOR pathway proteins (p-PI3K, PI3K, p-AKT, AKT, p-MTOR, MTOR) and apoptosis-related proteins (BCL-2, BAX, Caspase-3), with GAPDH as a loading control. Data were analyzed using one-way ANOVA; **P < 0.01; ***P < 0.001; ****P < 0.0001. |
MFGE8 Is Essential for ERC-Exos to Inhibit Apoptosis and Enhance Proliferation of Injured Intestinal Epithelial Cells
The reparative effects of MFGE8-expressing ERC-Exos on intestinal epithelial cells were further verified by in vitro proliferation and apoptosis assays. TUNEL (green) assays revealed that ERC-Exos reduced DSS-induced apoptosis by 4.5-fold (P < 0.0001, Figure 7C and D), but this reparative effect was reversed by knockdown of MFGE8 (P < 0.0001) or inhibition of αvβ5 (P < 0.0001). EdU (green) proliferation assays demonstrated that MFGE8 expressed in ERC-Exos significantly enhanced the regeneration of DSS-injured intestinal epithelial cells (P < 0.0001; Figure 7E and F). This restorative effect was markedly attenuated following MFGE8 knockdown (P < 0.0001) and αvβ5 integrin inhibition (P < 0.0001). CCK-8 assays demonstrated that ERC-Exos significantly enhanced the viability of DSS-induced NCM460 cells (P < 0.0001; Figure 7G), whereas this effect was attenuated by MFGE8 knockdown (P < 0.01) and αvβ5 inhibition (P < 0.01). These results demonstrate that MFGE8 expressed in ERC-Exos coordinately suppresses apoptosis and promotes proliferation of intestinal epithelial cells through αvβ5 integrin.
MFGE8-Expressing ERC-Exos Modulates PI3K/AKT Signaling Pathway in Intestinal Epithelial Cells
The PI3K/AKT/mTOR pathway regulates apoptosis inhibition, cell proliferation, and tissue repair.31 To clarify the molecular mechanisms, Western blotting analyzed the PI3K/AKT/mTOR axis and key apoptotic regulators (anti-apoptotic BCL-2, pro-apoptotic BAX; Figure 7H), with GAPDH as a loading control. ERC-Exos significantly increased PI3K and AKT phosphorylation compared to the Untreated group (total PI3K/AKT unchanged across groups), while MFGE8−/−-ERC-Exos or Cilengitide reduced this phosphorylation. mTOR phosphorylation and BCL-2/BAX expression was assessed. ERC-Exos elevated mTOR phosphorylation, upregulated BCL-2, and downregulated BAX—shifting the BCL-2/BAX ratio toward anti-apoptosis. In contrast, MFGE8−/−-ERC-Exos or Cilengitide reversed these effects: reduced p-mTOR, BCL-2 and increased BAX (total mTOR unchanged). To quantify the changes in signaling and apoptotic proteins, statistical analysis of Western blotting results was performed (Figure S2a–e). Among the PI3K/AKT/mTOR pathway-related indicators, p-PI3K/PI3K, p-AKT/AKT, and p-mTOR/mTOR exhibited high phosphorylation levels in the Control group compared to the Untreated group; ERC-Exos treatment significantly elevated their phosphorylation, while MFGE8−/−-ERC-Exos and Cilengitide attenuated this effect (p-PI3K/PI3K: P< 0.05, P< 0.01; p-AKT/AKT and p-mTOR/mTOR: P< 0.0001). For apoptotic regulators, the Control group had a high anti-apoptotic/pro-apoptotic BCL-2/BAX ratio and low Caspase-3/GAPDH expression, whereas the Untreated group showed a decreased BCL-2/BAX ratio and drastically increased Caspase-3/GAPDH levels; ERC-Exos significantly increased the BCL-2/BAX ratio and reduced Caspase-3 expression, while MFGE8−/−-ERC-Exos and Cilengitide reversed these effects (BCL-2/BAX ratio: P< 0.05, P< 0.01, P< 0.0001; Caspase-3/GAPDH: P< 0.001, P< 0.0001). These results indicate that MFGE8 expressed in ERC-Exos activates PI3K/AKT/mTOR signaling through αvβ5 integrin engagement, thereby inhibiting apoptosis and enhancing the proliferation of intestinal epithelial cells, ultimately ameliorating colitis.
