")
Back to Journals » International Journal of Nanomedicine » Volume 19
Oral Delivery of Photopolymerizable Nanogels Loaded with Gemcitabine for Pancreatic Cancer Therapy: Formulation Design, and in vitro and in vivo Evaluations
Authors Yugatama A , Huang YL, Hsu MJ, Lin JP, Chao FC , Lam JK, Hsieh CM
Received 2 December 2023
Accepted for publication 19 April 2024
Published 25 April 2024 Volume 2024:19 Pages 3753—3772
DOI https://doi.org/10.2147/IJN.S443610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Jie Huang
Adi Yugatama,1,2,* Ya-Lin Huang,1,* Ming-Jen Hsu,3 Jia-Pei Lin,1 Fang-Ching Chao,4 Jenny KW Lam,5 Chien-Ming Hsieh1,5,6
1School of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei, 11031, Taiwan; 2Department of Pharmacy, Sebelas Maret University, Surakarta, 57126, Indonesia; 3Department of Pharmacology, Taipei Medical University, Taipei, 11031, Taiwan; 4CNRS UMR 8612, Institut Galien Paris-Saclay, Université Paris-Saclay, Orsay, 91400, France; 5Department of Pharmaceutics, School of Pharmacy, University College, London, WC1N 1AX, UK; 6Ph.D. Program in Drug Discovery and Development Industry, College of Pharmacy, Taipei Medical University, Taipei, 11031, Taiwan
*These authors contributed equally to this work
Correspondence: Jenny KW Lam, Department of Pharmaceutics, School of Pharmacy, University College London, 29– 39 Brunswick Square, London, WC1N 1AX, UK, Email [email protected] Chien-Ming Hsieh, School of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei, 11031, Taiwan, Email [email protected]
Background: Gemcitabine (GEM) faces challenges of poor oral bioavailability and extensive first-pass metabolism. Currently, only injectable formulations are available for clinical use. Hence, there is an urgent demand for the development of advanced, efficacious, and user-friendly dosage forms to maintain its status as the primary treatment for pancreatic ductal adenocarcinoma (PDAC). Nanogels (NGs) offer a novel oral drug delivery system, ideal for hydrophilic compounds like GEM. This study aims to develop NGs tailored for GEM delivery, with the goal of enhancing cellular uptake and gastrointestinal permeability for improved administration in PDAC patients.
Methods: We developed cross-linked NGs via photopolymerization of methacryloyl for drug delivery of GEM. We reveal characterization, cytotoxicity, and cellular uptake studies in Caco-2 and MIA PaCa-2 cells. In addition, studies of in vitro permeability and pharmacokinetics were carried out to evaluate the bioavailability of the drug.
Results: Our results show NGs, formed via photopolymerization of methacryloyl, had a spherical shape with a size of 233.91± 7.75 nm. Gemcitabine-loaded NGs (NGs-GEM) with 5% GelMA exhibited efficient drug loading (particle size: 244.07± 19.52 nm). In vitro drug release from NGs-GEM was slower at pH 1.2 than pH 6.8. Cellular uptake studies indicated significantly enhanced uptake in both MIA PaCa-2 and Caco-2 cells. While there was no significant difference in GEM’s AUC and Cmax between NGs-GEM and free-GEM groups, NGs-GEM showed markedly lower dFdU content (10.07 hr∙μg/mL) compared to oral free-GEM (19.04 hr∙μg/mL) after oral administration (p< 0.01), highlighting NGs’ efficacy in impeding rapid drug metabolism and enhancing retention.
Conclusion: In summary, NGs enhance cellular uptake, inhibit rapid metabolic degradation of GEM, and prolong retention after oral administration. These findings suggest NGs-GEM as a promising candidate for clinical use in oral pancreatic cancer therapy.
Keywords: oral delivery, nanogel, gemcitabine, pancreatic cancer
Introduction
Pancreatic cancer remains one of the top ten causes of cancer deaths, which is possibly due to its late stage of recognition. In 2020, it was estimated that 495,773 new cases of pancreatic cancer, and the incidence varied by region and population.1 Gemcitabine (GEM) is a first-line drug for metastatic pancreatic cancer approved by the Food and Drug Administration (FDA). GEM has demonstrated tremendous efficacy along with clinical benefits over 5-fluorouracil for more than 20 years.2 The drug has demonstrated wide-spectrum application in various solid tumors.
Despite exhibiting considerable therapeutic efficacy, GEM is subject to various constraints, such as a relatively brief plasma half-life, as well as limited permeability.3–5 The brief duration of serum half-life can be ascribed to the swift and extensive process of deamination of GEM by cytidine deaminase (CDA), which is present in several tissues such as blood, liver, and kidneys.6,7 Cytidine deaminase enzymatically converts GEM into its inactive metabolite, known as 2’,2’-difluoro-2’-deoxyuridine (dFdU), which is then eliminated through urinary excretion. Consequently, the administration of GEM necessitates regular dosing intervals in order to sustain a therapeutic concentration of the drug within the systemic circulation.7
In addition, it is notable that GEM is frequently administered to patients through the intravenous (IV) route, leading to an increased risk of venous pain in the injection site and higher associated costs.8,9 After mediation by human nucleoside transporters that diffuse inside cells, GEM is phosphorylated twice to become monophosphate (dFdCMP), and the diphosphate form (dFdCDP), by deoxycytidine kinase and nucleoside monophosphate kinase. Notably, a higher binding affinity to cytidine deaminase makes it vulnerable to metabolization into its inactive form (dFdU), especially upon intravenous administration, which contributes to accumulating toxicity.10,11 Further, this enzymatic plasma deaminase causes GEM to have a short half-life (8 to 17 minutes).12 One way to increase the efficacy of GEM treatment in pancreatic ductal adenocarcinoma (PDAC) is to use nano-based formulations. Previous studies showed that other drugs, such as paclitaxel and irinotecan, can successfully be formulated into more efficacious treatments for PDAC by nanoparticle systems.13,14 Recently, some strategies were developed to enhance GEM delivery for cancer treatment through nanoparticle formulations, such as a prodrug-based approach,15,16 low-dimensional inorganic nanomaterial (ie, black phosphorus and graphene oxides)-assisted delivery,17,18 polymeric and lipid nanoparticle-based hydrogels,19–22 and combinative drug-based hydrogels23–27 which are formulated towards injectable administration forms15,16,24,28 and local delivery.16
Among drug delivery administration routes, oral delivery is the most favorable option due to its primary advantages in offering a non-invasive route and enabling self-administration.29 It eradicates any discomfort that might arise from an injection, thus being the most recommended choice to increase patient compliance with medication. Oral delivery provides the physiological benefit of a high surface area of epithelial cells (enterocytes), allowing drug compounds to be readily absorbed into the bloodstream.29–32 However, several drugs, including GEM, suffer from low oral bioavailability due to poor intestinal permeability and metabolic instability. Numerous studies have been conducted to confront these drawbacks by incorporating drug compounds into nanoparticle forms, manipulating the limited properties to increase residence times and facilitate better interactions with cells of the gastrointestinal (GI) tract.33–37
For example, polymeric-based nanoparticles are known to have stable characteristics while protecting the encapsulated drug during its stay in the GI tract. Its polymeric base possesses the capability to improve the physicochemical properties of the nanoparticles, turning them into a suitable scaffold for a controlled-release mechanism.30 Significant impacts of this approach were shown in high cellular uptake, greater efficient targeting, and improved bioavailability.23,38–40 Hydrogels also offer spatial and temporal control over numerous chemotherapeutic agents owing to their tunable physical properties, high drug-loading efficiency, excellent biocompatibility, controllable degradability, and programmable release profiles.41–45
Hydrogel nanoparticles (NGs) are three-dimensional hydrogel materials formed by a cross-linked expanded polymer network with high capacity at the nanoscale, enabling them to retain water without completely dissolving in aqueous media. This chemical composition can be manipulated to adjust various characteristics, including dimensions, charge, amphiphilicity, porosity, softness, and degradability. NGs possess several adjustable characteristics, such as flexibility, nanoscale size, dispersibility in biological fluids, high stability, biodegradability, and chemical activity. Furthermore, they exhibit controllable synthesis and swelling ratio, facilitate drug loading and release, and function as carriers responsive to environmental stimuli. NGs have the potential to be engineered as materials that exhibit responsiveness to a variety of external and internal stimuli, such as changes in the reductive environment, light, electricity, pH, temperature, enzyme activity, magnetic fields, and ultrasound.46–53 In most cases, these reactions induce conformational alterations in the NG structure and may lead to the “on-demand” discharge of the released cargo. They can transport and safeguard the active ingredients enclosed within from degradation during storage or blood circulation (eg, hydrolysis or enzymatic degradation), reduce toxic side effects, or deliver the medication exclusively to the intended site of action.54,55 Thus, NGs play an important role in maintaining cargo integrity and preventing its degradation and elimination. Additionally, they actively contribute to the delivery process, facilitating a controlled and triggered response at the desired target location.56
To our knowledge, the oral administration of gemcitabine remains challenging in clinical applications. Additionally, there is no reported formulation for an orally administered hydrogel-based GEM for pancreatic cancer. Consequently, there is a compelling demand to develop an oral delivery system that enhances both cellular uptake and gastrointestinal permeability. We, herein, propose oral NGs composed of polymer gelatin and methacrylic groups as a nanocarrier for GEM. This study was aimed at characterizing the physicochemical, drug release, permeability, and pharmacokinetics (PK) of NGs containing GEM (NGs-GEM). Coating GEM within the NGs system not only improved the drug release in the intestinal medium but also exhibited cellular uptake in MIA PaCa-2 and Caco-2 cell models compared to free-GEM. The bioavailability of the NGs-Gem was improved by 40% with less formation of the dFdU inactive metabolite.
