Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Optogenetics in preclinical neuroscience and psychiatry research: recent insights and potential applications
Authors McDevitt R, Reed S, Britt J
Received 10 April 2014
Accepted for publication 20 May 2014
Published 22 July 2014 Volume 2014:10 Pages 1369—1379
DOI https://doi.org/10.2147/NDT.S45896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Ross A McDevitt,1 Sean J Reed,2 Jonathan P Britt2,3
1Intramural Research Program, National Institute on Drug Abuse, National Institutes of Health, Baltimore, MD, USA; 2Integrated Program in Neuroscience, Montreal Neurological Institute, 3Department of Psychology, McGill University, Montreal, QC, Canada
Abstract: There have been significant advances in the treatment of psychiatric disease in the last half century, but it is still unclear which neural circuits are ultimately responsible for specific disease states. Fortunately, technical limitations that have constrained this research have recently been mitigated by advances in research tools that facilitate circuit-based analyses. The most prominent of these tools is optogenetics, which refers to the use of genetically encoded, light-sensitive proteins that can be used to manipulate discrete neural circuits with temporal precision. Optogenetics has recently been used to examine the neural underpinnings of both psychiatric disease and symptom relief, and this research has rapidly identified novel therapeutic targets for what could be a new generation of rational drug development. As these and related methodologies for controlling neurons ultimately make their way into the clinic, circuit-based strategies for alleviating psychiatric symptoms could become a remarkably refined approach to disease treatment.
Keywords: optogenetics, depression, anxiety, addiction, obsessive-compulsive disorder
Introduction
Within the last half century, there has been remarkable progress in the treatment of psychiatric disease. The first chemical antidepressants were discovered fortuitously in clinical trials designed to treat tuberculosis.1 Since then, researchers in both academia and industry have developed an impressive array of related compounds, which work through similar mechanisms but treat a host of different psychiatric conditions.2 These developments have led to an abundance of pharmacotherapies that have dramatically increased the number of available treatment options for psychiatrists. Unfortunately, currently available drugs still produce numerous deleterious side effects and, for a substantial number of patients, are largely ineffective in alleviating their disease symptoms. Consequently, there remains a strong need for more effective, targeted pharmacotherapies to treat psychiatric disease.
A better understanding of the neurobiological processes that underlie psychiatric disease states is expected to help drug development efforts. Accordingly, there has been a considerable effort to identify biological variables that consistently differ between healthy individuals and those with disease.3 Studies in humans have implicated various circuits in specific psychiatric disease states using neuroimaging, genomic, and post mortem tissue analyses.4 However, data produced from these studies are largely post hoc in nature and cannot be used to conclusively infer cause and effect. The attribution of causality to specific neuronal circuitry or reliable biomarkers has been hampered by technological limitations and overall etiological complexities.5 In order to determine if a specific biological process is integral to a psychiatric condition, it must be experimentally manipulated in preclinical models of the disease. To this end, numerous animal models of psychiatric disease states have been developed.6
One approach to modeling psychiatric disease in an animal is to use a behavioral task in which an animal’s performance is sensitive to drug treatments that have already proved to be effective in humans.7 When new drugs produce behavioral effects that are similar to those elicited with proven pharmacotherapies, they are likely to have similar mechanisms of action to the old drugs and be similarly effective in treating the disease. This strategy provides strong predictive validity in modeling treatment of disease states and has been successfully used to screen novel compounds. However, its utility may be limited to compounds that act via the same mechanisms as those used to initially develop the behavioral model. An additional caveat to this line of research is that predictive validity in modeling treatment does not necessarily mean that the etiology or mechanisms underlying the disease in humans are relevant to the given model. Thus, while animal models of psychiatric disease offer a powerful approach to understand symptomatology, they may be less able to provide information about the neural processes that are ultimately responsible for the initiation of disease states.
The unclear neuronal substrates of psychiatric diseases and correspondingly slow progress of novel drug development have resulted in pharmaceutical companies recently abandoning development of psychiatric compounds.8 To reinvigorate research, it may be necessary to take a more refined technological approach to the study of disease symptoms and treatment. The maladaptive behaviors associated with complex psychiatric disorders are likely the result of specific aberrant processes in discrete, albeit multiple, neural circuits. The development of optogenetics has provided researchers with a powerful set of tools to manipulate neural activity in genetically defined populations of cells in a pathway-specific manner.9 In applying optogenetic strategies to established animal models of psychiatric disease, investigators have made rapid progress in defining the neural circuitry responsible for both psychiatric disease and symptom relief.
Optogenetic tools
Optogenetic approaches enable researchers to activate or inhibit groups of neurons, which can be defined by genetic identity and/or projection target. This is made possible through the targeted expression of light-sensitive proteins known as rhodopsins, which are responsive to specific wavelengths of light. Rhopdopsins are protein complexes that contain an opsin (channel) and a light-sensitive cofactor (retinal).10 The opsins used in optogenetic strategies are generally derived from the type 1 class of prokaryotic rhodopsin proteins that support a covalent bond with an internalized retinal molecule.11 The ionic environment around this covalent bond influences the spectral sensitivity of the protein, whereas the amino acid residues around it play a significant role in channel kinetics.12 Photon absorption causes a conformational change in the rhodopsin protein, via retinal isomerization, which allows for ion translocation across the channel.