Discussion
UC is a refractory inflammatory bowel disease characterized by chronic relapse and unmet therapeutic needs.32 Stem cell-derived exosomes have emerged as a promising therapeutic strategy, functioning as natural nanoscale carriers of bioactive molecules with tissue regenerative and immunomodulatory capabilities.33 ERCs derived non-invasively from menstrual blood have been identified as a novel class of MSCs with distinct advantages and superior accessibility compared to traditional MSCs. (eg, adipose/bone marrow-derived).34 Furthermore, ERC-Exos preserve the anti-inflammatory effects from parental cell as well as possessing benefits of low immunogenicity and logistical feasibility.35 Previous work by Zhu et al established that ERC-Exos ameliorate experimental colitis through downregulation of intestinal ferritin deposition.36 This unique combination of biological properties and positions ERC-Exos as particularly promising candidates for UC therapy.37
Conventional delivery of ERC-Exos has been limited to intravenous administration due to their susceptibility to degradation by gastric acid and digestive enzymes, precluding their use via the more physiologically oral route.38 However, numerous existing oral exosome delivery strategies are beleaguered by inherent limitations that impede their translational potential,39 including inadequate colonic targeting specificity, which results in inefficient accumulation of bioactive exosomes at the pathological foci and futile dissemination to non-targeted tissues; and subpar encapsulation efficacy, a flaw that not only curtails the loading capacity of exosomes but also compromises their in vivo bioavailability and ultimately undermines the therapeutic efficacy of the delivered exosomes.40 To address this critical limitation, we developed an oral capsule formulation of ERC-Exos encapsulated in a chitosan-sodium alginate composite in this study (Figure 8). Natural polysaccharides, including sodium alginate, pectin, and chitosan, are extensively employed in hydrogel fabrication because of their favorable properties, such as mild gelation conditions, excellent biocompatibility, and resistance to acidic and enzymatic degradation.41 These characteristics make them particularly suitable for oral drug delivery applications in the gastrointestinal tract. For instance, Mohammadbaghban et al demonstrated that orally administered chitosan-alginate-catechin nanocarriers, synthesized via ionic gelation, effectively ameliorated Alzheimer’s disease models.42 Mohd H. M. Jaafar et al demonstrated that an oral insulin delivery system utilizing alginate-chitosan composite coatings effectively shields insulin from enzymatic degradation while enhancing intestinal absorption.43 The protective efficacy of the sodium alginate-chitosan polysaccharide platform for safeguarding protein therapeutics in the gastrointestinal environment demonstrates its versatility as a drug delivery system. The sodium alginate-chitosan polysaccharide platform has been regarded as a promising strategy based on its protective efficacy for safeguarding protein therapeutics in the gastrointestinal environment.
 |
Figure 8 Schematic representation of ERC-Exos isolation, CSE preparation, and the protective mechanism of MFGE8-expressing CSE in colitis. Orally administered CSE resists harsh gastric degradation (protected by the CS and SA matrix) while undergoing pH-responsive degradation in the colon for targeted ERC-Exos release. Mechanistically, MFGE8 on ERC-Exos binds to αvβ5 integrins on damaged intestinal epithelial cells, activating the PI3K/AKT/mTOR pathway. This upregulates anti-apoptotic BCL-2, reduces pro-apoptotic BAX/cleaved Caspase-3, and functionally promotes epithelial proliferation (blue ↑), inhibits apoptosis (red ↓), and restores mucosal barrier integrity.In parallel, MFGE8 inhibits colon dendritic cell (DC) maturation (red ↓) and CD4⁺ T cell proliferation (red ↓), which reduces pro-inflammatory cytokines (IL-6, IL-1β, TNF-α; red ↓) and increases anti-inflammatory IL-10 (blue ↑). Collectively, these dual effects (epithelial repair + immune homeostasis) synergistically alleviate intestinal inflammation and UC. |
Notably, our study represents the first successful application of this alginate-chitosan system for encapsulating ERC-Exos in UC treatment. We developed stable CSE nanocomposites by encapsulating ERC-Exos via the ionic crosslinking of SA and CS. CSEs presented as spherical microcapsules with smooth surfaces, demonstrating efficient encapsulation of ERC-Exos. The preservation of exosomal marker proteins post-encapsulation confirmed the maintenance of their structural and functional integrity. Critically, CSE exhibited an encapsulation efficiency of 89.2%±1.5%, which outperforms most reported chitosan-alginate-based exosome delivery systems (typically 70%–85%).44 This key modification enhances the carrier’s adhesion to the negatively charged colonic mucosa and facilitates drug retention at the target site.45 For oral delivery systems, stability represents a critical prerequisite for successful clinical translation. When incubated in phosphate-buffered saline (PBS) for 15 consecutive days, CSE maintained a consistent particle size distribution. The pH-dependent properties of CSE, which aligns with the need for targeted delivery in ulcerative colitis, enables the site-specific release of therapeutics and paves the way for clinical translation. These results indicated that the CSE encapsulation system effectively preserved ERC-Exos bioactivity, enabling them to exert a therapeutic effect in the colon and significantly ameliorate colitis symptoms. Notably, MFGE8 knockdown markedly attenuated the therapeutic efficacy of ERC-Exos in colitis treatment, indicating an irreplaceable role of MFGE8 in colitis treatment.
