Back to Journals » International Journal of Nanomedicine » Volume 16
Optimization and Pharmacokinetic Evaluation of Synergistic Fenbendazole and Rapamycin Co-Encapsulated in Methoxy Poly(Ethylene Glycol)-b-Poly(Caprolactone) Polymeric Micelles
Authors Shin HJ, Jo MJ, Jin IS, Park CW, Kim JS, Shin DH
Received 14 April 2021
Accepted for publication 2 July 2021
Published 16 July 2021 Volume 2021:16 Pages 4873—4889
DOI https://doi.org/10.2147/IJN.S315782
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Hee Ji Shin,1,* Min Jeong Jo,1,* Ik Sup Jin,1 Chun-Woong Park,1 Jin-Seok Kim,2 Dae Hwan Shin1
1College of Pharmacy, Chungbuk National University, Cheongju, 28160, Republic of Korea; 2Drug Information Research Institute (DIRI), College of Pharmacy, Sookmyung Women’s University, Seoul, 04310, Republic of Korea
*These authors contributed equally to this work
Correspondence: Dae Hwan Shin
College of Pharmacy, Chungbuk National University, Osongsaengmyeong 1-ro, Osong-eup, Heungdeok-gu, Cheongju, 28160, Republic of Korea
Tel +82 43 261 2820
Fax +82 43 268 2732
Email [email protected]
Purpose: We aimed to develop a nanocarrier formulation incorporating fenbendazole (FEN) and rapamycin (RAPA) with strong efficacy against A549 cancer cells. As FEN and RAPA are poorly soluble in water, it is difficult to apply them clinically in vivo. Therefore, we attempted to resolve this problem by encapsulating these drugs in polymeric micelles.
Methods: We evaluated drug synergy using the combination index (CI) values of various molar ratios of FEN and RAPA. We formed and tested micelles composed of different polymers. Moreover, we conducted cytotoxicity, stability, release, pharmacokinetic, and biodistribution studies to investigate the antitumor effects of FEN/RAPA-loaded mPEG-b-PCL micelles.
Results: We selected mPEG-b-PCL-containing FEN and RAPA at a molar ratio of 1:2 because these particles were consistent in size and had high encapsulation efficiency (EE, %) and drug loading (DL, %) capacity. The in vitro cytotoxicity was assessed for various FEN, RAPA, and combined FEN/RAPA formulations. After long-term exposures, both the solutions and the micelles had similar efficacy against A549 cancer cells. The in vivo pharmacokinetic study revealed that FEN/RAPA-loaded mPEG-b-PCL micelles had a relatively higher area under the plasma concentration–time curve from 0 to 2 h (AUC0– 2 h) and 0 to 8 h (AUC0– 8 h) and plasma concentration at time zero (Co) than that of the FEN/RAPA solution. The in vivo biodistribution assay revealed that the IV injection of FEN/RAPA-loaded mPEG-b-PCL micelles resulted in lower pulmonary FEN concentration than the IV injection of the FEN/RAPA solution.
Conclusion: When FEN and RAPA had a 1:2 molar ratio, they showed synergism. Additionally, using data from in vitro cytotoxicity, synergism between a 1:2 molar ratio of FEN and RAPA was observed in the micelle formulation. The FEN/RAPA-loaded mPEG-b-PCL micelle had enhanced bioavailability than the FEN/RAPA solution.