Opsins are broadly categorized based on their depolarizing or hyperpolarizing effects. Depolarizing opsins, or channelrhodopsins (ChRs), permit the translocation of cations into the intracellular space upon photon absorption and retinal isomerization. Channelrhodopsin-1 (ChR1) was the first of this type to be characterized, but it produces relatively small photocurrents and is not widely used.13 Channelrhodopsin-2 (ChR2) has a higher photocurrent than its predecessor and has proved to be quite effective at altering neuronal firing patterns in mammalian tissue.14,15 It is maximally activated by 470 nm wavelength light and can sustain reliable firing up to 40 Hz. Multiple variants of ChR1 and ChR2 have recently been developed through point mutations and chimeric complexes in an attempt to improve or alter ion permeability (CatCH), channel kinetics (ChETA), and wavelength selectivity (C1V1)12,16 (see Table 1 for a list of notable variants). The creation of red-shifted opsins, such as C1V1, has enabled independent excitation of two depolarizing opsins in the same tissue.17,18 In addition, longer wavelengths mitigate light scatter and absorption, thus promoting deeper tissue penetration.19
 | Table 1 Summary of noteworthy opsin proteins commonly used in optogenetic research |
In general, the photocurrent generated from opsin activation decays quickly following light cessation. This feature is an asset for most experimental designs, but certain behavioral paradigms benefit from prolonged manipulations. ChR2 variants known as stabilized-step function opsins significantly extend the open-state of the channel from a single pulse of blue light, which promotes a sustained depolarized state. This state increases the likelihood of firing in response to endogenous input, and can be inactivated with a single pulse of yellow light.20 Recent step function opsin variants can induce depolarized states that last as long as 30 minutes following a single activating pulse of light.21
Hyperpolarizing opsins, in contrast, are more limiting in both their overall characteristics and variety. Most hyperpolarizing opsins have an activation profile that is significantly red-shifted in comparison with the standard depolarizing ChRs. Potent inhibition can be achieved through the pumping of chloride ions into the neuron through halorhodopsin (eg, eNpHR3.022) or via expelling protons from the intracellular space using archaerhodopsin (eg, eArch3.016). However, despite their effectiveness, pumps are not energetically efficient, owing to their one proton to one ion photocycle,23 and do not alter the cell’s input resistance.24 Additionally, sustained halorhodopsin activation can disrupt chloride gradients, leading to a change in gamma-aminobutyric acid type A (GABAA) receptor reversal potential, thus eliciting a post-inhibitory excitatory state.25 Recently, however, it has been shown that point mutations in a chimeric ChR protein can convert it into a chloride conducting channel.26,27 The slow mutant variant, known as slow ChloC, possess a 10-second long open-state during which neurons are effectively silenced.26 Longer periods of neuronal inhibition can be achieved with a pharmacogenetic approach that utilizes designer receptors that are exclusively activated by designer drugs.28
In addition to the advances in opsin-mediated excitatory and inhibitory neuronal activity, there has been success in engineering chimeric opsin proteins that directly alter intracellular signaling cascades through G-protein coupled receptors.29 This class of proteins are known as OptoXRs, and they have been designed to incorporate bovine rhodopsin (type II opsin) into endogenously expressed adrenergic30 and serotonergic31 receptors. As a result of these developments, optogenetics provides a temporally refined and multifaceted toolkit of opsins allowing various controls on neuronal activity within genetically defined neuronal cell types.
To achieve selective opsin expression, DNA constructs encoding the opsins, often fused with a fluorescent protein marker (eg, yellow fluorescent protein), are introduced into neurons in an anatomically restricted manner, commonly by transgenic introduction, viral injection, or in vivo electroporation.32–35 Expression of opsins within neurons can be further restricted with the use of selective promoters or DNA recombination systems. For example, one popular expression protocol involves the use of Cre-recombinase, for its ability to invert gene orientations based on Cre-identifiable tags (lox sites) that flank opsin genes. Thus, cell-specific opsin expression can be achieved using transgenic animals that express Cre under a cell-specific gene promoter (eg, tyrosine hydroxylase), followed by viral infection carrying a payload with the opsin gene in an inverted orientation. As a result, only neurons expressing Cre will be able express the opsin gene since it will have been inverted to its correct orientation.
Once the proteins are expressed, they will diffuse within the membranes of cells and traffic to distal neural processes. This allows for the direct manipulation of spiking activity, either through stimulation of cell bodies or their distal processes. Thus, there are multiple dimensions by which photostimulation can be restricted, ie, anatomical location of DNA delivery, cell-type specific expression, and localized light delivery. When these approaches are used together, anatomically localized, genetically defined neural pathways can be repeatedly stimulated or inhibited, in vitro and in vivo.
Depression
Current treatments for clinical depression include pharmacotherapies that directly increase synaptic levels of serotonin and/or norepinephrine via reuptake inhibition. These two neurotransmitter systems have diffuse projections throughout the brain and are involved in a variety of functions, which may be the reason why individual patients respond to different antidepressant drugs in a highly variable manner. While antidepressant drugs act on these entire systems, it is presumed that only a subset of these projections are ultimately responsible for the therapeutic actions.36,37 This appears to be an area ripe for optogenetic analyses. However, antidepressant drugs typically require weeks of medication before symptoms alleviate, which complicates experimental designs and raises questions about the direct role of serotonin/norepinephrine in the therapeutic effects of antidepressant medications.
The initial optogenetic explorations of depression have instead attempted to dissect a nonpharmacological treatment for depression known as deep brain stimulation, a treatment that has been explored as an alternative therapeutic option for severely depressed individuals. Deep brain stimulation via implanted electrodes that target frontal cortical areas and the nucleus accumbens has been demonstrated to relieve symptoms of intractable depression in human subjects.38–42 It is unclear if there is a common neurobiological substrate mediating the therapeutic effects of electrical brain stimulation and antidepressant medication. Unlike drug-induced antidepressant effects, symptom relief following deep brain stimulation has a rapid onset. However, interpretation of the immediate effects of electrical stimulation is problematic, because it is not obvious how neural activity is altered by this manipulation. Electrical stimulation elicits nonspecific activation of all neuronal cell types and processes in a small area around the electrode. Since frontal cortical areas integrate information from throughout the brain and have projections that are similarly diffuse, it is difficult to attribute therapeutic effects to specific neural pathways. As such, electrical stimulation lacks the resolution needed to precisely define which local networks and cell types are responsible for the observed therapeutic changes.