MFGE8, a ubiquitously expressed ubiquitously in stem cells, serves as a critical mediator of intercellular communication and participates in diverse physiological and pathological processes.46 Growing evidence supports the therapeutic potential of MFGE8 in tissue repair and regeneration.47 Studies by Ashish Chogle et al have demonstrated that MFGE8 deficiency leads to increased susceptibility to colitis and delays mucosal healing, while exogenous administration of MFGE8 alleviates UC.48 Similarly, our results indicate that MFGE8 is the key factor for the therapeutic effect of ERC-Exos on colitis. Specifically, MFGE8 helped ERC-Exos repair DSS-induced damage to the intestinal epithelium. Intestinal epithelial cells constitute a selective dynamic barrier that efficiently defends the intestinal lumen against harmful substances. This intricate defense system is predominantly composed of two interdependent components, the mucus layer and epithelial TJs.49 These elements establish a cohesive mucosal barrier that strictly limits the penetration of harmful luminal contents, while maintaining essential absorptive and immune surveillance functions. Disruption of either component of the intestinal barrier compromises the barrier integrity, leading to pathological antigen translocation and sustained inflammation. The TJs are principally composed of ZO-1, Occludin, and Claudins, as well as junctional adhesion molecules (JAMs).50 The dysfunction of TJs increases intestinal permeability, permitting paracellular translocation of immunogenic luminal components that drive pathogenic immune responses in gastrointestinal disorders.49 Our detailed investigations demonstrated that oral CSE administration significantly reversed DSS-induced augment intestinal permeability. Mechanistically, MFGE8-expressing ERC-Exos upregulated the expression of key tight junction proteins (ZO-1, occludin, and claudin-1) impaired by DSS, indicating an essential role of MFGE8 in both the maintenance and repair of epithelial barrier integrity during colitis. The intestinal mucus barrier is primarily composed of MUC2, which is secreted by the goblet cells. This gel-forming mucin plays a pivotal role in microbial symbiosis and containment.51 The disruption of the balanced crosstalk between microbial symbiosis and microbial containment can lead to intestinal diseases. Notably, our study revealed that MFGE8-expressing ERC-Exos prevented DSS-induced goblet cell depletion, and maintained mucin secretion during inflammation. Moreover, MFGE8 exerted dual cytoprotective effects across colonic tissues, simultaneously inhibiting apoptosis and promoting cellular regeneration, as demonstrated by complementary TUNEL and EdU assays.
Homeostasis of the intestinal mucosal immune system promotes the maintenance of intestinal epithelial integrity, and its imbalance induces and exacerbates colitis.52 Pailin Chiaranunt’s research demonstrates that the activation of CD4+ T cell clones is critical in controlling intestinal inflammation and inhibiting the proliferation and differentiation of CD4⁺ T cells alleviated UC.53 Additionally, Song et al revealed that inhibiting the maturation of DCs relieves DSS-induced inflammatory colitis.54 Interestingly, our results revealed that ERC-Exo treatment effectively ameliorated the DSS-induced dysregulation of intestinal CD4+ T cells and DCs in the colon. In particular, we found that ERC-Exo treatment significantly regulated the cytokine profile in colitis mice by suppressing the expression of pro-inflammatory mediators (IL-6, TNF-α, and IL-1β) and enhancing the production of the anti-inflammatory mediator IL-10. Indeed, MFGE8 exerts immunomodulatory effects on multiple types of immune cells and ameliorate multiple immune-mediated diseases.54 Specifically, Praveen Vasudevan et al demonstrated that the immunomodulation of MFGE8 improves functional outcomes after cardiomyocyte transplantation.55 Crucially, our results revealed that the recovery of intestinal immune homeostasis in colitis mice was largely attributed to the expression of MFGE8 in ERC-Exos. However, given the cellular heterogeneity of the intestinal lamina propria and the complexity of immunoregulatory networks, the exact mechanisms underlying the immunomodulatory functions of MFGE8 require further investigation.
Collectively, our findings indicate that oral administration of CSE effectively delivered MFGE8-exopessing ERC-Exos to the inflamed sites of DSS-induced colitis, where they were internalized to exert multifunctional therapeutic effects. The encapsulated MFGE8 mediates comprehensive mucosal repair by strengthening tight junctions to coordinate restoration of intestinal epithelial barrier integrity, enhancing intestinal barrier function by regulating goblet cells while promoting intestinal cell proliferation and inhibiting apoptosis. MFGE8 alleviates UC inflammation primarily by attenuating the intestinal infiltration and maturation of DCs, suppressing the proliferation of CD4+ T cells, thereby reducing the release of pro-inflammatory cytokines. This comprehensive therapy simultaneously targets structural barrier defects and inflammatory dysregulation, thereby improving the pathological progression of UC.