Keywords: combination therapy, drug interaction, cytotoxicity, biodistribution, bioavailability
Introduction
Lung cancer is a leading cause of malignant tumor-related death. Non-small cell lung cancers (NSCLC) account for ≤ 80–85% of all lung cancers that reach advanced stages.1 Numerous treatment methods have been developed for NSCLC. However, radiation therapy and chemotherapy have limited efficacy and do not markedly improve survival.2 Therefore, novel lung cancer treatment strategies with lower toxicity and greater efficacy are needed.3
Combination therapy of drugs with various anticancer mechanisms has emerged as an effective treatment strategy for reducing chemoresistance, overcoming tumor heterogeneity, and obtaining synergistic anticancer efficacy.4 A typical example is “Triolimus,” a formulation made by encapsulating three drugs (paclitaxel, rapamycin, 17-AAG) in poly(ethylene glycol)-block-poly(d,l-lactic acid) (PEG-b-PLA). Triolimus simultaneously targets several major pathways using drugs with different mechanisms: paclitaxel, a microtubule stabilizer; rapamycin, a mammalian target of rapamycin (mTOR) inhibitor; and 17-AAG, a heat shock protein 90 (Hsp90) inhibitor. These Triolimus agents also exhibit synergistic cytotoxicity and improved aqueous solubility, as well as potent tumor growth inhibition in xenograft models, providing a strong basis for further preclinical and clinical development.5,6 In addition, several preclinical and clinical studies have been conducted on the relevance of this combination drug in nanocarrier formulations. Koot et al showed that Riminocelles™, a formulation of co-encapsulated paclitaxel and clofazimine, demonstrated superior efficacy in preclinical studies.7 Batist et al demonstrated for the first time, that CPX-1, a liposome formulation co-encapsulated with irinotecan and floxuridine, was well tolerated and exhibited antitumor activity in patients with advanced solid tumors in a Phase 1 study.8 Therefore, administering various chemotherapeutic drugs together as appropriate nanocarrier formulations may be a promising strategy for cancer treatment.
Fenbendazole (FEN; [5-(phenylthio)-1H-benzimidazol-2-yl] carbamic acid methyl ester) is a benzimidazole anthelmintic agent that is widely used against various parasites infecting different animal species.9,10 FEN binds tubulin which is an essential structural protein in microtubules. Hence, its anti-parasite action is based on interference with tubulin microtubule equilibrium.11,12 FEN has demonstrated very low toxicity in various animal species including dogs.13–16 Moreover, several studies conducted on cancer cell treatment reported positive effects of FEN. Chu et al demonstrated that benzimidazoles such as FEN and albendazole destroyed microtubules, induced apoptosis, and inhibited drug resistance in human ovarian cancer cells.17 Dogra et al showed that FEN exerts antitumor efficacy by disrupting microtubule dynamics, regulating genes involved in several cellular pathways, and activating p53.18 Aycock-Williams et al reported that FEN, when administered alone, inhibited the proliferation of human prostate cancer cell lines to a greater extent than vitamin E succinate (VES). Furthermore, there was a synergy of efficacy between FEN and VES.19 Han et al demonstrated that FEN elicited anticancer effects against HL-60 cells by producing reactive oxygen species (ROS).20 Dogra et al showed that FEN inhibited proteasomal function, activated the mitochondrial pathway, and caused apoptosis in human lung cancer cell lines.21 Despite its numerous anticancer effects, clinical application of FEN is difficult owing to its low solubility in water.22,23
Rapamycin (RAPA) is a carboxylic lactone-lactam macrolide antibiotic that was originally isolated from the bacterium Streptomyces hygroscopicus.24,25 It was initially developed as an antifungal agent. Nevertheless, it subsequently presented with potent anti-proliferative/anti-cancer and immunosuppressive efficacy.26–28 It was reported that RAPA inhibited tumor growth in several cancer types, including colon cancer,29,30 breast cancer,31,32 and lung cancer.33 RAPA inhibits the protein kinase mTOR (mammalian target of rapamycin) that plays a key role in cytokine-mediated cell proliferation. In the G1-S phase, RAPA induces cell cycle arrest, prevents IL-2-stimulated T cell proliferation, and causes immunosuppression.34–36 Several studies have also reported synergistic effects between RAPA and a drug with mechanisms resembling those of FEN. Shafer et al showed that RAPA potentiated paclitaxel efficacy against endometrial cancer by inducing apoptosis, inhibiting cell proliferation, and potentially increasing tubulin acetylation and neutralization.37 Marimpietri et al suggested that vinblastine and RAPA triggered dramatic G2-M phase cell cycle arrest and almost fully depleted cells actively synthesizing DNA in neuroblastoma.38 Zhou et al demonstrated that co-targeting with temsirolimus (mTOR inhibitor) and vinblastine (microtubule-destabilizing agent) was more effective against hepatocellular carcinoma than single targeting with temsirolimus or vinblastine.39 Based on the foregoing studies, we predicted that a combination of FEN and RAPA should effectively and synergistically inhibit cancer cells. However, RAPA is also poorly soluble in water and is difficult to convert into clinically and commercially acceptable injectable formulations.40
Polymeric micelles are potentially invaluable as they can overcome low water solubility.41–43 When a hydrophobic drug is encapsulated in a micelle, it can be completely dispersed in water and formulated for intravenous injection.44 Polymeric micelles have amphiphilic properties including hydrophobic cores and hydrophilic shells. Hence, they can enable the solubilization of hydrophobic drugs.45 In addition, they are biodegradable, biocompatible, nontoxic, and have high vascular permeability at the target site because they are small (< 100 nm).46,47 We used three candidate polymers to design an optimal micelle formulation. Methoxy poly(ethylene glycol)-b-poly(caprolactone) (mPEG-b-PCL) is a diblock copolymer with a hydrophobic PCL core and a hydrophilic PEG shell. Degradation products of these copolymers are easily excreted, nontoxic, and do not accumulate in vivo.48,49 Moreover, as mPEG-b-PCL has a simple structure and is biocompatible, it has been widely used in drug delivery systems to increase drug solubility.50,51 Methoxy poly(ethylene glycol)-b-poly(
In the present study, we used micelles to solubilize a combination of the microtubule inhibitor FEN and the mTOR inhibitor RAPA. We then evaluated the combination index (CI) of FEN and RAPA and selected the optimal drug ratio. We also investigated the physicochemical properties of micelles, in vitro cytotoxicity, in vitro release, and pharmacokinetic profiles.