Researchers’ attempts to dissect stimulation-induced antidepressant effects with optogenetic tools have focused on alleviating depression in animal models of the disease. One of these animal models is known as the forced swim test, where laboratory rodents are placed in tanks of water and scored for time spent actively struggling versus passively floating.43,44 Antidepressant treatments increase the amount of time that animals spend struggling. Other models of interest expose animals to stressful events, which creates a behavioral phenotype that is sensitive to chemical antidepressant treatments.45 Optogenetic studies employing these animal models (summarized in Table 2) demonstrate that photostimulation of neurons in the medial prefrontal cortex can elicit antidepressant-like behavior in both forced swim and chronic social defeat models of depression.46–49 Additional research has demonstrated that optogenetic stimulation directed to the specific medial prefrontal cortex projections that innervate the dorsal raphe nucleus is sufficient to produce antidepressant-like effects.48 The dorsal raphe nucleus is the principal source of ascending serotonin projections and the neurotransmitter system that most antidepressant pharmacological treatments act upon.50 Thus, convergent lines of evidence implicate the dorsal raphe nucleus and its regulation by the medial prefrontal cortex in the treatment of depression.
 | Table 2 Contribution of various brain regions and circuits to the expression of depression-like symptoms |
Despite the strong evidence implicating the medial prefrontal cortex-dorsal raphe nucleus circuit in treating symptoms of depression, many ambiguities remain regarding its precise role in this process. This pathway appears to preferentially excite GABAergic interneurons in the dorsal raphe nucleus,46,51–53 suggesting that excitatory medial prefrontal cortex input to the dorsal raphe nucleus may act to depress serotonin levels via feed-forward inhibition. Consistent with this idea, pharmacological inhibition of the medial prefrontal cortex has been shown to enhance both stress-induced serotonin activity and “learned helplessness” behavior.54 Collectively, these optogenetic and pharmacological studies support the provocative conclusion that acute decreases in serotonin function are capable of producing antidepressant-like effects. Similar behavioral effects have been demonstrated by reducing serotonin function via dorsal raphe nucleus negative-feedback autoreceptor activation55 or overexpression.56 These findings go against studies in humans in which acute depletion of serotonin synthesis decreases mood in people with a history of major depressive disorder. Thus, “antidepressant” responses in some behavioral models might be produced by any perturbation of serotonin signaling, regardless of directionality of effect.
In contrast with the forced swim test, optogenetic research exploiting the chronic social defeat model of depression has supported the more conventional conclusion that enhancing serotonin neuron activity in the dorsal raphe nucleus reduces depression-like behavior. Specifically, social defeat experiences have been demonstrated to sensitize GABAergic interneuron function in the dorsal raphe nucleus, and photoinhibition of this population of neurons prevents the acquisition of defeat-induced social avoidance.57 Further, bidirectional manipulations of medial prefrontal cortex-dorsal raphe nucleus circuitry can modulate defeat behavior in positive or negative ways, accordingly.46 A greater understanding of these behavioral models and the microcircuitry of the dorsal raphe nucleus will help explain how different neural inputs are integrated in this region of the brain.
Other medial prefrontal cortex projection targets, in addition to the dorsal raphe nucleus, have recently been explored as well. Optogenetic stimulation to the medial prefrontal cortex projections to the nucleus accumbens have been shown to produce antidepressant-like effects in mice that exhibited depressive symptomatology following cholecystokinin infusions into the medial prefrontal cortex.58 This work highlights a potential role for the nucleus accumbens in depression, which is a region that has been somewhat overlooked in relation to this disease state. While the nucleus accumbens does receive serotonin and norepinephrine input,59,60 it is most prominently innervated by dopaminergic fibers. Photoactivation of midbrain dopamine neurons that innervate the nucleus accumbens has been shown to elicit antidepressant-like effects in both the tail suspension and forced swim tests.61 On the other hand, activation of accumbens-projecting dopamine cell bodies exacerbates the effects of social defeat stress,62 so this system may not have a simple role in depressive symptomatology. Indeed, midbrain dopamine neuron stimulation has been reported to produce opposite effects on sucrose preference in different studies, which suggests that specific conclusions may be more attributable to experimental parameters than depression in general.61–63 More research is needed to clarify how dopamine and its downstream effectors regulate mood in relation to chronic depression.
Anxiety
Anxiety disorders are generally characterized by excessive feelings of apprehension in the absence of any immediate threat. This category of disorders is broad, as it includes generalized anxiety, obsessive-compulsive, and post-traumatic stress disorders.64 Modern medications used to treat anxiety disorders largely target serotonin and GABAergic neurotransmitter systems; however, drugs that block norepinephrine function appear to have particular utility in post-traumatic stress disorder.65,66
Research into the neurobiology of anxiety has largely focused on the amygdala and its extended compartment in the bed nucleus of the stria terminalis (BNST), as these regions are critical for the manifestation of fear. Early studies in laboratory animals found that electrical stimulation of the amygdala produced behavioral and autonomic responses that resemble a state of fear, whereas lesion or inactivation of this region inhibited the expression of experimentally induced anxiety and conditioned fear.67 The basolateral amygdala, where multiple pathways converge, appears to be the locus of associative fear learning. Neural activity here is acutely sensitive to the presentation of previously learned, fear-evoking stimuli.68 Neurons in the basolateral complex innervate the central nucleus of the amygdala, which is the primary output structure of the amygdala nuclei. This region regulates the behavioral and physiological manifestations of fear.69,70 The BNST, which primarily receives input from within the amygdala, serves as an additional output relay station.71