Materials and Methods
Materials
The cell lines and related chemicals in this study were purchased from the Taiwan Biological Resources Conservation and Research Center (Bioresource Collection and Research Center, BCRC, Hsinchu, Taiwan) with a complete culture medium. The cell lines were inoculated in 75T culture flasks, and cultivated in an incubator (37 °C, 5% CO2).
The following products were purchased commercially: 2,2’-azobis(2-methyl-N-(2-hydroxyethyl)propionamide) (VA-086, Wako Chemicals, Osaka, Japan), 2’,2’-difluorodeoxyuridine (dFdU, Carbosynth, Compton, UK), antibiotic-antimycotic solution (100×), Dulbeccos modified Eagles medium (Corning® DMEM), trypsin-EDTA (Corning, NY, USA), GEM (dFdC, Scinopharm Taiwan, Tainan, Taiwan), 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT), 2’-deoxyuridine (dU), gelatin from porcine skin (type A, 300 g bloom of gel strength), hydrochloric acid (HCl), lucifer yellow, methacrylic anhydride, pancreatin, pepsin from porcine stomach mucosa (800–2500 units/mg of protein), sodium carbonate, sodium bicarbonate, sodium hydroxide, Span 80, human transferrin (Sigma-Aldrich, St. Louis, MO, USA), Dulbecco’s cell line BCRC no. MIA PaCa-2 60,139 human pancreatic cancer cell line, C2BBe1 (Caco-2) 60,182 human colorectal adenocarcinoma cell line, modified Eagle’s medium (MEM 12800–017), fetal bovine serum (FBS), horse serum (HS) (Gibco, Invitrogen, Grand Island, NY, USA), acetonitrile, methanol (LiChrosolv® Reag. Ph Eur.), Tween 80 (Merck, Billerica, MA, USA), dimethylformamide (DMF), sodium chloride, sodium acetate, tetrahydrofuran (JT Baker, Center Valley, PA, USA), and n-octane, (Alfa Aesar, Heysham, England).
Synthesis of Methacrylate-Esterified Gelatin (GelMA)
The method used is a modification of previous research to obtain a higher degree of substitution.57 The utilization of a carbonate-bicarbonate (CB) buffer has been found to result in a greater level of substitution (DS) in comparison to the utilization of phosphate-buffered saline (PBS). Briefly, 10% gelatin type-A was fully dissolved in 0.1 M CB buffer (3.18 g Na2CO3 and 5.86 g NaHCO3 in 1 L of deionized water preheated to 50 °C) under magnetic stirring at 50 °C. Subsequently, the pH was modified to 9, followed by the gradual addition of methacrylic anhydride (MA) to the gelation solution in a 10:1 ratio. The methacrylation reaction was conducted with continuous stirring at 50 °C for 90 minutes. The reaction is halted when the pH reaches 7.4. Following that, the mixture was dialyzed against pure water (12~14 kDa cut off) to remove excess methacrylic acid and salts at 40 °C for 5 days. Finally, the GelMA floc was obtained after lyophilization, and the degree of methacrylation was further characterized by nuclear magnetic resonance (NMR).
GelMA-Labeled fluorescein Isothiocyanate (FITC) synthesis
GelMA was dissolved in 100 mM NaHCO3 buffer (5 mL) at 40 °C to obtain a concentration of 20 mg/mL. A 2 mg/mL FITC solution in DMF (1 mL) was added to the previous solution and stirred for 6 h at 40 °C. The mixture was then dialyzed against distilled water at 40 °C for 7 days followed by freeze-drying to obtain solid FITC-GelMA.
Determination of the GelMA Degree of Substitution (DS)
The quantification of methacryloyl functionalization was conducted using a 500 MHz 1H NMR instrument (Bruker Avance DRX 500, Burladingen, Germany), using a previously established protocol.58 Prior to the analysis, GelMA samples were dissolved in a 5 mg/mL solution of deuterium oxide (D2O). The quantification of the DS in GeIMA involved determining the proportion of amino groups (namely lysine and hydroxylysine) inside the gelatin that underwent modification. The lysine methylene (LM) signals of the GeIMA and unmodified gelatin spectra at 2.8–2.95 ppm were then integrated to determine the respective areas. Subsequently, to calculate DS the following equation is used.

Preparation of GelMA NGs
The methods of preparation of GelMA NGs were adapted from a previous report with slight modifications.59 GelMA NGs were produced through the process of photopolymerization of GelMA within a water-in-oil (w/o) emulsion. First, 5% GelMA was prepared in ultrapure water by heating it at 40 °C. Once it had dissolved, 1.5% VA-086 (the initiator) was added as the aqueous phase. Next, the mixture was introduced into an organic (octane) and surfactants (Tween 80: Span 80 = 3:2) mixture. According to a prior investigation, the composition of the organic, surfactants, and aqueous phase was reported as 66.7:16.7:16.7.60 The mixture was homogenized using a homogenizer (T 25 digital ULTRA-TURRAX®, IKA, Staufen, Germany) for 5 minutes at 8000 rpm. Subsequently, it was homogenized using an ultrasonic probe sonicator (Q700, QSonica, Newtown, CT, USA) with an amplitude of 10% (70 W) at 4°C for 15 minutes. The GelMA gel in the emulsion was formed by subjecting UV light at a wavelength of 385 nm to an intensity of 10 W/cm2 for 30 minutes. Following photopolymerization, tetrahydrofuran (THF) was introduced with a ratio of 1:9 and centrifugated (8000 g, 10 minutes) to eliminate the organic phase and surfactants. Subsequently, the precipitate was reconstituted with deionized water (DIW) or PBS pH 7.4, before freeze-drying to obtain dry NGs in the form of a powder.
Characterization of NGs-GEM
Characterization of NGs-GEM included particle size, polydispersity index (PDI), and zeta potential utilizing a Malvern Zetasizer NanoZS (Malvern Instruments, Worcestershire, UK). Following dilution with DIW, the samples were examined at 25 °C under a dispersion angle of 90°C. Furthermore, an analysis of the structure and surface characteristics of the NGs and NGs-GEM was conducted using transmission electron microscopy (TEM, HT7700 Hitachi, Tokyo, Japan).
Previously, a dilution of 50-fold was carried out using DIW and stained with 2% uranyl acetate, a positively charged dye, before being subjected to imaging. A drop of NGs or NGs-GEM sample was placed on a carbon-coated copper grid for observation.