Mechanistically, MFGE8 is a structurally and functionally unique secretory protein that orchestrates critical biological processes through its modular domains. Its NH2-terminal EGF-like domain harbors an evolutionarily conserved RGD motif for specific αvβ3/αvβ5 integrin binding, while COOH-terminal C1/C2 domains mediate phosphatidylserine-dependent membrane binding with nanomolar affinity.56 Remarkably, H. Kemperman et al identified that both αVβ5 and αVβ6 integrins are co-expressed in colonic epithelial cells, with αVβ5 demonstrating significantly higher expression levels than αVβ6.57 Our supplementary mechanistic studies demonstrate that MFGE8 expressed in ERC-Exos mediates the phagocytosis of ERC-Exos by colonic cells via the αvβ5 integrin and facilitates the protective effects of MFGE8 on intestinal epithelial cells. Moreover, the αvβ5 integrin receptor mediates complex intracellular signaling pathways to regulate cellular metabolism and proliferation.58 Céline Defilles et al demonstrated that the αvβ5 integrin receptor activates the PI3K/AKT signaling pathway, thereby promoting cellular proliferation, metabolic activity, and cell migration. Substantial evidence has demonstrated that PI3K/AKT signaling significantly enhances cellular proliferative capacity and metabolic activity, which are critical for maintaining intestinal epithelial homeostasis.59,60 For instance, Zhou et al demonstrated that the activation of the PI3K/AKT pathway can promote wound re-epithelialization by inducing keratinocyte migration and epithelial mesenchymal transition.61 Extending this mechanistic framework to our study, we observed that ERC-Exos expressing MFGE8 serves to abrogate the impaired phosphorylation of PI3K and AKT in damaged intestinal epithelial cells, while concurrently orchestrating the activation of mammalian target of rapamycin (mTOR) — a key effector of the PI3K/AKT pathway that regulates cell proliferation, metabolic homeostasis, and protein synthesis critical for intestinal mucosal repair.62 The regulation of cellular apoptosis relies on the coordinated action of key molecules: B-cell lymphoma 2 (BCL2) is a classic anti-apoptotic protein that maintains cell survival by inhibiting mitochondrial membrane permeability; BCL2-associated X protein (BAX) is a pro-apoptotic protein that promotes the release of apoptosis-related factors from mitochondria; and Caspase-3 serves as the central executor of the apoptotic cascade, whose activation directly initiates and executes the apoptotic program.63 Together, these three molecules form the core molecular axis governing cellular apoptosis regulation. Consistent with the activation of the PI3K/AKT/mTOR pathway, our results further revealed tha MFGE8-expressing ERC-Exos modulate the balance of apoptotic regulators: they upregulate the expression of the anti-apoptotic protein BCL2 and downregulate the pro-apoptotic protein BAX, shifting the BCL2/BAX ratio toward an anti-apoptotic phenotype. Concomitantly, this pathway activation suppresses the cleavage and activation of Caspase-3—a central executor of the apoptotic cascade—thereby reducing intestinal epithelial cell apoptosis induced by inflammatory stress. Notably, inhibition of αvβ5 integrin via cilengitide not only suppresses the phosphorylation levels of PI3K and AKT but also abrogates the downstream activation of mTOR. This integrin-dependent pathway disruption further reverses the regulatory effects on the apoptotic molecular axis: It decreases BCL2 expression, increases BAX levels, and promotes Caspase-3 activation—ultimately disrupting the balance of cellular apoptosis and impairing the repair capacity of intestinal epithelial cells. Mechanistically, MFGE8-mediated activation of the PI3K/AKT/mTOR pathway, coupled with the modulation of BCL2/BAX balance and Caspase-3 activity, constitutes the core mechanism underlying the therapeutic effects of ERC-Exos in intestinal barrier repair. However, given the structural complexity of MFGE8 and its multiple receptor interactions, αvβ5 may represent one of several molecular pathways responsible for its pleiotropic actions. The spectrum of receptors and downstream signaling networks in the biological activities of MFGE8 requires systematic exploration in future research.
This study elucidated the remarkable bioprotective efficacy of our sodium alginate-chitosan oral delivery platform and demonstrated the therapeutic potential of orally administered ERC-Exos as a clinically viable nanobiologic for inflammatory bowel disease treatment. However, we must objectively recognize the key challenges in the translation of CSE from basic research to clinic. Although the current ionic crosslinking-based CSE preparation enables stable lab-scale production, scaling to GMP-compliant industrial production needs further research. Additionally, while CSE (formulated with ERC-Exos) shows relative stability in experiments, subtle batch-to-batch variations in ERC-Exos remain, requiring stricter batch quality control.64 Moreover, the residual MFGE8 following knockdown might lead to an underestimation of its function. In future research, a complete knockout model will be adopted for verification. For human safety, this study used mice rather than non-human primates; further validation with human intestinal organoids is needed. Moreover, individual differences in exosome uptake and unresolved questions about intestinal microbiota interactions also demand additional research. However, the regulatory effect of CSE on the intestine is consistent with the research results of Huang et al, who reported that “the exosome-microbiota interaction can promote intestinal barrier repair.65 Song et al confirmed the safety of exosome-based nanomedicines in gastrointestinal diseases,66 which further supports the translational potential of our research. Importantly, our findings highlight the pivotal contribution of MFGE8 in mediating both the tissue-reparative and anti-inflammatory functions of ERC-Exos. The identification of the aforementioned translational challenges and the preliminary exploration of corresponding strategies collectively lay a solid mechanistic and applied foundation for their future pharmaceutical development and clinical translation in mucosal repair therapies.