Recent optogenetic studies have extended this model of amygdala circuitry and highlighted a role for this structure in behavioral measures of anxiety (Figure 1). Photostimulation of the basolateral amygdala is sufficient to induce fear, and any stimuli paired with this stimulation can become fear-inducing.72 In addition, activity in this region is critical for the consolidation of fear memories.73 These studies have supported the classical view of the amygdala as a critical site of fear learning. Other optogenetic studies have broadened this view by showing that neighboring projections out of the basolateral amygdala can mediate opposing effects on fear and anxiety. Photostimulation of basolateral complex projections to the central lateral amygdala reduces anxiety in the elevated plus maze,74 whereas projections to the CA1 region of the hippocampus increase these and other measures of anxiety.75,76 This work underscores the importance of characterizing the role of adjacent structures and discrete projections. Indeed, the hippocampus is similarly nonuniform, as photostimulation of the ventral, but not dorsal, dentate gyrus area elicits a robust anxiolytic effect.77,78
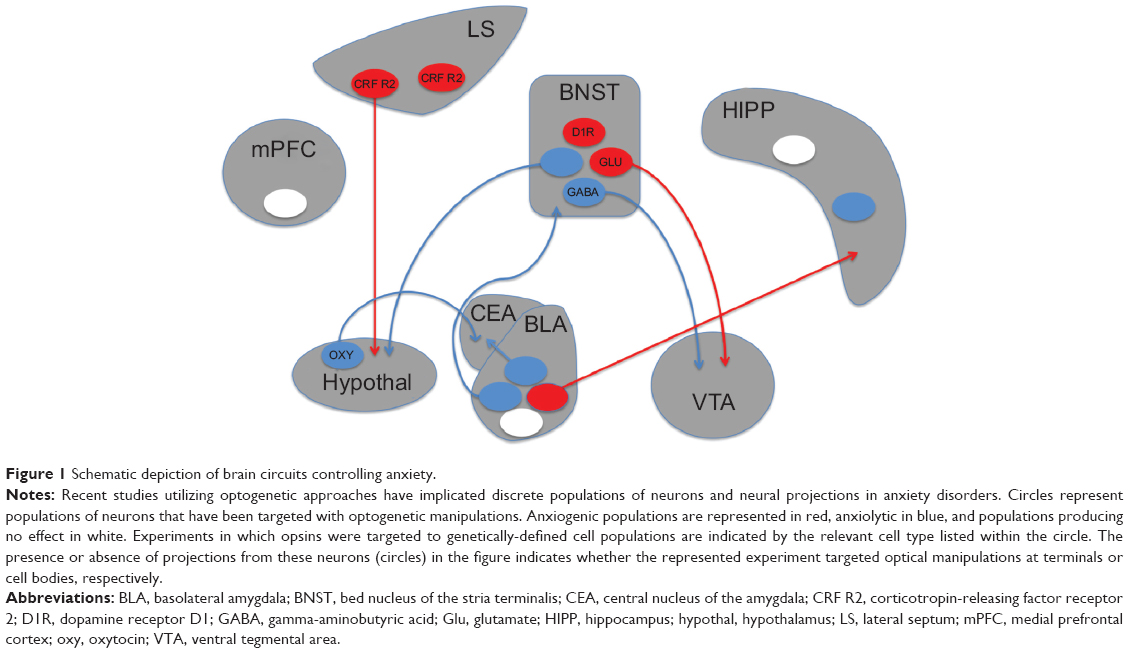 | Figure 1 Schematic depiction of brain circuits controlling anxiety. |
Anxiety regulation within the extended amygdala is also complex. The BNST contains two parallel output pathways to the ventral tegmental area (VTA), ie, a GABAergic pathway that is anxiolytic and a glutamatergic pathway that is anxiogenic.79 The behavioral and physiological components of anxiety can be dissociated via other BNST projections as well. Stimulation of BNST to lateral hypothalamus projections produces avoidance of open arms in an elevated plus maze, but has no effect on respiration rate. Conversely, stimulation of BNST to parabrachial nucleus projections increases respiratory rate without altering plus maze behavior.80 Stimulation of BNST cell bodies directly can produce various responses that depend on the exact location and genetic cell type targeted by the stimulation, factors that likely influence which output pathways are preferentially recruited.
Beyond the extended amygdala, a role for the lateral septum in anxiety has also been demonstrated with optogenetics. Although this structure has been classically associated with anxiolytic responses,81 optogenetic stimulation of cells expressing corticotropin-releasing factor receptor type 2 produces anxiogenic effects.82 This effect was demonstrated to occur via GABAergic projections to the anterior hypothalamus. The hypothalamus contains oxytocin-producing neurons that project to the amygdala and participate in the regulation of anxiety. Selective stimulation of this pathway reduced expression of conditioned fear by directly activating GABAergic neurons in the central lateral amygdala.83
Obsessive-compulsive (OCD) disorder is a specific subset of anxiety disorders that is characterized by repetitive behaviors aimed at relieving anxiety related to intrusive, irrational thoughts. As with general anxiety disorders, obsessive-compulsive disorder can be treated with antidepressant pharmacotherapies. Research into OCD has revealed a role of striatal circuitry in the expression of stereotyped, repetitive actions. A genetic mouse model of OCD has been developed in which mice lack the postsynaptic scaffolding protein Sapap3.84 This gene is expressed strongly in the striatum, and its deletion results in defective corticostriatal circuitry. Mice lacking the gene exhibit high levels of anxiety and excessive grooming behavior, both of which are reversed with chronic antidepressant treatment. Dorsal striatal medium spiny neurons in these mutant mice exhibit exaggerated responses to the presentation of cues associated with grooming.85 Photostimulation of lateral orbitofrontal cortical projections to the dorsal striatum attenuated this striatal neuron hyperactivity by activating fast-spiking interneurons in the striatum. Other recent studies that have examined the orbitofrontal projections to the ventral striatum in wild-type mice found that photostimulation of this pathway can increase grooming.86 While this phenomenon was only produced following chronic stimulation, it persisted for 2 weeks and could be reversed by chronic antidepressant treatment. These studies highlight the importance of corticostriatal circuitry in OCD, but future studies need to clarify where stimulation could be targeted to ultimately alleviate symptoms of the disorder. Indeed, a recent study implicates the more lateral orbitofrontal cortex in controlling the shift from habitual to goal-directed actions.87
Addiction
Drug abuse disorders are characterized by compulsive drug use in the face of adverse consequences. Highly addictive substances seem to disrupt the processes of self-control associated with normal reward-seeking behavior. Initial research mapping out regions of the brain involved in reward processing and motivation began over a half century ago with electrical intracranial self-stimulation experiments.88 Recent studies that have used optogenetic techniques to extend this research have already confirmed that stimulation of midbrain dopamine neurons in the VTA and substantia nigra pars compacta is highly reinforcing.89–92 Although the predominant effect of VTA dopamine neuron stimulation is to reinforce and motivate operant behavior, an aversive subcircuit has also been identified, that consists of VTA dopaminergic neurons receiving input from the lateral habenula and projecting to the medial prefrontal cortex.93 In addition, photoactivation of GABAergic interneurons in the VTA has proved to be aversive.94,95 This research has largely confirmed ideas that have been developed over the preceding years, but a new level of detail is starting to emerge.