Drug Loading Efficiency of NGs-GEM
To study the drug loading efficiency (DLE), 3 mg/mL of a GEM solution was first homogenously mixed with the NGs at a ratio of 3:1 and incubated for complexation at 4 °C for 24 h. Ultrafiltration (10 kDa Ultra-50 Centrifugal Filter Units) was then used to collect the NGs-GEM precipitate. After centrifugation, the precipitate obtained was analyzed using high-performance liquid chromatographic (HPLC, Waters e2695 Separation Module, Milford, MA, USA) with UV-visible detector at 268 nm, C18 Column (4.6 × 250 mm, 5 µm, ZORBAX Extend-C18, Agilent, Santa Clara, CA, USA). Methanol and 0.1% formic acid (5:95% v/v) as mobile phase, after 0.22 μm membrane filtration for ultrasonic shock degassing to complete the mobile phase preparation. The DLE (%) was calculated using the following equation.

where D0 is the GEM added, and D1 is the GEM in the supernatant.
In vitro Drug Release of NGs-GEM
The release kinetics of GEM from NGs-GEM and free-GEM were analyzed by weighing the sample (equivalent to 0.4 mg GEM) and dissolving it in 1 mL of simulated gastric fluid (SGF) or simulated intestinal fluid (SIF). The solution was then added to a cellulose membrane with 3.5 kDa MWCO containing 20 mL of SGF or SIF and slowly homogenized at 100 rpm and 37 ± 1 °C. Samples were taken at several times (5, 10, 30, 60, 120, 240, and 360 min) with 1 mL of volume replacement each time. Prior to the HPLC analysis, samples were 0.22 μm membrane filtered. Results were compiled as an in vitro drug release graph. The compositions of SGF and SIF are described in the following two sections. SGF was prepared by preparing 34 mM NaCl and 84 mM HCl, adjusting the pH to 1.2 with 0.2 M HCl, and then adding pepsin to obtain a concentration of 3.2 g/L. SIF was prepared using a similar method: a solution of 50 mM KH2PO4 and 15 mM NaOH was prepared, then adjusting the pH to 6.8 with 0.2 M HCl, and pancreatin was added to obtain a concentration of 10 g/L.
Cytotoxicity Assessments
The cytotoxicity of NGs-GEM was evaluated by conducting MTT tests on Caco-2 and MIA PaCa-2 cells. Briefly, the cells were distributed into 48-well culture plates at a seeding density of 2×104 cells per well. Following a day incubation period at 37 °C with 5% CO2, the adherent cell culture media was substituted with a serum-free culture media. Subsequently, the cells were treated with varying concentrations (0.001 to 1000 µg/mL) of GEM, NGs, and NGs-Gem. After a 48-hour incubation period, 300 µL of 1 mg/mL MTT solution in PBS pH 7.4 was introduced and incubated for 1 hour at 37 °C. Subsequently, the extraction process was carried out by adding 100 µL DMSO. The measurement of optical density (OD) was conducted at a wavelength of 540 nm using a microplate reader (Cytation™ 3 Cell Imaging Multi-Mode Reader, BioTek, Winooski, VT, USA).
Cellular Uptake
Caco-2 and MIA PaCa-2 cells were first seeded under a constant temperature of 37 °C and 5% CO2 for 1 day at the density of 5×105 cells/well. To assess the uptake of NGs-GEM, cells were incubated with NGs-FITC (concentration 2.5 mg/mL) at interval times of 0.5, 1, 2, and 4 hours. After the incubation period, cells were washed 3 times with cold 1× PBS to remove excess NGs-FITC. Subsequently, trypsinization was performed. The fluorescent signal of FITC was assessed using a flow cytometer (FACS Canto-II, BD Biosciences, San Jose, CA, USA) in order to assess the cellular uptake of NGs.
In vitro Permeability
The permeability assay was conducted using monolayer cultures cultivated in a 24-well plate with a PET membrane featuring 0.4 µm pores (Falcon®, Corning, located in Glendale, AZ, USA).
The Caco-2 cultures were seeded at a density of 105 cells/well into each well insert and thereafter incubated at 37 °C and a carbon dioxide 5% for 96 hours. Transepithelial electrical resistance (TEER) values were measured using an epithelial volt/ohmmeter (EVOM, World Precision Instruments, Sarasota, FL, USA) to assess the integrity of the cell monolayer before the study (greater than 400 Ω cm2). Additionally, the apparent permeability coefficient (Papp) at 485 nm and 530 nm for the yellow lucifer marker was also monitored to ensure cell integrity (less than 10−6 cm/s).
For the permeability study, 300 µL of a GEM or NGs-GEM solution (with an initial drug concentration of 0.1 mg/mL) dissolved in DMEM was added to the apical chamber of each well, while the basolateral side contained 0.6 mL of transport buffer. Samples (100 µL) were extracted at time intervals of 0.5, 1, 1.5, and 2 hours. Following this, the collected samples were retained at 4 °C until the HPLC analysis. The permeability coefficient (PE) is calculated using reciprocal correlation as determined by the following equation.61

where PEcell is the PE through the cell layer only, PEall is the PE through the coated membrane insert and cell layer, and PEblank is the PE across the coated membrane insert without the cell layer.
In vivo PK Studies
In the PK study, 8~10 week old male Sprague Dawley rats were divided into 3 groups (n=6) randomly to investigate PK profiles of (1) the oral free-GEM solution, (2) oral NGs-GEM, and (3) GEM intravenously (iv) at doses 25, 25, and 10 mg/kg BW, respectively. The procedures used complied with the Taiwan Animal Welfare Law and were approved by the TMU Institutional Animal Care and Use Committee (approval number: LAC-2017-0282). Blood samples of 0.5 mL were sampled at 5, 10, 45, 60, 120, 240, 480, 720, and 960 minutes after treatment. Blood samples were placed in heparinization tubes and then centrifuged (4000 rpm, 10 mins, 4 °C). The supernatant obtained was then stored (−80 °C) for further HPLC-UV analysis.
GEM and dFdU in rat plasma were quantitatively analyzed by HPLC-UV at 268 nm, sample injection of 5 μL, column thermostat at 30 °C, and flow rate of 0.8 mL/min. This experiment was done using the following conditions: methanol and 0.1 M acetate buffer (10:90% v/v) as mobile phase (2 L of sodium acetate (16.4 g) buffer was prepared and the pH adjusted to 5.0 with 5 N HCl).
PK parameters were analyzed using non-compartmental analysis using the mean ± standard deviation (SD) values of each treatment group. The terminal elimination rate constant (Ke), half-life (T1/2), the area under the plasma drug concentration-time curve (AUC) from the beginning to the endpoint (AUC0–last), and AUC0→∞ were calculated and presented as mean ± SD. Metabolite ratios (dFdU/GEM) were computed by dividing the AUC0→∞ of dFdU by AUC0→∞ of GEM.
Statistical Analysis
All data are expressed as mean ± SD unless otherwise stated. Statistical evaluation used a Student’s t-test or one-way analysis of variance (ANOVA) and differences between groups were considered significant when the probability (p-value) was less than 0.01.
Results and Discussion
Synthesis and Characterization of GelMA
The GelMA macromer was successfully synthesized via a methacrylation reaction. GelMA is synthesized through the reaction of methacrylic anhydride (MAA) and gelatin, resulting in the attachment of methacryloyl to the amino groups on the side chain of the amino acids. This reaction also produces methacrylic acid as a byproduct. The synthesized GeIMA macomer was characterized by a 1H-NMR spectrum. The 1H-NMR spectrum of the GeIMA confirmed the confirmed the structure of the desired polymer (Figure 1). The presence of two signals of the vinyl methacrylate group of MA (δ = 5.4 and 5.7 ppm) and a decrease in the signal of methylene lysine (δ = 3.05 ppm) confirms the free-MA modification.62 In addition, enrichment of the signal methyl group in MA (δ = 1.97 ppm) supported the substitution of MA. The DS was calculated by comparing the proton signals methylene lysine of unmodified gelatin and GelMA. This result suggested the successful grafting of MA onto gelatin promoted by a cross-linking mechanism to form a three-dimensional structure of GelMA.
 |
Figure 1 Synthesis of gelatin methacrylate (GelMA) (a) and 1H nuclear magnetic resonance spectra of gelatin and GelMA (b). |
Variations in the added MA volume can change the DS of GEIMA. The DS can affect the size, porosity, compressive modulus, and swelling ability of the hydrogel. In this study, we modified the previous method to obtain a high DS. In addition, based on previous research, CB buffer was used to obtain GEIMA with a high level of DS.63
Optimization of NGs
GelMA NGs can be created by exposing a GelMA solution with a photoinitiator added under UV light. The mechanical properties of the GelMA hydrogels can be regulated by changing the concentration of MA and UV exposure time. The degrees of substitution may affect the porosity, pore size, compressive modulus, and swelling behavior of the hydrogels.64 Indeed, the shape and materials within this type of NP play key roles in determining the final properties of the hydrogel NPs. Harnessing GelMA in this case was able to improve the hydrogel NP formulation and control their size and chemistry.