Materials and Methods
Material and Reagents
Methoxy poly(ethylene glycol)-b-poly(D,L-lactide) (mPEG [4000]-b-PLA [2200]) was purchased from Advanced Polymer Materials, Inc. (Montreal, QC, Canada). The mPEG-b-PCL (MW 2000:2000 Da) was purchased from PolySciTech® (West Lafayette, IN, USA). Soluplus® [Polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer (PCL-PVAc-PEG)] was obtained from BASF (Ludwigshafen, Rhineland-Palatinate, Germany). RAPA and genistein were purchased from LC Laboratories® (Woburn, MA, USA). FEN, dimethyl sulfoxide (DMSO), thiazolyl blue tetrazolium bromide (MTT), tert-butanol, cremophor EL®, and dimethylacetamide (DMA) were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). Distilled water (DW) was purchased from Tedia (Fairfield, OH, USA). Acetonitrile (ACN) and ethanol (EtOH) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Methanol (MeOH) was purchased from Honeywell, Burdick & Jackson (Ulsan, Korea). All other reagents were used as received without additional purification and were at least HPLC grade.
Cell Line and Cell Culture
The A549 cells used here were human alveolar basal epithelial adenocarcinoma cells and were purchased from the American Type Culture Collection (Manassas, VA, USA). Roswell Park Memorial Institute medium (RPMI 1640), Dulbecco’s phosphate-buffered saline (DPBS), penicillin-streptomycin solution, trypsin, and fetal bovine serum (FBS) were purchased from Corning Inc. (Corning, NY, USA). The A549 cell medium consisted of RPMI 1640 medium supplemented with 10% (v/v) FBS and 1% (w/v) penicillin-streptomycin solution. The cells were grown at 37°C under a 5% CO2 atmosphere.
Combination Index (CI) Analysis
The CI analysis was performed using the Chou-Talalay method to evaluate drug-drug interactions.56 CI was calculated as follows:

where (D)1 and (D)2 are the 50% inhibitory concentrations (IC50) values of each drug in the combination and (Dx)1 and (Dx)2 are the IC50 values of the drugs. This equation defines antagonism (CI > 1), additivity (CI = 1), and synergism (CI < 1).
Preparation of FEN- and RAPA-Loaded Polymeric Micelles
FEN/RAPA-loaded polymeric micelles were prepared from three polymers by the freeze-drying method (Figure 1).57,58 Various FEN/RAPA ratios were dissolved along with the polymers in 1 mL tert-butanol and stirred in water at 60°C for 3 min. Then 1 mL DW was added and the FEN/RAPA-polymer mixture was vortexed, quickly frozen at −70°C for 1 h, put in a freeze-dryer (Advantage Pro; SP Scientific, Warminster, PA, USA), and lyophilized for 24 h. Then 1 mL DW at 60°C was added to rehydrate the mixture. The FEN/RAPA-loaded micelle solution was centrifuged at 16,600 x g for 5 min at 4°C (Hanil Science Inc., Gimpo, Korea) and the supernatant was collected and sterilized and purified using a non-pyrogenic sterile syringe filter with 0.2 μm pore size (Corning, NY, USA).