Conclusion
In conclusion, this study demonstrated that the oral administration of CSE represents a novel and effective therapeutic strategy for UC. The development of an oral delivery system for exosomes addresses the limitations of traditional intravenous administration by protecting exosomes from degradation in the gastrointestinal tract and enabling their targeted release into the colon. Our findings revealed that CSE significantly alleviated experimental colitis in mice. The therapeutic efficacy of CSE is largely mediated by MFGE8, a key protein expressed in ERC-exosomes, which enhances exosome uptake by intestinal epithelial cells through the αvβ5 integrin receptor and activates the PI3K/AKT/mTOR signaling pathway. This activation leads to suppression of apoptosis, promotion of epithelial cell proliferation, and restoration of tight junction integrity, thereby repairing the damaged intestinal barrier. The critical role of MFGE8 in mediating the therapeutic effects of CSE underscores its potential as a therapeutic target for UC and other inflammatory conditions. However, as previously stated, this research is subject to certain limitations. These include the standardization of CSE industrial production, the batch - to - batch quality control of ERC -Exos, and the necessity to validate the safety and efficacy of CSE in human intestinal organoids. All of these aspects necessitate further investigation. Collectively, these findings pave the way for the development of exosome-based therapies as promising alternatives to conventional treatments for inflammatory bowel disease.
Abbreviations
PI3K, Phosphatidylinositol 3-Kinase; AKT, Serine/Threonine Protein Kinase B; TNF-α, Tumor Necrosis Factor-α; IL-1β, Interleukin-1β; IL-6, Interleukin-6; IL-10, Interleukin-10; EdU, 5-ethynyl-2′-deoxyuridine; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; DSS, Dextran Sulfate Sodium; ANOVA, Analysis of Variance; H&E, Hematoxylin and eosin.
Data Sharing Statement
Data will be made available on request.
Ethics Approval and Consent to Participate
All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals: Eighth Edition and the Basel Declaration, and were approved by the Animal Ethics Committee of Tianjin Medical University (Approval No. IRB2024-DW-07). Human biological samples were collected in compliance with ethical standards under the approval of the Institutional Review Board of Tianjin Medical University General Hospital (Approval No. IRB2024-YX-013-01). Informed consent was obtained from all participants, and the study adhered strictly to the principles of the Declaration of Helsinki.
Funding
This work was supported by grants to Hao Wang from the National Natural Science Foundation of China (No. 82270794), Natural Science Foundation of Tianjin (No. 25JCYBJC00500), Horizontal Research Project of Tianjin Medical University General Hospital (No. 2023048), and Tianjin Key Medical Discipline Construction Project (No. TJYXZDXK-3-001B, and TJYXZDXK-3-013C).
Disclosure
The authors declare no actual or potential conflicts of interest, whether financial, personal, or professional, which could be perceived as influencing the research presented in this work.
References
1. Bruner LP, White AM, Proksell S. Inflammatory bowel disease. Prim Care. 2023;50(3):411–23. doi:10.1016/j.pop.2023.03.009
2. Du L, Ha C. Epidemiology and pathogenesis of ulcerative colitis. Gastroenterol Clin North Am. 2020;49(4):643–654. doi:10.1016/j.gtc.2020.07.005
3. Vasovic M, Gajovic N, Brajkovic D, Jovanovic M, Zdravkovaic N, Kanjevac T. The relationship between the immune system and oral manifestations of inflammatory bowel disease: a review. Cent Eur J Immunol. 2016;41(3):302–310. doi:10.5114/ceji.2016.63131
4. Lautenschläger C, Schmidt C, Fischer D, Stallmach A. Drug delivery strategies in the therapy of inflammatory bowel disease. Adv Drug Deliv Rev. 2014;71:58–76. doi:10.1016/j.addr.2013.10.001
5. Zhou C, Zhang B, Yang Y, et al. Stem cell-derived exosomes: emerging therapeutic opportunities for wound healing. Stem Cell Res Ther. 2023;14(1):107. doi:10.1186/s13287-023-03345-0
6. Murphy MP, Wang H, Patel AN, et al. Allogeneic endometrial regenerative cells: an “Off the shelf solution” for critical limb ischemia? J Transl Med. 2008;6:45. doi:10.1186/1479-5876-6-45