Further, the NGs consisted of an aqueous phase (GelMA), surfactants, and organic phases (Tween 80: span 80: octane at 0.6: 0.9: 6). The effects of different parameters of the NGs are discussed below, such as different concentrations of GelMA, comparison of two organic phases, and parameters of the ultrasonicator.
GelMA (at 5%, 10%, or 15%) was first examined for solubility in organic solvents. As shown in Figure 2, GelMA seemed to precipitate in heptane. Meanwhile, good solubility was indicated in octane (Figure 3) composed of a 5% hydrogel in a water and octane mixture. Since this was indicated as the best potential solvent, this composition was used for further experiments.
 |
Figure 2 Image of NGs (heptane): (a) 15%, (b) 10%, and (c) 5% gelatin methacrylate (GelMA). |
 |
Figure 3 Image of NGs prepared in different organic solvents: (a) heptane and (b) octane. |
An ultrasonicator was employed to disperse the GelMA in the organic phase. UV-visible light was utilized to induce VA086 to generate free radicals (N=N; double-bond breakage) and initiate covalent linkages within gelMAs, thus forming hydrogel NPs (NGs). The ultrasonic oscillation time was parameterized to evaluate its effect on particle size formation. As shown in Figure 4, the particle size along with the polydispersity index (PDI) decreased as the oscillation time increased (within the range of 10 to 30 min). For instance, a lower particle size (< 250.0 nm) and PDI (< 1.000) were obtained after 15 min of ultrasonication. Further, particle sizes of NGs and NGs-Gem were 233.91 ± 7.75 and 244.07 ± 19.52 nm, respectively, suggesting good compatibility of the drug with the NG elements (Table 1). This was shown by the slight increase in particle size compared to native NGs. Interestingly, the surface potential of native NGs was negatively charged, while the NGs-Gem were positively charged. The free hydrogen of GEM hydrochloride was likely bound to the NG surface, thus forming a positively charged surface of NGs-Gem.
 |
Table 1 Characterization of 5% Gelatin Methacrylate (GelMA) Nanogels (NGs) with 15 Min of Sonication Time (Mean ± Standard Deviation) |
 |
Figure 4 Particle size of 5% gelatin methacrylate (GeIMA) NGs. |
Characterization of NGs
We next evaluated the appearance of native NGs, the interface level, and the encapsulated formation of NGs-GEM by TEM. The results showed that the structures of NG and NGs-GEM were spherical, and particle sizes were in accordance with those demonstrated using the particle size analyzer (Figure 5).
 |
Figure 5 Characterization of NGs and NGs-GEM. (a) TEM image of NGs, (b) TEM image of NGs-GEM. (scale bar = 200 nm). |
At the same time, drug concentrations (ranging 1~5 mg/mL) were prepared and loaded in 1 mg NGs to determine the drug loading efficiency (DLE, %). As shown in Table 2, it was obvious that the DLE increased with an increment in the drug concentration, indicating that the NG system was able to handle concentrations of GEM of 1~3 mg/mL. Therefore, the drug loading test was conducted at 3 mg/mL. The DLE of NGs in this experiment was 37.2%±9.3%. This result was in accordance with the theory that cross-linked NGs can have high DLE capacities up to 50% by weight because of their structural properties. This benefit also made NGs a relatively high DLE among other nanocarriers.65
 |
Table 2 Drug Loading Efficiency of 5% Gelatin Methacrylate (GelMA) NGs with 15 Min of Sonication Time (Mean ± Standard Deviation) |
In vitro Release Test
The in vitro release behavior was evaluated by testing the NG system in SGF and SIF environments at 37±1 °C. An in vitro dissolution test was used to simulate the release characteristics of the drug in the body and observe its effects in different digestive fluids. As shown in Figure 6, the release of free-GEM in the first hour was 63.1% in SGF and 61.6% in SIF. Release extents reached 93.6% and 100.8% after 6 h in the two environments. Free-GEM was completely released after 6 h in both high- and low-acid environments. Interestingly, NGs-GEM in both SGF and SIF solutions were released more slowly, reaching 21.4% and 54.6% in the first hour and continuing to 34.3% and 76.6% at 6 h, respectively. Comparing the curves of the NP system in the two different environments, NGs-GEM seemed to obtain higher release rates in an intestinal environment (SIF), which had a higher pH.
 |
Figure 6 Drug release profile of Free-GEM and NGs-GEM in (a) simulated gastric fluid (SGF) and (b) simulated intestinal fluid (SIF). |
Free GEM was fully released after 6 h in both the high- and low-acidic environments. Comparing the in vitro release curves of NGs-Gem in SGF and SIF, results suggested that the release percentage of the NGs-Gem group in SIF medium was higher than that in SGF, which also indicates that NGs-gem group tended to be released more slowly in the more-acidic environment. This was likely due to the presence of -COOH groups on the NG surface. When NGs are in a medium with a pH greater than the pKa value of the acidic side groups on the polymer chain, acidic groups are ionized, resulting in a swelling of the polymeric system, thereby releasing drug molecules into the SIF environment. In addition, the release speed and total amount of drug released in the NGs-gem group significantly improved in SIF. A greater amount of negatively charged ions in the SIF medium and greater repulsion effects among hydrogel polymer chains caused a higher degree of swelling.66 Consequently, if the surface of the hydrogel has cationic groups, such as amino groups (-NH2), the colloidal network would swell in a lower pH (more acidic) environment.67 The use of NGs in this regard, seemed to be capable of circumventing drug degradation and maintaining drug stability during transport through the acidic environment (gastric fluid).68
Cytotoxicity Test
To examine the cytotoxic effects of the NGs system, an MTT assay was carried out on MIA PaCa-2 and Caco-2 cells. Blank-NGs (containing no drugs) at certain concentrations (ranging from 0.001~1000 μg/mL) were incubated with cancerous cells for 48 h. Cell survival was determined using the MTT reduction reaction. As shown in Figure 7a, high rates of cell survival (> 80%) were indicated in both cell types, which is in accordance with ISO 10993–5 (survival rates greater than 80% are classified as non-toxic activity of a material). The effects of different drug concentrations were also compared to the NGs-GEM system in Figure 7b. It showed that the NGs-GEM system could potentially more strongly suppress cancerous cells than blank-NGs.