 |
Figure 1 Fenbendazole (FEN)/rapamycin (RAPA)-loaded micelle preparation using freeze-dryer. |
Physicochemical Micelle Characterization
The particle size, polydispersity index (PDI), and zeta potential of the FEN/RAPA-loaded micelles were measured with a dynamic light scattering (DLS) device (Anton Paar, Litesizer 500, Graz, Austria). The angle of the size measurement was automatically selected between side scatter (90°) and back scatter (175°). FEN/RAPA-loaded micelle encapsulation efficiency (EE, %) and drug loading (DL, %) were obtained by HPLC and calculated as follows:


Data for the micelle sample analyses are expressed as means ± SD of three separate experiments.
Transmission Electron Microscopy Study
Transmission electron microscopy (TEM) (JEOL Ltd., Tokyo, Japan) was used to observe the ultrastructure of the FEN/RAPA-loaded mPEG-b-PCL micelles. The micelle samples were diluted in DW, dropped onto copper grids, dried at room temperature for 2 d, and examined at accelerating voltage of 200 kV.59,60
In vitro Drug Release Assay
The dialysis method was used to measure the in vitro rates of encapsulated FEN and RAPA release from micelles.61 FEN/RAPA-loaded mPEG-b-PCL micelles were placed in a dialysis membrane bag (MW cutoff = 20 kDa) and immersed in 2.0 L phosphate-buffered saline (PBS; pH 7.4). The PBS was maintained at 37°C, constantly stirred at 200 rpm with a magnetic bar, and replaced with fresh medium at 8, 24, 72, 168, and 240 h to maintain the sink. Twenty-microliter samples were collected at 0, 2, 4, 6, 8, 24, 48, 72, 168, 240, and 336 h and diluted 10x with ACN. The FEN and RAPA concentrations were measured by HPLC. Data were processed in SigmaPlot v. 10.0 using the Peppas model.
In vitro Stability of FEN/RAPA-Loaded mPEG-b-PCL Micelle
Stability tests were performed for 2 wk at 4°C (refrigeration), 25°C (room temperature), and 37°C (water bath) conditions. Twenty-microliter micelle samples were collected on days 0, 1, 2, 5, 7, and 14. Their particle sizes and PDI were measured with a DLS device. Data were processed in SigmaPlot v 10.0 (Systat Software, San Jose, CA, USA).
In vitro Cytotoxicity Assay
Cytotoxicity is a biological index and confirms drug-induced cell death.62 Here, cytotoxicity was assessed by the MTT assay.63 A549 cells were seeded in 96-well plates at 5000/well density. After incubation at 37°C and under 5% CO2 for 24 h, the medium was removed, and the cells were treated with free FEN, free RAPA, free FEN/RAPA, FEN-loaded mPEG-b-PCL micelles, RAPA-loaded mPEG-b-PCL micelles, or FEN/RAPA-loaded mPEG-b-PCL micelles. The free drugs were dissolved in 100 µL DMSO, diluted 1000x with RPMI 1640 medium, and used as the initial concentration. The drug-loaded mPEG-b-PCL micelle solutions were diluted 10x with RPMI 1640 medium and used as the initial concentration. After 48 h, the medium was removed and 100 µL MTT solution (0.5 mg/mL) was added. After incubation for 4 h, the MTT solution was removed and 100 µL of DMSO was added followed by gentle shaking for 10 min at 200 rpm on an orbital shaker (N-BIOTEK, NB-101S, Bucheon, Korea). A microplate reader (Molecular Devices, Spectra Max ID3, San Jose, CA, USA) measured the absorbance at 540 nm. Data were processed in GraphPad Prism v. 5 (GraphPad Software, La Jolla, CA, USA).