The reinforcing effects of midbrain dopamine neuron activity are believed to be mediated primarily via projections to the striatum,96–98 although it has been recently shown that VTA-habenula projections also contribute to this effect.99 Within the striatum, multiple glutamatergic inputs, in addition to the dopaminergic pathway, can reinforce instrumental behavior.100–102 Projections out of the striatum are segregated into two populations based on the expression of certain receptor proteins in the efferent neurons as well as projection targets.103–105 Optogenetic activation of these neuronal populations produces opposing behavioral effects, as photostimulation of dopamine D1 receptor-expressing neurons (direct pathway neurons) produces reward-related behavior and photostimulation of dopamine D2 receptor-expressing neurons (indirect pathway neurons) produces aversion.106 More refined behavioral studies are necessary to expand upon this simple reward/aversion dichotomy.
As individuals repeatedly abuse addictive substances, addictive behaviors develop that are marked by a loss of self-control. Research into the mechanisms of this process has focused on the neural plasticity that addictive drugs produce within reward circuitry.107–110 Within the striatum, drug exposure appears to preferentially potentiate glutamatergic inputs onto direct pathway striatal neurons.109,111 Individual animals with greater potentiation onto the other projection neurons in the striatum, ie, the indirect pathway neurons, show resistance to compulsive drug use.111 Following prolonged withdrawal, potentiation of formerly silent basolateral amygdala-striatum synapses is prominent.112 Pharmacological reversal of this potentiation reverses behavioral manifestation of incubation of drug craving. Numerous pathways, however, appear to be sensitive to drug exposure. Within the dorsomedial nucleus accumbens, drug-induced synaptic potentiation has also been shown to occur within the glutamatergic hippocampus-striatum pathway.101 In addition, direct pathway striatal neurons (dopamine D1 receptor-expressing), which project back to the VTA and target GABAergic interneurons, are also potentiated by repeated drug use.113
Several studies have focused on plasticity occurring within the medial prefrontal cortex. Individual rats that exhibit compulsive drug-seeking show intrinsic hypoactivity of medial prefrontal cortex neurons,114 while presynaptic enhancement occurs in the prefrontal cortex-accumbens projection.115 The causal role of cortical areas in addiction appears to be highly complex. Whereas 1 Hz stimulation of prefrontal cortex cell bodies inhibits compulsive drug-seeking,114 inhibition of the prefrontal cortex-accumbens pathway reduces both reinstatement of cocaine-seeking116 and compulsive alcohol self-administration.117 Furthermore, depotentiation of the prefrontal cortex-accumbens pathway reverses cocaine-induced psychomotor sensitization.109 Thus, while optogenetics have allowed for the ability to study pathway specificity in drug-induced plasticity, future studies are crucial for determining the role of parameters such as drug type and schedule in mediating this plasticity.
Schizophrenia, autism, and beyond
Schizophrenia and autism involve a constellation of symptoms that are not easy to model in rodents. One avenue of research into schizophrenia has focused on neurobiological markers seen within human populations, rather than attempting to recapitulate behavioral models of specific symptoms.118 For example, cortical gamma oscillations (30–80 Hz), which are altered in schizophrenic patients, can be driven by optogenetic stimulation of fast-spiking cortical interneurons.119,120 This manipulation alters the gating of sensory information, which is a common symptom of schizophrenia. A related line of research examines how disruptions in the cellular balance of excitation and inhibition within neural networks underlie deficits in social behavior.121 Recent research has found that increasing excitatory, but not inhibitory, input to pyramidal neurons of the prefrontal cortex causes social and cognitive disturbances in rodents.21 This experiment utilized a step function opsin in conjunction with red-shifted opsins to simultaneously alter excitatory and inhibitory components in cortical microcircuits.
Autism spectrum disorders can manifest as repetitive actions and reduced social interaction, which are symptoms that animal studies have tried to address in relation to OCD85–87 and depression.46–49 As the symptomatology of many psychiatric diseases overlap, some rodent behavioral studies have the potential to address common underlying circuit disturbances. However, there are numerous and obvious differences between people suffering from autism spectrum disorders and those with OCD. For example, people with autism are generally not bothered by their repetitive behaviors, whereas compulsions in OCD are a significant source of anxiety. Additional studies are needed to delineate the neural circuit disruptions that may be common across disorders versus those that are unique to specific psychiatric diseases.
Significant progress has also been made with optogenetic tools in counteracting neural circuit disruptions that underlie certain neurological disorders, such epilepsy and Parkinson’s disease, which are comparatively easy to model in animals. Epileptiform activity can be completely shut down with photoinhibitions targeted to excitatory neurons as well as with photostimulations targeted to inhibitory neurons.122–124 This work is comparable with the advances seen in Parkinson’s disease, in which movement deficits have been minimized in animal models through photostimulations and inhibitions targeted to discrete pathways and groups of neurons.125–127 The research on neurological disorders benefits from having straightforward behavioral assays that are directly relevant to the treatment of human disease.
Conclusion
Optogenetics offers valuable tools for studying the neurobiology of psychiatric disease in animal models. The use of genetically-encoded, light-sensitive proteins, which can activate and inhibit discrete neural circuits, is revolutionizing behavioral neuroscience. When integrated with established techniques, ie, brain imaging, genetic manipulations, behavioral assays, and electrophysiology, optogenetics can help uncover the circuitry responsible for complex psychiatric disease states. To date, these tools have been used preclinically to advance our understanding of these phenomena in both rodent models and in nonhuman primates. There has been early success with this research in delineating previously overlooked neural pathways that should be a target in future drug discovery efforts. However, a great deal of clarification is still needed in this work, particularly in the cases where different publications have supported disparate conclusions. As our understanding of the neurobiology of various psychiatric disease states advances, optogenetic tools may eventually be used as therapeutic agents in human patients. This research promises to advance therapeutic drug development and to help refine the groupings of diverse symptomatology present within many psychiatric diseases.