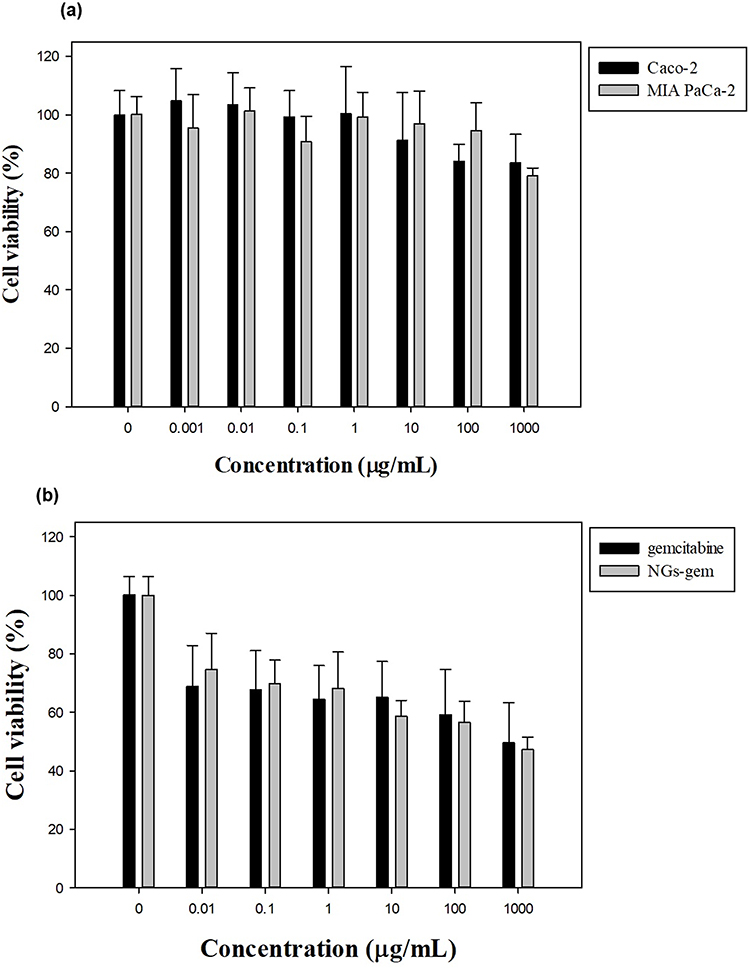 |
Figure 7 MTT assay of NGs in MIA PaCa-2 and Caco-2 cells (a); MTT assay of NGs-GEM in MIA PaCa-2 cells (b). |
Cell Uptake
GelMa prior to NG preparation was first FITC-labeled to facilitate visualization and quantification of NG cellular internalization. The particle size of NG labeled with FITC and then freeze-dried increased to 276.1 ± 3.77 nm with a PDI of 0.494 ± 0.056. MIA PaCa-2 and Caco-2 cells were cultured for different time points (0.5~4 h) and then further analyzed through flow cytometry to observe cellular behavior toward the NGs. According to Figure 8, more NGs were taken up by both types of cells as the incubation time increased. Uptake by MIA PaCa-2 cells increased from 25.84% to 74.55% in 4 h (Figure 9), while Caco-2 cells increased from 15.68% to 69.20% (Figure 10). These results imply that the NP system successfully delivered the drug to cells. Therefore, with an increase in exposure time, there was an increment in the cellular uptake of NGs, which showed similar trends for both types of cells. However, higher uptake was found by Mia PaCa-2 cells. These results are similar to previous studies, where conventional chemotherapeutic drugs (such as gemcitabine) had better ability to inhibit cell proliferation in Mia PaCa-2 cells than other cells.69,70
 |
Figure 8 Fluorescence intensity from FACS measurements in (a) MIA PaCa-2 and (b) Caco-2 cells incubated with NGs for 1 and 4 h. Average fluorescence intensity normalized to the control upon incubation of cells with NGs (c). Data are expressed as the mean ± standard deviation (n=3). ** p < 0.01, *** p < 0.001, **** p < 0.0001, when incubated with NGs for 1 to 4 h compared to the control. #p < 0.05, ###p < 0.01, in MIA PaCa-2 compared to Caco-2 cells. |
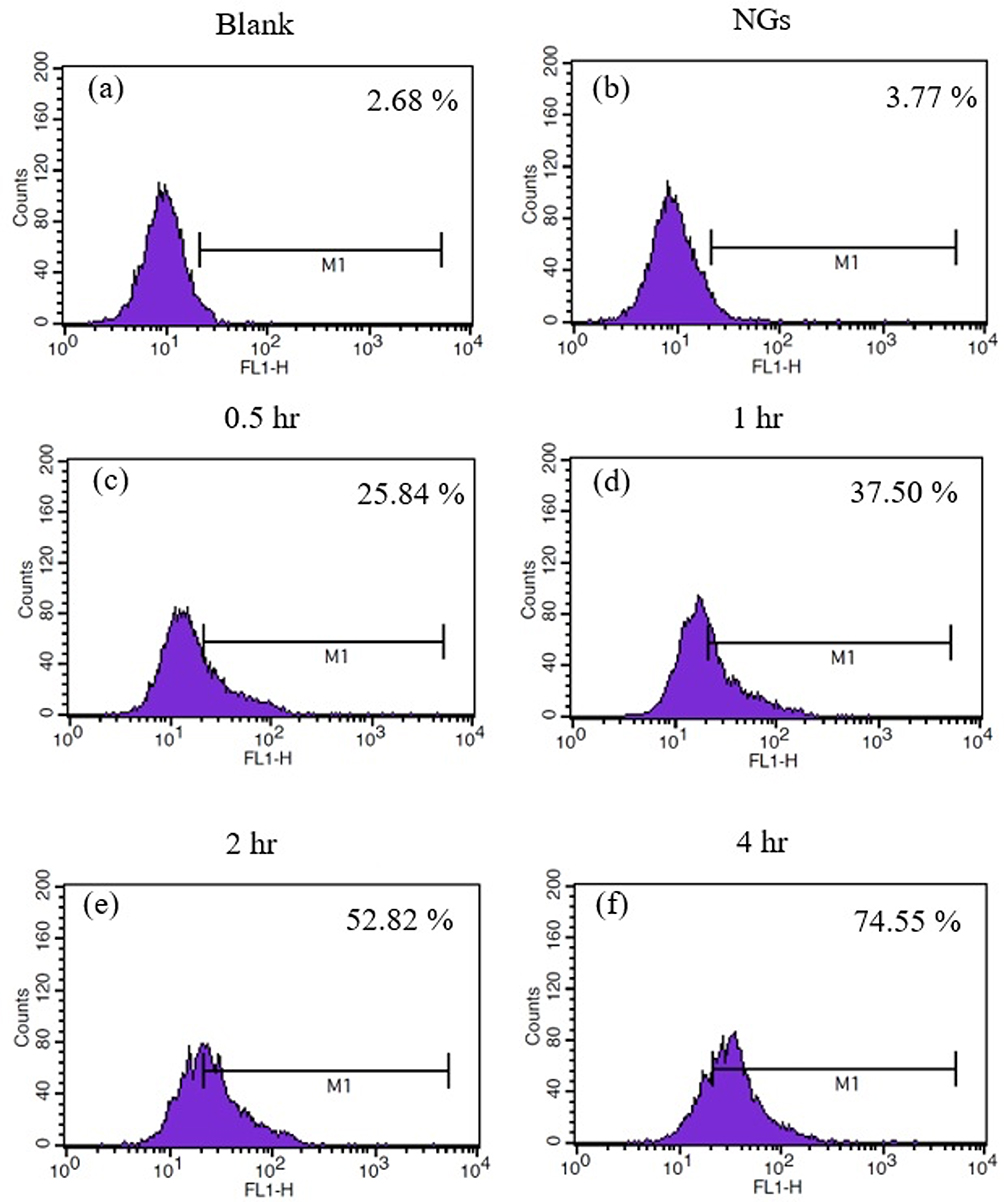 |
Figure 9 Cell uptake of NGs-FITC incubated with MIA PaCa-2 cells. (a) Blank, (b) NGs, (c) 0.5, (d) 1, (e) 2, and (f) 4 hours. |
 |
Figure 10 Cell uptake of NGs-FITC incubated with Caco-2 cells. (a) Blank, (b) NGs, (c) 0.5, (d) 1, (e) 2, (f) 4 hours. |
Permeability Test
This experiment was conducted to evaluate the integrity of the cell barrier by measuring TEER values. Caco-2 cells were first seeded in inserts at different cell densities (5×104, 10×104, and 15×104 cells/well) and measured every 24 h (for 7 days) to confirm the integrity of single-layer cells. Caco-2 cells were used as it is a well-known cell line in developing in vitro GI tract models.71 Before evaluating the transport of the NG system, we tested for confluent formation and polarized layers among three cell densities. The 5×104 cells/well density had the lowest polarized layers, typically at 96 h. Meanwhile, the integrity of 10×104 and 15×104 cells/well demonstrated similar TEER values (~500 Ω cm2). In addition, the TEER value dropped rapidly at time points beyond 96 h, possibly due to cell apoptosis. To evaluate TEER values of the barrier function and integrity of the monolayer of cells, the paracellular marker, Lucifer yellow, was employed. Since the restriction value of substance diffusion of the single-layer Caco-2 cells was 150~400 Ω cm2, we used a 10×104 cell density for 96 h for a further experiment. This caused the TEER value to exceed 400 Ω cm2 before the permeability test. As shown in Table 3, the permeability of free GEM and NGs-GEM were (17.22 ± 2.29) ×10−6 and (25.34 ± 3.63) ×10−6 cm/s, respectively. This result suggests that both systems allowed transport into the cell barrier. Interestingly, the NGs-GEM group facilitated more significant penetration (Student’s t-test analysis, p<0.01) than the free drug, suggesting the potential of the NG system to improve GEM penetration across the intestinal barrier. A gastrointestinal permeability was also measured, where the permeability constant was 7.5-fold greater for NGs-GEM than for free-GEM using the Caco-2 cell model.