In vitro Clonogenic Assay
A clonogenic assay is conducted to evaluate in vitro cell survival based on the growth of a single cell into a colony.64 A549 cells were seeded in six-well plates at 200/well density. After 24 h, the cells were treated with free FEN, free RAPA, free FEN/RAPA, FEN-loaded mPEG-b-PCL micelles, RAPA-loaded mPEG-b-PCL micelles, or FEN/RAPA-loaded mPEG-b-PCL micelles. After 2 wk incubation, the medium was removed and the colonies were stained with 1 mL crystal violet solution (0.5% w/v). After 30 min incubation, the plates were rinsed with clean water. The number of colonies with more than 50 cells was counted. Colony formation was calculated as follows:65

In vivo Pharmacokinetic Study
The animal testing and experimentation protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Chungbuk National University, Korea (No. CBNUA-1502-21-01; approval date: February 10, 2021). Sprague-Dawley rats (male; age 7–8 wk) were obtained from Samtako Bio Korea (Osan, Korea) and used in the in vivo studies. The rats were maintained in cages containing aspen shavings and provided with sufficient water and food. Free FEN and RAPA were dissolved in Cremophor EL®:DMA:EtOH:DPBS (10:10:20:60, v/v/v/v) and used as controls.66 The rats were divided into six groups administered intravenous (IV) injections of FEN solution, FEN-loaded mPEG-b-PCL micelles, RAPA solution, RAPA-loaded mPEG-b-PCL micelles, FEN/RAPA solution, or FEN/RAPA-loaded mPEG-b-PCL micelles in a tail. FEN was administered at 5 mg/kg and RAPA was administered at 30 mg/kg. About 500 µL of blood was collected from the infraorbital plexus at 5, 15, 30, 60, 120, 240, and 480 min after IV injection, centrifuged at 2450 x g for 5 min to obtain plasma, and stored at −70°C in a deep freezer. The FEN and RAPA pharmacokinetic parameters were calculated in SigmaPlot v. 10.0 (Systat Software, Inc., San Jose, CA, USA). A one-compartment model was used for FEN, and a two-compartment model was used for RAPA. In addition, the AUC value for a specific time period was calculated using the trapezoidal rule.
Biodistribution Study
A biodistribution study was conducted to analyze the tissue distribution patterns of FEN and RAPA at 8 h after IV administration of drug. The rats were euthanized with CO2 and their livers, spleens, kidneys, hearts, lungs, and muscles were excised. Each sample was washed in PBS and stored at −70°C in a deep freezer.
High-Performance Liquid Chromatography (HPLC) Analysis
Assay Conditions
The HPLC system (Waters, Milford, MA, USA) consisted of a 2695 separation module and a 2996 photodiode array detector. It measured FEN, RAPA, and genistein (internal standard, IS) sample concentrations. The temperature of the Fortis C18 chromatography column (5 µm; 4.6 mm x 250 mm) (Fortis Technologies Ltd., Cheshire, UK) was maintained at 30°C. FEN, RAPA, and genistein were eluted in isocratic mode. The sample injection volume was 10 µL and the flow rate of the mobile phase (ACN:water (70:30, v/v)) was 1.0 mL/min. FEN, RAPA, and genistein were detected at 295 nm, 277 nm, and 259 nm, respectively, and their retention times were 5.33 min, 20.3 min, and 3.5 min, respectively. A calibration curve was plotted by correlating the concentration of each drug with its corresponding peak area and the sample concentrations were interpolated from it.
Preparation of Biological Samples
Samples stored at −70°C were thawed at room temperature before measuring their FEN and RAPA concentrations. Then 100 µL plasma sample, 200 µL MeOH, and 25 µL IS were mixed in a 1.5-mL microtube (Corning, Axygen® 1.5 MaxyClear Microtube, NY, USA) and centrifuged at 16,600 x g for 5 min at 4°C. Supernatants were passed through a 0.2-µm filter and their FEN and RAPA concentrations were measured by HPLC. To establish the distribution patterns of the FEN and RAPA remaining in the tissues after administration, 0.2 g of each tissue was taken, combined with 0.8 mL PBS, and pulverized with a Teflon pestle in a glass Potter-Elvehjem-type homogenizer (IKA Works Inc., Ultra Turrax T-25, Staufen, Germany). Then 200 µL plasma, 400 µL MeOH, and 50 µL IS were put in a microtube and the FEN and RAPA concentrations in the samples were analyzed as previously described.
Statistical Analysis
ANOVA was run using GraphPad Prism v. 5.0 (GraphPad Software, La Jolla, CA, USA) to identify statistically significant differences between experimental data. To obtain the P-value, Tukey’s test was used as a post hoc test. P < 0.05 indicated statistical significance.