Acknowledgments
This research was supported by a grant from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2014-05069) as well as with funding from the National Institute on Drug Abuse Intramural Research Program at the National Institutes of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
Loomer HP, Saunders JC, Kline NS. A clinical and pharmacodynamic evaluation of iproniazid as a psychic energizer. Psychiatr Res Rep Am Psychiatr Assoc. 1957;8:129–141. | ||
Kapur S, Mamo D. Half century of antipsychotics and still a central role for dopamine D2 receptors. Proc Neuropsychopharmacol Biol Psychiatry. 2003;27:1081–1090. | ||
Wiedemann K. Biomarkers in development of psychotropic drugs. Dialogues Clin Neurosci. 2011;12:225–234. | ||
Hyman SE. Revitalizing psychiatric therapeutics. Neuropsychopharmacology. 2014;39:220–229. | ||
Krystal JH, State MW. Psychiatric disorders: diagnosis to therapy. Cell. 2014;157:201–214. | ||
Razafsha M, Behforuzi H, Harati H, et al. An updated overview of animal models in neuropsychiatry. Neuroscience. 2013;240:204–218. | ||
Chadman KK, Yang M, Crawley JN. Criteria for validating mouse models of psychiatric diseases. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:1–11. | ||
Miller G. Is pharma running out of brainy ideas? Science. 2010;329:502–504. | ||
Tye KM, Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012;13:251–266. | ||
Zhang F, Wang LP, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Methods. 2006;3:785–792. | ||
Rothschild KJ, Argade PV, Earnest TN, et al. The site of attachment of retinal in bacteriorhodopsin. A resonance raman study. J Biol Chem. 1982;10:8592–8595. | ||
Gunaydin LA, Yizhar O, Berndt A, Sohal VS, Deisseroth K, Hegemann P. Ultrafast optogenetic control. Nat Neurosci. 2010;13:387–392. | ||
Nagel G, Ollig D, Fuhrmann M, et al. Channelrhodopsin-1: a light-gated proton channel in green algae. Science. 2002;28:2395–2398. | ||
Nagel G, Szellas T, Kateriya S, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;25:13940–14945. | ||
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-time scale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. | ||
Mattis J, Tye KM, Ferenczi EA, et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods. 2012;9:159–172. | ||
Prigge M, Schneider F, Tsunoda SP, et al. Color-tuned channelrhodopsins for multiwavelength optogenetics. J Biol Chem. 2012;287:31804–31812. | ||
Erbguth K, Prigge M, Schneider F, Hegemann P, Gottschalk A. Bimodal activation of different neuron classes with the spectrally red-shifted channelrhodopsin chimera C1V1 in Caenorhabditis elegans. PLoS One. 2012;7:e46827. | ||
Yaroslavsky AN, Schulze PC, Yaroslavsky IV, Schober R, Ulrich F, Schwarzmaier HJ. Optical properties of selected native and coagulated human brain tissues in vitro in the visible and near infrared spectral range. Phys Med Biol. 2002;47:2059–2073. | ||
Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. Bi-stable neural state switches. Nat Neurosci. 2008;12:229–234. | ||
Yizhar O, Fenno LE, Prigge M, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. | ||
Gradinaru V, Zhang F, Ramakrishnan C, et al. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. | ||
Bamberg E, Tittor J, Osterhelt D. Light-driven proton or chloride pumping by halorhodopsin. Proc Natl Acad Sci U S A. 1993;90:639–643. | ||
Zhang F, Wang LP, Brauner M, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. | ||
Raimondo JV, Kay L, Ellender TJ, Akerman CJ. Optogenetic silencing strategies differ in their effects on inhibitory synaptic transmission. Nat Neurosci. 2012;15:1102–1104. | ||
Wietek J, Wiegert S, Adeishvili N, et al. Conversion of channelrhodopsin into a light-gated chloride channel. Science. 2014;344:409–412. | ||
Berndt A, Lee SY, Ramakrishnan C, Deisseroth K. Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science. 2014;344:420–424. | ||
Rogan SC, Roth BL. Remote control of neuronal signalling. Pharmacol Rev. 2011;63:291–513. | ||
Moreno JL, Hollowat T, González-Maeso J. G protein-coupled receptor heterocomplexes in neuropsychiatric disorders. Prog Mol Biol Transl Sci. 2013;117:187–205. | ||
Airan RD, Thompson KR, Fenno L, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458:1025–1029. | ||
Oh E, Maejima T, Liu C, Deneris E, Herlitze S. Substitution of 5-HT1A receptor signaling by a light-activated G protein-coupled receptor. J Biol Chem. 2010;285:30825–30836. | ||
Britt JP, McDevitt RA, Bonci A. Use of channelrhodopsin for activation of CNS neurons. Curr Protoc Neurosci. 2012;2:2–16. | ||
Sparta DR, Stamatakis AM, Phillips JL, Hovelso N, van Zessen R, Stuber GD. Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat Protoc. 2012;7:12–23. | ||
Mattis J, Tye KM, Ferenczi EA, et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods. 2012;9:159–172. | ||
Ting JT, Feng G. Recombineering strategies for developing next generation BAC transgenic tools for optogenetics and beyond. Front Behav Neurosci. 2014;8:111. | ||
Invernizzi RW, Parini S, Sacchetti G, et al. Chronic treatment with reboxetine by osmotic pumps facilitates its effect on extracellular noradrenaline and may desensitize a2-adrenoreceptors in the prefrontal cortex. Br J Pharmacol. 2001;132:183–188. | ||
Bach-Mizrachi H, Underwood MD, Tin A, Ellis SP, Mann JJ, Arango V. Elevated expression of tryptophan hydroxylase-2 mRNA at the neuronal level in the dorsal and median raphe nuclei of depressed suicides. Mol Psychiatry. 2008;13:507–513. | ||
Nahas Z, Anderson BS, Borckardt J, et al. Bilateral epidural prefrontal cortical stimulation for treatment-resistant depression. Biol Psychiatry. 2010;67:101–109. | ||
Bewernick BH, Hurlemann R, Matusch A, et al. Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol Psychiatry. 2010;67:110–116. | ||
Schlaepfer TE, Cohen MX, Frick C, et al. Deep brain stimulation to reward circuitry alleviates anhedonia in refractory major depression. Neuropsychopharmacology. 2008;33:368–377. | ||
Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. | ||
Grubert C, Hurlemann R, Bewernick BH, et al. Neuropsychological safety of nucleus accumbens deep brain stimulation for major depression: effects of 12-month stimulation. World J Biol Psychiatry. 2011;12:516–527. | ||
Cryan JF, Valentino RJ, Lucki I. Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Biobehav Rev. 2005;29:547–569. | ||
Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. | ||
Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52:90–110. | ||
Challis C, Beck SG, Berton O. Optogenetic modulation of descending prefrontocortical inputs to the dorsal raphe bidirectionally bias socioaffective choices after social defeat. Front Behav Neurosci. 2014;8:43. | ||
Kumar S, Black SJ, Hultman R, et al. Cortical control of affective networks. J Neurosci. 2013;33:1116–1129. | ||
Warden MR, Selimbeyoglu A, Mirzabekov JJ, et al. A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. 2012;492:428–432. | ||
Covington HE 3rd, Lobo MK, Maze I, et al. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. 2010;30:16082–16090. | ||
Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol Rev. 1992;72:165–229. | ||
Hajos M, Richards CD, Szekely AD, Sharp T. An electrophysiological and neuroanatomical study of the medial prefrontal cortical projection to the midbrain raphe nuclei in the rat. Neuroscience. 1998;87:95–108. | ||
Jankowski MP, Sesack SR. Prefrontal cortical projections to the rat dorsal raphe nucleus: ultrastructural features and associations with serotonin and gamma-aminobutyric acid neurons. J Comp Neurol. 2004;468:518–529. | ||
Celada P, Puig MV, Casanovas JM, Guillazo G, Artigas F. Control of dorsal raphe serotonergic neurons by the medial prefrontal cortex: involvement of serotonin-1A, GABA(A), and glutamate receptors. J Neurosci. 2001;21:9917–9929. | ||
Amat J, Baratta MV, Paul E, Bland ST, Watkins LR, Maier SF. Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat Neurosci. 2005;8:365–371. | ||
Schreiber R, De Vry J. Neuroanatomical basis for the antidepressant-like effects of the 5-HT(1A) receptor agonists 8-OH-DPAT and ipsapirone in the rat forced swimming test. Behav Pharmacol. 1993;4:625–636. | ||
McDevitt RA, Hiroi R, Mackenzie SM, et al. Serotonin 1B autoreceptors originating in the caudal dorsal raphe nucleus reduce expression of fear and depression-like behavior. Biol Psychiatry. 2011;69:780–787. | ||
Challis C, Boulden J, Veerakumar A, et al. Raphe GABAergic neurons mediate the acquisition of avoidance after social defeat. J Neurosci. 2013;33:13978–13988. | ||
Vialou V, Bagot RC, Cahill ME, et al. Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: role of DeltaFosB. J Neurosci. 2014;34:3878–3887. | ||
Berridge CW, Stratford TL, Foote SL, Kelley AE. Distribution of dopamine beta-hydroxylase-like immunoreactive fibers within the shell subregion of the nucleus accumbens. Synapse. 1997;27:230–241. | ||
Bockstaele EJ, Biswas A, Pickel VM. Topography of serotonin neurons in the dorsal raphe nucleus that send axon collaterals to the rat prefrontal cortex and nucleus accumbens. Brain Res. 1993;624:188–198. | ||
Tye KM, Mirzabekov JJ, Warden MR, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493:537–541. | ||
Chaudhury D, Walsh JJ, Friedman AK, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–536. | ||
Lammel S, Tye KM, Warden MR. Progress in understanding mood disorders: optogenetic dissection of neural circuits. Genes Brain Behav. 2014;13:38–51. | ||
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry. 2005;62:593–602. | ||
Skolnick P. Axioselective anxiolytics: on a quest for the holy grail. Trends Pharmacol Sci. 2012;33:611–620. | ||
Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170:1003–1010. | ||
Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. | ||
Fanselow MS, LeDoux JE. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23:229–232. | ||
Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. | ||
Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. | ||
Weller KL, Smith DA. Afferent connections to the bed nucleus of the stria terminalis. Brain Res. 1982;232:255–270. | ||
Johansen JP, Hamanaka H, Monfils MH, et al. Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci U S A. 2010;107:12692–12697. | ||
Huff ML, Miller RL, Deisseroth K, Moorman DE, LaLumiere RT. Posttraining optogenetic manipulations of basolateral amygdala activity modulate consolidation of inhibitory avoidance memory in rats. Proc Natl Acad Sci U S A. 2013;110:3597–3602. | ||
Tye KM, Prakash R, Kim SY, et al. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. | ||
Felix-Ortiz AC, Beyeler A, Seo C, Leppla CA, Wildes CP, Tye KM. BLA to vHPC inputs modulate anxiety-related behaviors. Neuron. 2013;79:658–664. | ||
Felix-Ortiz AC, Tye KM. Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci. 2014;34:586–595. | ||
Kheirbek MA, Hen R. Dorsal vs ventral hippocampal neurogenesis: implications for cognition and mood. Neuropsychopharmacology. 2011;36:373–374. | ||
Kheirbek MA, Drew LJ, Burghardt NS, et al. Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron. 2013;77:955–968. | ||
Jennings JH, Sparta DR, Stamatakis AM, et al. Distinct extended amygdala circuits for divergent motivational states. Nature. 2013;496:224–228. | ||
Kim SY, Adhikari A, Lee SY, et al. Diverging neural pathways assemble a behavioural state from separable features in anxiety. Nature. 2013;496:219–223. | ||
Sheehan TP, Chambers RA, Russell DS. Regulation of affect by the lateral septum: implications for neuropsychiatry. Brain Res Brain Res Rev. 2004;46:71–117. | ||
Anthony TE, Dee N, Bernard A, Lerchner W, Heintz N, Anderson DJ. Control of stress-induced persistent anxiety by an extra-amygdala septohypothalamic circuit. Cell. 2014;156:522–536. | ||
Knobloch HS, Charlet A, Hoffmann LC, et al. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron. 2012;73:553–566. | ||