 |
Table 3 Permeability Value of GEM (96 h, n=3) |
Analysis of GEM and dFdU in Serum
Gemcitabine is a prodrug requiring phosphorylation within tumor cells to attain its active form. The phosphorylation process generates certain metabolites crucial for the antineoplastic effect. However, the primary metabolite produced through deamination, known as dFdU, lacks significant relevance to the cytotoxic effect of GEM. Elevated dFdU plasma concentration suggests rapid conversion of GEM into dFdU.72 In order to evaluate GEM and dFdU present in the blood, an HPLC-UV analysis was conducted to measure a series of concentrations (of 0.078125~10 μg/mL). Intra- and inter-day experiments were then conducted to determine the precision, accuracy, and linear relationship of the analytical method.
As shown in Supplementary Material 1: Supplementary Figure 1, retention times of GEM and dFdU were 5.5 and 7.8 min, respectively, with acceptable regression coefficient values (r2) in the intra- and inter-day experiments (r2 ≥ 0.995). The coefficient of variation (CV) and relative standard error (RSE) were both < 15%, indicating that all analytical parameters met standardized criteria of precision, accuracy, and reproducibility (Supplementary Figure 2, Supplementary Tables 1 and 2).
PK Study
The experimental groups were divided into (1) oral NGs-GEM, (2) oral free-GEM, and (3) iv free-GEM. The PK profile and plasma drug concentration were monitored at various time points. As shown in Figures 11 and 12, the PK curve of the GEM in the NGs-GEM group confirmed that the drug was transported by the carrier, absorbed through the gastrointestinal tract, and delivered to the blood. However, compared with the free-GEM group, there was no significant difference. Nevertheless, according to the drug dynamics graph of dFdU, the AUC of the NGs-Gem group was lower than the free-GEM group, suggesting that the hydrogel form likely reduced the catabolism of GEM into its inactivated element.
 |
Figure 11 Pharmacokinetic profile of GEM in Sprague-Dawley rats after oral and intravenous administrations of NGs-GEM and free-GEM (n=6). |
 |
Figure 12 Pharmacokinetic profile of 2’,2’-difluorodeoxyuridine (dFdU) in Sprague-Dawley rats after oral and intravenous administrations of NGs-GEM and free-GEM (n=6). |
In addition, we conducted a PK analysis of GEM and dFdU using Phoenix® and WinNonlin® PK software, with a non-compartment model method using the experimental results (doses, plasma concentrations, and sampling times). Results of the GEM analysis of the NGs-GEM group showed that the Cmax was 2.69 μg/mL and tmax was 1.13 h. Meanwhile, Cmax of the oral GEM group was 2.89 μg/mL and tmax was 1.13 h. Values of t1/2 of the NGs-GEM, oral GEM, and i.v. GEM groups were 3.37 ± 1.24, 4.87 ± 2.43, and 3.37 ± 0.78 h, respectively (Table 4). Although the half-life of the oral GEM group was slightly longer than that of the other two groups, an independent one-way ANOVA suggested that the three groups did not significantly differ.
 |
Table 4 Pharmacokinetic Parameters of NGs-GEM (n=6, Mean ± Standard Deviation) |
Regarding dFdU contents, Cmax of the NGs-GEM group was 0.84 μg/mL and that of the oral gemcitabine group was 2.52 μg/mL, which significantly differed (p < 0.0001 by a one-way ANOVA). The tmax of the NGs-GEM group was 3.67 h and that of the oral GEM group was 2.17 h, which confirmed that the NGs-GEM group lowered the rate of GEM being metabolized into its inactive metabolites. AUC results showed that the NGs-GEM group was 10.07 hr∙μg/mL and oral GEM was almost two times higher at 19.04 hr∙μg/mL. As mentioned earlier, the majority of GEM is inactivated mainly by CDA-mediated conversion to dFdU. The NGs-GEM group exhibited a significant reduction (40%) in the formation of dFdU compared to the free-GEM group, as evidenced by a decrease in the AUC for dFdU. The substantial difference in metabolite ratios (NGs-GEM vs oral GEM; 0.62:0.99) between these two groups (p < 0.01) further confirmed that the use of NGs incorporated with GEM seemed to successfully prevent the drug from being rapidly metabolized, thereby increasing the effect of drug retention in the body, which is in accordance with the antitumor efficacy of GEM. This aligns with the previous observations, where the deamination of GEM occurred at a significantly slower rate, indicating that micellar nanocarriers offer additional protection for GEM against enzymatic degradation.73 Furthermore, our result is in accordance with the theory previously found that nanogels could prevent the quick metabolism of biomolecules as long as they can avoid opsonization followed by clearance.74 Nevertheless, a comprehensive analysis and further investigation into the specific metabolic pathways and factors influencing GEM metabolism are essential to provide a detailed explanation for the observed discrepancy in the pharmacokinetic data.
Conclusion
In this study, NGs-GEM hydrogel NPs with a particle size of approximately 250 nm for oral delivery were successfully prepared. The in vitro drug release results showed an effective slow-release rate of the NGs-GEM system in an acidic environment, which has the potential to reduce compound degradation during transport through the GI tract. The cell permeability study indicated higher induction of the NGs-Gem across the intestinal barrier compared to free GEM in the Caco-2 cell model. NGs-GEM also demonstrated higher cell uptake compared to free GEM in MIA PaCa-2 and Caco-2 cells. In the context of the in vivo study, despite the absence of observable distinctions in the bioavailability of GEM between the oral NGs-GEM and free GEM groups, the NGs-GEM oral delivery system demonstrates a notable suppression of inactive metabolite formation. This is substantiated by a significantly lower dFdU content in NGs-GEM compared to oral free-GEM (10.07 hr∙μg/mL and 19.04 hr∙μg/mL, respectively; p<0.01). Overall, results showed that the NGs-GEM is a potential antitumor candidate for oral drug delivery. Further antitumor studies and drug safety assessments of NGs-GEM are required to evaluate deeper functions of NGs-GEM in cancer therapeutic applications.
Acknowledgments
This research was conducted partially funded by the National Science and Technology Council, Taiwan (NSTC112-2221-E-038-014) and Taipei Medical University (TMU106-AE1-B23).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre Phase III trial. Lancet Oncol. 2005;6(6):369–376. doi:10.1016/S1470-2045(05)70175-3
3. Abbruzzese JL, Grunewald R, Weeks EA, et al. A Phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol. 1991;9(3):491–498. doi:10.1200/JCO.1991.9.3.491
4. Reid JM, Qu W, Safgren SL, et al. Phase I trial and pharmacokinetics of gemcitabine in children with advanced solid tumors. J Clin Oncol. 2004;22(12):2445–2451. doi:10.1200/JCO.2004.10.142
5. Moog R, Burger A, Brandl M, et al. Change in pharmacokinetic and pharmacodynamic behavior of gemcitabine in human tumor xenografts upon entrapment in vesicular phospholipid gels. Cancer Chemother Pharmacol. 2002;49(5):356–366. doi:10.1007/s00280-002-0428-4
6. Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100(3):331–346. doi:10.1016/j.jconrel.2004.09.001
7. Derakhshandeh K, Fathi S. Role of chitosan nanoparticles in the oral absorption of Gemcitabine. Int J Pharm. 2012;437(1–2):172–177. doi:10.1016/j.ijpharm.2012.08.008
8. Kawazoe H, Mori N, Ido S, et al. Liquid formulation of gemcitabine increases venous pain in patients with cancer: a retrospective study. Clin Ther. 2020;42(4):712–719. doi:10.1016/j.clinthera.2020.02.010
9. Arciero V, Luo J, Parmar A, et al. Real-world cost-effectiveness of first-line gemcitabine plus nab-paclitaxel vs FOLFIRINOX in patients with advanced pancreatic cancer. JNCI Cancer Spectr. 2022;6(4). doi:10.1093/jncics/pkac047
10. Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22(4):3–10.