Results
Evaluation of Synergy Between FEN and RAPA
The IC50 values of FEN, RAPA, and their combinations at various molar ratios were determined in A549 cells (Table 1). The IC50 value of FEN was 11.7 μM. The viability of A549 cells exposed to the highest concentration of RAPA was > 50%. Hence, the IC50 value surpassed that concentration. The CI for various FEN:RAPA molar ratios were calculated using the IC50 values for FEN and RAPA (Table 2).67 When the FEN:RAPA molar ratio was 1:1, the CI was < 1.14. Therefore, synergy between FEN and RAPA could neither be ruled out nor confirmed. Other drug combinations showed synergism as their CIs were < 1.
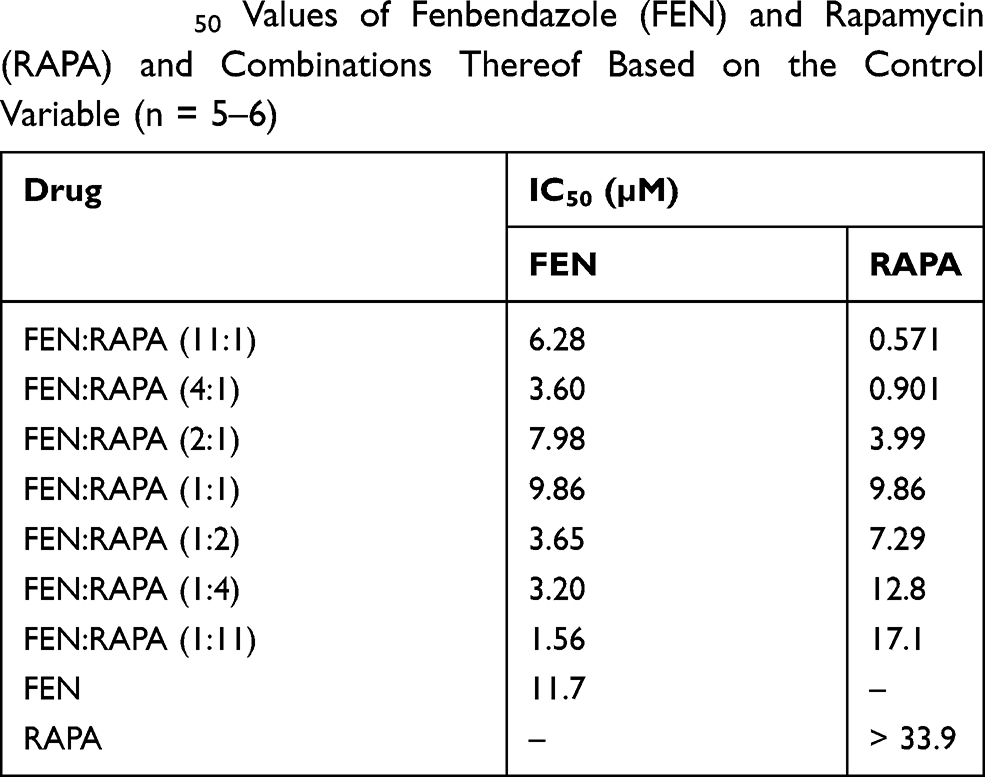 |
Table 1 IC50 Values of Fenbendazole (FEN) and Rapamycin (RAPA) and Combinations Thereof Based on the Control Variable (n = 5–6) |
 |
Table 2 CI Values Based on Fenbendazole (FEN):rapamycin (RAPA) Rations (n = 5–6) |
Physicochemical Characterization of FEN/RAPA-Loaded Micelles
To determine the optimal FEN:RAPA molar ratio to enclose in the micelle, we assessed the antitumor efficacy of RAPA and the solubilities of FEN and RAPA in various polymers.6 Considering the CI, the molar ratios of FEN and RAPA were set to 1:1 and 1:2. EE (%), DL (%), particle size, PDI, and zeta potential of micelles synthesized from various polymers were calculated (Table 3). When micelles were prepared by fixing the polymer quantity to 100 mg, the mPEG-b-PLA micelles containing FEN and RAPA at a molar ratio of 1:2 had the best EE (%) and DL (%). Nevertheless, the sizes of the micelles fabricated from mPEG-b-PLA did not converge to a single peak. Therefore, we selected mPEG-b-PCL micelle with FEN and RAPA at a molar ratio of 1:2, as they were uniform in size and had high EE (%) and DL (%). We compared FEN/RAPA-loaded mPEG-b-PCL micelles containing different amounts of polymer and determined that 100 mg polymer was ideal. The size distribution graph showed that mPEG-b-PCL encapsulated with FEN and RAPA in a 1:2 ratio had a size of less than 100 nm (Figure 2A), and in the TEM image, the micelles showed a uniform spherical shape (Figure 2B).