Welch JM, Lu J, Rodriguiz RM, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448:894–900. | ||
Burguiere E, Monteiro P, Feng G, Graybiel AM. Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science. 2013;340:1243–1246. | ||
Ahmari SE, Spellman T, Douglass NL, et al. Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science. 2013;340:1234–1239. | ||
Gremel CM, Costa RM. Orbitofrontal and striatal circuits dynamically encode the shift between goal-directed and habitual actions. Nat Commun. 2013;4:2264. | ||
Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol. 1954;47:419–427. | ||
Witten IB, Steinberg EE, Lee SY, et al. Recombinase-driver rat lines: tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron. 2011;72:721–733. | ||
Ilango A, Kesner AJ, Keller KL, Stuber GD, Bonci A, Ikemoto S. Similar roles of substantia nigra and ventral tegmental dopamine neurons in reward and aversion. J Neurosci. 2014;34:817–822. | ||
Kim KM, Baratta MV, Yang A, Lee D, Boyden ES, Fiorillo CD. Optogenetic mimicry of the transient activation of dopamine neurons by natural reward is sufficient for operant reinforcement. PLoS One. 2012;7:e33612. | ||
Rossi MA, Sukharnikova T, Hayrapetyan VY, Yang L, Yin HH. Operant self-stimulation of dopamine neurons in the substantia nigra. PLoS One. 2013;8:e65799. | ||
Lammel S, Lim BK, Ran C, et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012;491:212–217. | ||
Tan KR, Yvon C, Turiault M, et al. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012;73:1173–1183. | ||
van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron. 2012;73:1184–1194. | ||
Stuber GD, Britt JP, Bonci A. Optogenetic modulation of neural circuits that underlie reward seeking. Biol Psychiatry. 2012;71:1061–1067. | ||
Britt JP, McGehee DS. Presynaptic opioid and nicotinic receptor modulation of dopamine overflow in the nucleus accumbens. J Neurosci. 2008;28:1672–1681. | ||
Cagniard B, Beeler JA, Britt JP, McGehee DS, Marinelli M, Zhuang X. Dopamine scales performance in the absence of new learning. Neuron. 2006;51:541–547. | ||
Stamatakis AM, Jennings JH, Ung RL, et al. A unique population of ventral tegmental area neurons inhibits the lateral habenula to promote reward. Neuron. 2013;80:1039–1053. | ||
Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–625. | ||
Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron. 2012;76:790–803. | ||
Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30:8229–8233. | ||
Kheirbek MA, Britt JP, Beeler JA, Ishikawa Y, McGehee DS, Zhuang X. Adenylyl cyclase type 5 contributes to corticostriatal plasticity and striatum-dependent learning. J Neurosci. 2009;29:12115–12124. | ||
Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. | ||
Bertran-Gonzalez J, Herve D, Girault JA, Valjent E. What is the degree of segregation between striatonigral and striatopallidal projections? Front Neuroanat. 2010;4:136. | ||
Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15:816–818. | ||
Britt JP, Bonci A. Optogenetic interrogations of the neural circuits underlying addiction. Curr Opin Neurobiol. 2013;23:539–545. | ||
Jayanthi S, McCoy MT, Chen B, et al. Methamphetamine downregulates striatal glutamate receptors via diverse epigenetic mechanisms. Biol Psychiatry. 2014;76:47–56. | ||
Pascoli V, Turiault M, Luscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–75. | ||
Jacobs B, Creswell J, Britt J, Ford K, Bogen J, Zaidel E. Quantitative analysis of cortical pyramidal neurons after corpus callosotomy. Ann Neurol. 2003;54:126–130. | ||
Bock R, Shin JH, Kaplan AR, et al. Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat Neurosci. 2013;16:632–638. | ||
Lee BR, Ma YY, Huang YH, et al. Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat Neurosci. 2013;16:1644–1651. | ||
Bocklisch C, Pascoli V, Wong JC, et al. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science. 2013;341:1521–1525. | ||
Chen BT, Yau HJ, Hatch C, et al. Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature. 2013;496:359–362. | ||
Suska A, Lee BR, Huang YH, Dong Y, Schluter OM. Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proc Natl Acad Sci U S A. 2013;110:713–718. | ||
Stefanik MT, Moussawi K, Kupchik YM, et al. Optogenetic inhibition of cocaine seeking in rats. Addict Biol. 2013;18:50–53. | ||
Seif T, Chang SJ, Simms JA, et al. Cortical activation of accumbens hyperpolarization-active NMDARs mediates aversion-resistant alcohol intake. Nat Neurosci. 2013;16:1094–1100. | ||
Wang X, Carlen M. Optogenetic dissection of cortical information processing-shining light on schizophrenia. Brain Res. 2012;1476:31–37. | ||
Cardin JA, Carlen M, Meletis K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. | ||
Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. | ||
Rubenstein JL. Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr Opin Neurol. 2010;23:118–123. | ||
Ledri M, Madsen MG, Nikitidou L, Kirik D, Kokaia M. Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci. 2014;34:3364–3377. | ||
Paz JT, Davidson TJ, Frechette ES, et al. Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci. 2013;16:64–70. | ||
Krook-Magnuson E, Armstrong C, Oijala M, Soltesz I. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun. 2013;4:1376. | ||
Kravitz AV, Freeze BS, Parker PR, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. | ||
Yoon HH, Park JH, Kim YH, et al. Optogenetic inactivation of the subthalamic nucleus improves forelimb akinesia in a rat model of Parkinson disease. Neurosurgery. 2014;74:533–541. | ||
Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. | ||
Kleinlogel S, Feldbauer K, Dempski R, et al. Ultra light-sensitive and fast neuronal activation with the Ca2+ permeable channelrhodopsin CatCh. Nat Neurosci. 2011;14:513–520. | ||
Chow BY, Han X, Dobry AS, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463:98–102. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.