7. Fathi-Kazerooni M, Fattah-Ghazi S, Darzi M, et al. Safety and efficacy study of allogeneic human menstrual blood stromal cells secretome to treat severe COVID-19 patients: clinical trial Phase I & II. Stem Cell Res Ther. 2022;13(1):96. doi:10.1186/s13287-022-02771-w
8. Heidari N, Abbasi-Kenarsari H, Namaki S, et al. Adipose-derived mesenchymal stem cell-secreted exosome alleviates dextran sulfate sodium-induced acute colitis by Treg cell induction and inflammatory cytokine reduction. J Cell Physiol. 2021;236(8):5906–5920. doi:10.1002/jcp.30275
9. Harrell CR, Jovicic N, Djonov V, Arsenijevic N, Volarevic V. Mesenchymal stem cell-derived exosomes and other extracellular vesicles as new remedies in the therapy of inflammatory diseases. Cells. 2019;8(12):1605. doi:10.3390/cells8121605
10. Ha DH, Kim HK, Lee J, et al. Mesenchymal stem/stromal cell-derived exosomes for immunomodulatory therapeutics and skin regeneration. Cells. 2020;9(5). doi:10.3390/cells9051157
11. Wiklander OP, Nordin JZ, O’Loughlin A, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316. doi:10.3402/jev.v4.26316
12. Niu M, Pei Y, Jin T, et al. Colon-specific controlled release of oral liposomes for enhanced chemo-immunotherapy against colorectal cancer. Acta Pharm Sin B. 2024;14(11):4977–4993. doi:10.1016/j.apsb.2024.09.015
13. Zhang Y, Wang Y, Lu Y, et al. Advanced oral drug delivery systems for gastrointestinal targeted delivery: the design principles and foundations. J Nanobiotechnol. 2025;23(1):400. doi:10.1186/s12951-025-03479-8
14. Sen S, Xavier J, Kumar N, Ahmad MZ, Ranjan OP. Exosomes as natural nanocarrier-based drug delivery system: recent insights and future perspectives. 3 Biotech. 2023;13(3):101. doi:10.1007/s13205-023-03521-2
15. Yuan N, Shao K, Huang S, Chen C. Chitosan, alginate, hyaluronic acid and other novel multifunctional hydrogel dressings for wound healing: a review. Int J Biol Macromol. 2023;240:124321. doi:10.1016/j.ijbiomac.2023.124321
16. Deng C, Hu Y, Conceição M, et al. Oral delivery of layer-by-layer coated exosomes for colitis therapy. J Control Release. 2023;354:635–650. doi:10.1016/j.jconrel.2023.01.017
17. Guimarães D, Cavaco-Paulo A, Nogueira E. Design of liposomes as drug delivery system for therapeutic applications. Int J Pharm. 2021;601:120571. doi:10.1016/j.ijpharm.2021.120571
18. Luo W, Liu J, Algharib SA, Chen W. Antibacterial activity of enrofloxacin loaded gelatin-sodium alginate composite nanogels against intracellular Staphylococcus aureus small colony variants. J Vet Sci. 2022;23(3):e48. doi:10.4142/jvs.21292
19. Silvestre JS, Théry C, Hamard G, et al. Lactadherin promotes VEGF-dependent neovascularization. Nat Med. 2005;11(5):499–506. doi:10.1038/nm1233
20. Liu B, Zhang B, Qi J, et al. Targeting MFGE8 secreted by cancer-associated fibroblasts blocks angiogenesis and metastasis in esophageal squamous cell carcinoma. Proc Natl Acad Sci U S A. 2023;120(42):e2307914120. doi:10.1073/pnas.2307914120
21. Das A, Ghatak S, Sinha M, et al. Correction of MFG-E8 resolves inflammation and promotes cutaneous wound healing in diabetes. J Immunol. 2016;196(12):5089–5100. doi:10.4049/jimmunol.1502270
22. Zhao QJ, Yu YB, Zuo XL, Dong YY, Li YQ. Milk fat globule-epidermal growth factor 8 is decreased in intestinal epithelium of ulcerative colitis patients and thereby causes increased apoptosis and impaired wound healing. Mol Med. 2012;18(1):497–506. doi:10.2119/molmed.2011.00369
23. Gao Y, Wang C, Wang K, He C, Hu K, Liang M. The effects and molecular mechanism of heat stress on spermatogenesis and the mitigation measures. Syst Biol Reprod Med. 2022;68(5–6):331–347. doi:10.1080/19396368.2022.2074325
24. Zhang B, Wei X, Ding M, Luo Z, Tan X, Zheng Z. Daidzein protects Caco-2 cells against lipopolysaccharide-induced intestinal epithelial barrier injury by suppressing PI3K/AKT and P38 pathways. Molecules. 2022;27(24):8928. doi:10.3390/molecules27248928
25. Betker JL, Angle BM, Graner MW, Anchordoquy TJ. The potential of exosomes from cow milk for oral delivery. J Pharm Sci. 2019;108(4):1496–1505. doi:10.1016/j.xphs.2018.11.022
26. Lau SY, Kang M, Hisey CL, Chamley LW. Studying exogenous extracellular vesicle biodistribution by in vivo fluorescence microscopy. Dis Model Mech. 2023;16(8). doi:10.1242/dmm.050074
27. Liu J, Ren H, Zhang C, et al. Orally-delivered, cytokine-engineered extracellular vesicles for targeted treatment of inflammatory bowel disease. Small. 2023;19(50):e2304023. doi:10.1002/smll.202304023