 |
Table 3 Characteristics of Fenbendazole (FEN) and Rapamycin (RAPA) Encapsulated in Various Polymers (n = 3) |
 |
Figure 2 (A) Particle size distributions of fenbendazole (FEN)/rapamycin (RAPA)-loaded micelles according to feeding polymer type. (B) Transmission electron microscopy (TEM) images of final FEN/RAPA-loaded mPEG-b-PCL micelle formulation. (n = 3). |
Stability Test of FEN/RAPA-Loaded mPEG-b-PCL Micelles
The size and PDI of the FEN/RAPA-loaded mPEG-b-PCL micelles were measured at 4°C, 25°C, and 37°C for 2 wk (Figure 3). At 4°C, the mean micelle size was < 50 nm until day 7 but had increased to ~100 nm by day 14. At 20°C and 37°C, the micelle size had increased to ≥ 1000 nm after 1 d incubation.
 |
Figure 3 Changes in (A) size and (B) PDI of fenbendazole (FEN)/rapamycin (RAPA)-loaded mPEG-b-PCL micelles after 2 wk at various temperatures. (n = 4). |
In vitro Cytotoxicity Assay
The IC50 values of free FEN and the FEN-loaded mPEG-b-PCL micelle were 2245 nM and 8695 nM, respectively (Figure 4A and B). The IC50 values of free RAPA and RAPA-loaded mPEG-b-PCL micelle were 18,904 nM and 668,291 nM, respectively, and this difference was statistically significant (p < 0.001) (Figure 4C and D). The IC50 values of free FEN/RAPA and FEN/RAPA mPEG-b-PCL micelle were 1384 nM and 5535 nM, respectively (Figure 4E and F). The CIs of free FEN/RAPA and FEN/RAPA-loaded mPEG-b-PCL micelle were 0.25 and 0.22, respectively. These findings indicate a synergistic effect between the two free drugs and between the two drugs in micelles.
 |
Figure 4 In vitro cytotoxicity assay of A549 cells treated with (A) free fenbendazole (FEN), (B) FEN-loaded mPEG-b-PCL micelles, (C) free rapamycin (RAPA), (D) RAPA-loaded mPEG-b-PCL micelles, (E) free FEN/RAPA, and (F) FEN/RAPA-loaded mPEG-b-PCL micelles. (n = 6). |
In vitro Clonogenic Assay
A clonogenic assay was performed to evaluate the inhibitory efficacy of drugs against cancer cell proliferation after prolonged exposure. We compared colony formation after the free FEN and FEN-loaded mPEG-b-PCL micelle treatments for 2 wk and found that no colonies developed at 2090 nM in either treatment (Figure 5A). At 209 nM and 20.9 nM, however, the free FEN inhibited colony formation slightly more effectively than the FEN-loaded mPEG-b-PCL micelle. Both the free RAPA and the RAPA-loaded mPEG-b-PCL micelle treatments almost equally inhibited colony formation at all tested concentrations (Figure 5B). The FEN/RAPA-loaded mPEG-b-PCL micelle further inhibited colony formation unlike the free FEN/RAPA at 80 nM, 8 nM, and 0.8 nM (Figure 5C). However, all data showed no statistically significant difference.
 |
Figure 5 In vitro clonogenic assay of various formulations encapsulating fenbendazole (FEN), rapamycin (RAPA), or combination of FEN plus RAPA. A549 cell colony formation in (A) Free FEN and FEN-loaded mPEG-b-PCL micelle, (B) Free RAPA and RAPA-loaded mPEG-b-PCL micelle, (C) Free FEN/RAPA and FEN/RAPA-loaded mPEG-b-PCL micelle. (n = 3). |
In vitro Drug Release Profile
The drug release profiles of FEN and RAPA in FEN/RAPA-loaded mPEG-b-PCL micelles are shown in Figure 6. There was 17% and 45% FEN release at 24 h and 72 h, respectively. There was 36% and 64% RAPA release at 24 h and 72 h, respectively. After 240 h, > 90% of both drugs had been released from their micelles.