11. Samanta K, Setua S, Kumari S, Jaggi M, Yallapu MM, Chauhan SC. Gemcitabine combination nano therapies for pancreatic cancer. Pharmaceutics. 2019;11(11):574. doi:10.3390/pharmaceutics11110574
12. Tan M, Luo J, Tian Y. Delivering curcumin and gemcitabine in one nanoparticle platform for colon cancer therapy. RSC Adv. 2014;4(106):61948–61959. doi:10.1039/C4RA10431E
13. Semaan A, Maitra A. Rebooting pancreatic cancer knowledge and treatment options. Nat Rev Gastroenterol Hepatol. 2018;15(2):76–78. doi:10.1038/nrgastro.2017.182
14. Adiseshaiah PP, Crist RM, Hook SS, McNeil SE. Nanomedicine strategies to overcome the pathophysiological barriers of pancreatic cancer. Nat Rev Clin Oncol. 2016;13(12):750–765. doi:10.1038/nrclinonc.2016.119
15. Bastiancich C, Vanvarenberg K, Ucakar B, et al. Lauroyl-gemcitabine-loaded lipid nanocapsule hydrogel for the treatment of glioblastoma. J Control Release. 2016;225:283–293. doi:10.1016/j.jconrel.2016.01.054
16. Bastiancich C, Bianco J, Vanvarenberg K, et al. Injectable nanomedicine hydrogel for local chemotherapy of glioblastoma after surgical resection. J Control Release. 2017;264:45–54. doi:10.1016/j.jconrel.2017.08.019
17. Qin L, Ling G, Peng F, et al. Black phosphorus nanosheets and gemcitabine encapsulated thermo-sensitive hydrogel for synergistic photothermal-chemotherapy. J Colloid Interface Sci. 2019;556:232–238. doi:10.1016/j.jcis.2019.08.058
18. Schneible JD, Shi K, Young AT, et al. Modified graphene oxide (GO) particles in peptide hydrogels: a hybrid system enabling scheduled delivery of synergistic combinations of chemotherapeutics. J Mater Chem B. 2020;8(17):3852. doi:10.1039/D0TB00064G
19. Moysan E, González-Fernández Y, Lautram N, Béjaud J, Bastiat G, Benoit JP. An innovative hydrogel of gemcitabine-loaded lipid nanocapsules: when the drug is a key player of the nanomedicine structure. Soft Matter. 2014;10(11):1767–1777. doi:10.1039/c3sm52781f
20. Bilalis P, Skoulas D, Karatzas A, et al. Self-healing pH- and enzyme stimuli-responsive hydrogels for targeted delivery of gemcitabine to treat pancreatic cancer. Biomacromolecules. 2018;19(9):3840–3852. doi:10.1021/acs.biomac.8b00959
21. Wang J, Leng Q, Li Y, et al. Injectable hyaluronic acid hydrogel for the co-delivery of gemcitabine nanoparticles and cisplatin for malignant ascites therapy. J Biomed Nanotechnol. 2020;16(12):1727–1739. doi:10.1166/jbn.2020.3002
22. Wu W, Dai Y, Liu H, et al. Local release of gemcitabine via in situ UV-crosslinked lipid-strengthened hydrogel for inhibiting osteosarcoma. Drug Deliv. 2018;25(1):1642. doi:10.1080/10717544.2018.1497105
23. Wang W, Wang B, Liu S, et al. Bioreducible polymer nanocarrier based on multivalent choline phosphate for enhanced cellular uptake and intracellular delivery of doxorubicin. ACS Appl Mater Interfaces. 2017;9(19):15986–15994. doi:10.1021/acsami.7b03317
24. Bastiancich C, Bozzato E, Luyten U, Danhier F, Bastiat G, Préat V. Drug combination using an injectable nanomedicine hydrogel for glioblastoma treatment. Int J Pharm. 2019;559:220–227. doi:10.1016/j.ijpharm.2019.01.042
25. Zhuang B, Chen T, Xiao Z, Jin Y. Drug-loaded implantable surgical cavity-adaptive hydrogels for prevention of local tumor recurrence. Int J Pharm. 2020;577:119048. doi:10.1016/j.ijpharm.2020.119048
26. Schneible JD, Young AT, Daniele MA, Menegatti S. Chitosan hydrogels for synergistic delivery of chemotherapeutics to triple negative breast cancer cells and spheroids. Pharm Res. 2020;37(7). doi:10.1007/s11095-020-02864-2
27. Shabana AM, Kambhampati SP, ching HR, Kannan RM, Kokkoli E. Thermosensitive and biodegradable hydrogel encapsulating targeted nanoparticles for the sustained co-delivery of gemcitabine and paclitaxel to pancreatic cancer cells. Int J Pharm. 2021;593:120139. doi:10.1016/j.ijpharm.2020.120139
28. Wang C, Zhang G, Liu G, Hu J, Liu S. Photo- and thermo-responsive multicompartment hydrogels for synergistic delivery of gemcitabine and doxorubicin. J Control Release. 2017;259:149–159. doi:10.1016/j.jconrel.2016.11.007
29. Date AA, Hanes J, Ensign LM. Nanoparticles for oral delivery: design, evaluation and state-of-The-art. J Control Release. 2016;240:504–526. doi:10.1016/j.jconrel.2016.06.016
30. Des Rieux A, Fievez V, Garinot M, Schneider YJ, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116(1):1–27. doi:10.1016/j.jconrel.2006.08.013
31. Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2012;64(6):557–570. doi:10.1016/j.addr.2011.12.009
32. Kim KS, Suzuki K, Cho H, Youn YS, Bae YH. Oral nanoparticles exhibit specific high-efficiency intestinal uptake and lymphatic transport. ACS Nano. 2018;12(9):8893–8900. doi:10.1021/acsnano.8b04315
33. Beloqui A, Des Rieux A, Préat V. Mechanisms of transport of polymeric and lipidic nanoparticles across the intestinal barrier. Adv Drug Deliv Rev. 2016;106(Pt B):242–255. doi:10.1016/j.addr.2016.04.014
34. Jin L, Wang Q, Chen J, Wang Z, Xin H, Zhang D. Efficient delivery of therapeutic siRNA by Fe3O4 magnetic nanoparticles into oral cancer cells. Pharmaceutics. 2019;11(11):615. doi:10.3390/pharmaceutics11110615
35. Zhang W, Zheng N, Chen L, et al. Effect of shape on mesoporous silica nanoparticles for oral delivery of indomethacin. Pharmaceutics. 2019;11(1):4.
36. Lamson NG, Berger A, Fein KC, Whitehead KA. Anionic nanoparticles enable the oral delivery of proteins by enhancing intestinal permeability. Nat Biomed Eng. 2020;4(1):84–96. doi:10.1038/s41551-019-0465-5
37. Du X, Yin S, Xu L, et al. Polylysine and cysteine functionalized chitosan nanoparticle as an efficient platform for oral delivery of paclitaxel. Carbohydr Polym. 2020;229:115484. doi:10.1016/j.carbpol.2019.115484
38. Sorasitthiyanukarn FN, Muangnoi C, Ratnatilaka Na Bhuket P, Rojsitthisak P, Rojsitthisak P. Chitosan/alginate nanoparticles as a promising approach for oral delivery of curcumin diglutaric acid for cancer treatment. Mater Sci Eng C Mater Biol Appl. 2018;93:178–190. doi:10.1016/j.msec.2018.07.069
39. Khan AA, Mudassir J, Akhtar S, Murugaiyah V, Darwis Y. Freeze-dried lopinavir-loaded nanostructured lipid carriers for enhanced cellular uptake and bioavailability: statistical optimization, in vitro and in vivo evaluations. Pharmaceutics. 2019;11(2):97. doi:10.3390/pharmaceutics11020097
40. Hanurry EY, Mekonnen TW, Andrgie AT, et al. Biotin-decorated PAMAM G4.5 dendrimer nanoparticles to enhance the delivery, anti-proliferative, and apoptotic effects of chemotherapeutic drug in cancer cells. Pharmaceutics. 2020;12(5):443. doi:10.3390/pharmaceutics12050443
41. Peppas NA, Bures P, Leobandung W, Ichikawa H. Hydrogels in pharmaceutical formulations. Eur J Pharm Biopharm. 2000;50(1):27–46. doi:10.1016/S0939-6411(00)00090-4
42. Qiu Y, Park K. Environment-sensitive hydrogels for drug delivery. Adv Drug Deliv Rev. 2001;53(3):321–339. doi:10.1016/S0169-409X(01)00203-4
43. Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Adv Mater. 2006;18(11):1345–1360. doi:10.1002/adma.200501612
44. Hoare TR, Kohane DS. Hydrogels in drug delivery: progress and challenges. Polymer. 2008;49(8):1993–2007. doi:10.1016/j.polymer.2008.01.027
45. Li J, Mooney DJ. Designing hydrogels for controlled drug delivery. Nature Rev Mater. 2016;1(12):1–17. doi:10.1038/natrevmats.2016.71
46. Chen W, Zou Y, Zhong Z, Haag R. Cyclo(RGD)-decorated reduction-responsive nanogels mediate targeted chemotherapy of integrin overexpressing human glioblastoma in vivo. Small. 2017;13(6):1601997.