28. Han X, Wu Y, Zhuang Y, Zhang J. Dexmedetomidine reduces dextran sulfate sodium (DSS)-induced NCM460 cell inflammation and barrier damage by inhibiting RhoA/ROCK signaling pathway. Allergol Immunopathol. 2022;50(3):85–92. doi:10.15586/aei.v50i3.569
29. Shabbir A, Cox A, Rodriguez-Menocal L, Salgado M, Van Badiavas E. Mesenchymal stem cell exosomes induce proliferation and migration of normal and chronic wound fibroblasts, and enhance angiogenesis in vitro. Stem Cells Dev. 2015;24(14):1635–1647. doi:10.1089/scd.2014.0316
30. Wong SK, Lawrencia D, Supramaniam J, et al. In vitro digestion and swelling kinetics of thymoquinone-loaded pickering emulsions incorporated in alginate-chitosan hydrogel beads. Front Nutr. 2021;8:752207. doi:10.3389/fnut.2021.752207
31. Li JY, Ren KK, Zhang WJ, et al. Human amniotic mesenchymal stem cells and their paracrine factors promote wound healing by inhibiting heat stress-induced skin cell apoptosis and enhancing their proliferation through activating PI3K/AKT signaling pathway. Stem Cell Res Ther. 2019;10(1):247. doi:10.1186/s13287-019-1366-y
32. Jeong DY, Kim S, Son MJ, et al. Induction and maintenance treatment of inflammatory bowel disease: a comprehensive review. Autoimmun Rev. 2019;18(5):439–454. doi:10.1016/j.autrev.2019.03.002
33. de Pedro M, López E, González-Nuño FM, et al. Menstrual blood-derived mesenchymal stromal cells: impact of preconditioning on the cargo of extracellular vesicles as potential therapeutics. Stem Cell Res Ther. 2023;14(1):187. doi:10.1186/s13287-023-03413-5
34. Chen L, Qu J, Cheng T, Chen X, Xiang C. Menstrual blood-derived stem cells: toward therapeutic mechanisms, novel strategies, and future perspectives in the treatment of diseases. Stem Cell Res Ther. 2019;10(1):406. doi:10.1186/s13287-019-1503-7
35. Chen Q, Shao B, Xu YN, et al. IGF2 contributes to the immunomodulatory effects of exosomes from endometrial regenerative cells on experimental colitis. Int Immunopharmacol. 2024;140:112825. doi:10.1016/j.intimp.2024.112825
36. Zhu Y, Qin H, Sun C, et al. Endometrial regenerative cell-derived exosomes attenuate experimental colitis through downregulation of intestine ferroptosis. Stem Cells Int. 2022;2022:3014123. doi:10.1155/2022/3014123
37. Sacks D, Baxter B, Campbell BCV, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke. 2018;13(6):612–632. doi:10.1177/1747493018778713
38. Wu Q, Xing Z, Luo S, et al. Assembly and operation of an easy-to-make portable device for facilitating mouse lateral tail-vein injection. Lab Anim. 2022;51(1):11–21. doi:10.1038/s41684-021-00889-7
39. Guo C, Lin L, Wang Y, Jing J, Gong Q, Luo K. Nano drug delivery systems for advanced immune checkpoint blockade therapy. Theranostics. 2025;15(11):5440–5480. doi:10.7150/thno.112475
40. Ding H, Tan P, Fu S, et al. Preparation and application of pH-responsive drug delivery systems. J Control Release. 2022;348:206–238. doi:10.1016/j.jconrel.2022.05.056
41. Chehreara A, Tabandeh F, Otadi M, Alihosseini A, Partovinia A. Enhanced survival of Lacticaseibacillus rhamnosus in simulated gastrointestinal conditions using layer-by-layer encapsulation. Biotechnol Lett. 2022;44(11):1277–1286. doi:10.1007/s10529-022-03289-0
42. Mohammadbaghban E, Taravati A, Najafzadehvarzi H, Khaleghzadeh-Ahangar H, Tohidi F. Oral administration of encapsulated catechin in chitosan-alginate nanoparticles improves cognitive function and neurodegeneration in an aluminum chloride-induced rat model of Alzheimer’s disease. Physiol Rep. 2024;12(13):e16095. doi:10.14814/phy2.16095
43. Jaafar MHM, Hamid KA. Chitosan-coated alginate nanoparticles enhanced absorption profile of insulin via oral administration. Curr Drug Deliv. 2019;16(7):672–686. doi:10.2174/1567201816666190620110748
44. Evers MJW, van de Wakker SI, de Groot EM, et al. Functional siRNA delivery by extracellular vesicle-liposome hybrid nanoparticles. Adv Healthc Mater. 2022;11(5):e2101202. doi:10.1002/adhm.202101202
45. Zhang M, Xu X, Su L, et al. Oral administration of Sophora Flavescens-derived exosomes-like nanovesicles carrying CX5461 ameliorates DSS-induced colitis in mice. J Nanobiotechnol. 2024;22(1):607. doi:10.1186/s12951-024-02856-z