47. Cuggino JC, Molina M, Wedepohl S, Igarzabal CIA, Calderón M, Gugliotta LM. Responsive nanogels for application as smart carriers in endocytic pH-triggered drug delivery systems. Eur Polym J. 2016;78:14–24. doi:10.1016/j.eurpolymj.2016.02.022
48. Cuggino JC, Gatti G, Picchio ML, Maccioni M, Gugliotta LM, Alvarez Igarzabal CI. Dually responsive nanogels as smart carriers for improving the therapeutic index of doxorubicin for breast cancer. Eur Polym J. 2019;116:445–452. doi:10.1016/j.eurpolymj.2019.04.031
49. Hajebi S, Rabiee N, Bagherzadeh M, et al. Stimulus-responsive polymeric nanogels as smart drug delivery systems. Acta Biomater. 2019;92:1. doi:10.1016/j.actbio.2019.05.018
50. Liu B, Thayumanavan S. Substituent effects on the pH sensitivity of acetals and ketals and their correlation with encapsulation stability in polymeric nanogels. J Am Chem Soc. 2017;139(6):2306–2317. doi:10.1021/jacs.6b11181
51. Liechty WB, Peppas NA. Expert opinion: responsive polymer nanoparticles in cancer therapy. Eur J Pharm Biopharm. 2012;80(2):241–246. doi:10.1016/j.ejpb.2011.08.004
52. Maya S, Sarmento B, Nair A, Rejinold N, Nair S, Jayakumar R. Smart stimuli sensitive nanogels in cancer drug delivery and imaging: a review. Curr Pharm Des. 2013;19(41):7203–7218. doi:10.2174/138161281941131219124142
53. Oishi M, Nagasaki Y. Stimuli-responsive smart nanogels for cancer diagnostics and therapy. Nanomedicine. 2010;5(3):451–468. doi:10.2217/nnm.10.18
54. Li C, Obireddy SR, Lai WF. Preparation and use of nanogels as carriers of drugs. Drug Deliv. 2021;28(1):1594. doi:10.1080/10717544.2021.1955042
55. Cuggino JC, Blanco ERO, Gugliotta LM, Alvarez Igarzabal CI, Calderón M. Crossing biological barriers with nanogels to improve drug delivery performance. J Control Release. 2019;307:221–246. doi:10.1016/j.jconrel.2019.06.005
56. Kersey FR, Merkel TJ, Perry JL, Napier ME, Desimone JM. Effect of aspect ratio and deformability on nanoparticle extravasation through nanopores. Langmuir. 2012;28(23):8773–8781. doi:10.1021/la301279v
57. Shirahama H, Lee BH, Tan LP, Cho NJ. Precise tuning of facile one-pot gelatin methacryloyl (GelMA) synthesis. Sci Rep. 2016;6(1):1–11. doi:10.1038/srep31036
58. Hoch E, Hirth T, Tovar GEM, Borchers K. Chemical tailoring of gelatin to adjust its chemical and physical properties for functional bioprinting. J Mater Chem B. 2013;1(41):5675–5685. doi:10.1039/c3tb20745e
59. Kang MG, Lee MY, Cha JM, et al. Nanogels derived from fish gelatin: application to drug delivery system. Mar Drugs. 2019;17(4):246. doi:10.3390/md17040246
60. Kim J, Gauvin R, Yoon HJ, et al. Skin penetration-inducing gelatin methacryloyl nanogels for transdermal macromolecule delivery. Macromol Res. 2016;24(12):1115–1125. doi:10.1007/s13233-016-4147-9
61. Novakova I, Subileau EA, Toegel S, et al. Transport rankings of non-steroidal antiinflammatory drugs across blood-brain barrier in vitro models. PLoS One. 2014;9(1):e86806. doi:10.1371/journal.pone.0086806
62. Krishnamoorthy S, Noorani B, Xu C. Effects of encapsulated cells on the physical–mechanical properties and microstructure of gelatin methacrylate hydrogels. Int J Mol Sci. 2019;20(20):5061. doi:10.3390/ijms20205061
63. Lee BH, Shirahama H, Cho NJ, Tan LP. Efficient and controllable synthesis of highly substituted gelatin methacrylamide for mechanically stiff hydrogels. RSC Adv. 2015;5(128):106094–106097. doi:10.1039/C5RA22028A
64. Sun M, Sun X, Wang Z, Guo S, Yu G, Yang H. Synthesis and properties of gelatin methacryloyl (GelMA) hydrogels and their recent applications in load-bearing tissue. Polymers. 2018;10(11):1290. doi:10.3390/polym10111290
65. Pinelli F, Ferracin F, Perale G, Rossi F. Synthesis and applications of nanogels via covalent cross-linking strategies. Adv Chem Engineer. 2023;62:35–58.
66. Rizwan M, Yahya R, Hassan A, et al. pH sensitive hydrogels in drug delivery: brief history, properties, swelling, and release mechanism, material selection and applications. Polymers. 2017;9(4):137.
67. Bashir S, Hina M, Iqbal J, et al. Fundamental concepts of hydrogels: synthesis, properties, and their applications. Polymers. 2020;12(11):1–60. doi:10.3390/POLYM12112702
68. Jacob S, Nair AB, Shah J, Sreeharsha N, Gupta S, Shinu P. Emerging role of hydrogels in drug delivery systems, tissue engineering and wound management. Pharmaceutics. 2021;13(3):357. doi:10.3390/pharmaceutics13030357
69. Cheng G, Zielonka J, McAllister D, Tsai S, Dwinell MB, Kalyanaraman B. Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation. Br J Cancer. 2014;111(1):85–93. doi:10.1038/bjc.2014.272
70. Jaidev LR, Krishnan UM, Sethuraman S. Gemcitabine loaded biodegradable PLGA nanospheres for in vitro pancreatic cancer therapy. Mater Sci Eng C Mater Biol Appl. 2015;47:40–47. doi:10.1016/j.msec.2014.11.027
71. Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. TEER measurement techniques for in vitro barrier model systems. J Lab Autom. 2015;20(2):107–126. doi:10.1177/2211068214561025
72. Derissen EJB, Huitema ADR, Rosing H, Schellens JHM, Beijnen JH. Intracellular pharmacokinetics of gemcitabine, its deaminated metabolite 2′,2′‐difluorodeoxyuridine and their nucleotides. Br J Clin Pharmacol. 2018;84(6):1279. doi:10.1111/bcp.13557
73. Wang Y, Fan W, Dai X, et al. Enhanced tumor delivery of gemcitabine via PEG-DSPE/TPGS mixed micelles. Mol Pharm. 2014;11(4):1140–1150. doi:10.1021/mp4005904
74. Soni KS, Desale SS, Bronich TK. Nanogels: an overview of properties, biomedical applications and obstacles to clinical translation. J Control Release. 2016;240:109. doi:10.1016/j.jconrel.2015.11.009
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.