46. Motegi SI, Ishikawa O. Mesenchymal stem cells: the roles and functions in cutaneous wound healing and tumor growth. J Dermatol Sci. 2017;86(2):83–89. doi:10.1016/j.jdermsci.2016.11.005
47. Li H, Zhang T, Wang K, et al. MFGE8 protects against CCl(4) -induced liver injury by reducing apoptosis and promoting proliferation of hepatocytes. J Cell Physiol. 2019;234(9):16463–16474. doi:10.1002/jcp.28314
48. Chogle A, Bu HF, Wang X, Brown JB, Chou PM, Tan XD. Milk fat globule-EGF factor 8 is a critical protein for healing of dextran sodium sulfate-induced acute colitis in mice. Mol Med. 2011;17(5–6):502–507. doi:10.2119/molmed.2010.00074
49. Li J, Zhang L, Wu T, Li Y, Zhou X, Ruan Z. Indole-3-propionic acid improved the intestinal barrier by enhancing epithelial barrier and mucus barrier. J Agric Food Chem. 2021;69(5):1487–1495. doi:10.1021/acs.jafc.0c05205
50. Zhang Y, Wang T, Sun M, et al. Advanced nanomedicine: redefining therapeutic paradigms for inflammatory bowel disease. Adv Healthc Mater. 2023;12(19):e2300069. doi:10.1002/adhm.202300069
51. Fang J, Wang H, Zhou Y, Zhang H, Zhou H, Zhang X. Slimy partners: the mucus barrier and gut microbiome in ulcerative colitis. Exp Mol Med. 2021;53(5):772–787. doi:10.1038/s12276-021-00617-8
52. Li YY, Wang XJ, Su YL, et al. Baicalein ameliorates ulcerative colitis by improving intestinal epithelial barrier via AhR/IL-22 pathway in ILC3s. Acta Pharmacol Sin. 2022;43(6):1495–1507. doi:10.1038/s41401-021-00781-7
53. Chiaranunt P, Tometich JT, Ji J, Hand TW. T cell proliferation and colitis are initiated by defined intestinal microbes. J Immunol. 2018;201(1):243–250. doi:10.4049/jimmunol.1800236
54. Song HY, Han JM, Kim WS, et al. Deinococcus radiodurans R1 lysate induces tolerogenic maturation in lipopolysaccharide-stimulated dendritic cells and protects dextran sulfate sodium-induced colitis in mice. J Microbiol Biotechnol. 2022;32(7):835–843. doi:10.4014/jmb.2203.03008
55. Vasudevan P, Wolfien M, Lemcke H, et al. CCR2 macrophage response determines the functional outcome following cardiomyocyte transplantation. Genome Med. 2023;15(1):61. doi:10.1186/s13073-023-01213-3
56. Mai J, Wang K, Liu C, Xiong S, Xie Q. αvβ3-targeted sEVs for efficient intracellular delivery of proteins using MFG-E8. BMC Biotechnol. 2022;22(1):15. doi:10.1186/s12896-022-00745-7
57. Kemperman H, Wijnands YM, Roos E. alphaV Integrins on HT-29 colon carcinoma cells: adhesion to fibronectin is mediated solely by small amounts of alphaVbeta6, and alphaVbeta5 is codistributed with actin fibers. Exp Cell Res. 1997;234(1):156–164. doi:10.1006/excr.1997.3599
58. Kumar B, Chandran B. KSHV entry and trafficking in target cells-hijacking of cell signal pathways, actin and membrane dynamics. Viruses. 2016;8(11):305. doi:10.3390/v8110305
59. Cai N, Dai SD, Liu NN, Liu LM, Zhao N, Chen L. PI3K/AKT/mTOR signaling pathway inhibitors in proliferation of retinal pigment epithelial cells. Int J Ophthalmol. 2012;5(6):675–680. doi:10.3980/j.issn.2222-3959.2012.06.05
60. Zhou J, Guo Z, Peng X, et al. Chrysotoxine regulates ferroptosis and the PI3K/AKT/mTOR pathway to prevent cervical cancer. J Ethnopharmacol. 2025;338(Pt 3):119126. doi:10.1016/j.jep.2024.119126
61. Zhou C, Guan D, Guo J, et al. Correction: human parathyroid hormone analog (3-34/29-34) promotes wound re-epithelialization through inducing keratinocyte migration and epithelial-mesenchymal transition via PTHR1-PI3K/AKT activation. Cell Commun Signal. 2023;21(1):243. doi:10.1186/s12964-023-01318-7
62. Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143(17):3050–3060. doi:10.1242/dev.137075
63. Kirsch DG, Doseff A, Chau BN, et al. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274(30):21155–21161. doi:10.1074/jbc.274.30.21155
64. Thakur A, Rai D. Global requirements for manufacturing and validation of clinical grade extracellular vesicles. J Liq Biopsy. 2024;6:100278. doi:10.1016/j.jlb.2024.100278
65. Huang B, An H, Gui M, et al. Qingjie Fuzheng Granule prevents colitis-associated colorectal cancer by inhibiting abnormal activation of NOD2/NF-κB signaling pathway mediated by gut microbiota disorder. Chin Herb Med. 2025;17(3):500–512. doi:10.1016/j.chmed.2025.04.001
66. Song G, Zeng C, Li J, et al. Exosome-based nanomedicines for digestive system tumors therapy. Nanomedicine. 2025;20(10):1167–1180. doi:10.1080/17435889.2025.2493037
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.