 |
Figure 6 Fenbendazole (FEN) and rapamycin (RAPA) release profiles from FEN/RAPA-loaded mPEG-b-PCL micelles. (n = 3). |
In vivo Pharmacokinetic Study
Rat plasma concentration-time profiles after IV injection of various FEN and RAPA formulations are shown in Figure 7. RAPA was detected in plasma until 8 h after IV injection whereas FEN decreased to a level below the limit of detection (LOD) after 4 h. The mean plasma FEN concentration after FEN-loaded mPEG-b-PCL micelle injection was 1.6x greater than it was at 5 min (p < 0.01) and 15 min (p < 0.05) after injection of FEN solution. The plasma RAPA concentration after RAPA-loaded mPEG-b-PCL micelle injection was 2x greater than it was at 5 min after injection of RAPA solution (p < 0.001), 1.7x higher than it was at 15 min after injection of RAPA solution (p < 0.001), and 1.6x than it was at 30 min after injection of RAPA solution (p < 0.001). Plasma RAPA concentration after FEN/RAPA-loaded mPEG-b-PCL micelle injection was 1.4x greater than the plasma RAPA concentration at 5 min after injection of FEN/RAPA solution (p < 0.05). However, the plasma FEN concentration after FEN/RAPA-loaded mPEG-b-PCL micelle injection did not significantly differ from the plasma FEN concentration after injection of FEN/RAPA solution. The plasma concentrations of both drugs were higher at ≤ 60 min after IV micelle administration than they were at ≤ 60 min after injection of the IV solution. We calculated parameters related to FEN and RAPA (Tables 4 and 5). The area under the plasma concentration-time curve from 0 to 8 h (AUC0–8h) for RAPA after RAPA-loaded mPEG-b-PCL micelle injection was 1.3x higher than that after the injection of the RAPA solution (p < 0.001). The AUC0–8h of RAPA after FEN/RAPA-loaded mPEG-b-PCL micelle injection was 1.2x higher than AUC0–8h of RAPA after injection of the FEN/RAPA solution (p < 0.05). The area under the plasma concentration-time curve from 0 to 2 h (AUC0–2h) of FEN after administration of FEN micelle was 1.6x higher than AUC0–2h of FEN after the injection of FEN-loaded mPEG-b-PCL solution (p < 0.01). However, the AUC0–2h of FEN after the injection of FEN/RAPA solution and after FEN/RAPA-loaded mPEG-b-PCL micelle administration did not significantly differ.
 |
Table 4 Pharmacokinetic Parameters of Fenbendazole (FEN) After Intravenous (IV) Injection of FEN Solution, FEN-Loaded mPEG-b-PCL Micelle, FEN/RAPA Solution, and FEN/RAPA-Loaded mPEG-b-PCL Micelle into Rats (n = 3) |
 |
Table 5 Pharmacokinetic Parameters of Rapamycin (RAPA) After Intravenous (IV) Injection of RAPA Solution, RAPA-Loaded mPEG-b-PCL Micelle, FEN/RAPA Solution, and FEN/RAPA-Loaded mPEG-b-PCL Micelle into Rats (n = 3) |
 |
Figure 7 Plasma concentration-time profiles of fenbendazole (FEN) and rapamycin (RAPA) in rats. (A) FEN concentration in rat plasma after 5 mg/kg intravenous (IV) injection of FEN solution and FEN-loaded mPEG-b-PCL micelle. (B) RAPA concentration in rat plasma after 30 mg/kg IV injection of RAPA solution and RAPA-loaded mPEG-b-PCL micelle. (C) FEN concentration in rat plasma after 5 mg/kg IV injection of FEN/RAPA solution and FEN/RAPA-loaded mPEG-b-PCL micelle. (D) RAPA concentration in rat plasma after 30 mg/kg IV injection of FEN/RAPA solution and FEN/RAPA-loaded mPEG-b-PCL micelle. (n = 3). |
Biodistribution Study
FEN and RAPA distributions in specific organs were observed 8 h after IV injection. The FEN concentration in the lungs at 8 h after the administration of FEN solution was higher than the FEN concentration in the lungs at 8 h after the injection of FEN-loaded mPEG-b-PCL micelle (p < 0.05) (Figure 8A). Furthermore, the pulmonary FEN concentration at 8 h after IV injection of FEN/RAPA solution was higher than the pulmonary FEN concentration at 8 h after the administration of FEN/RAPA-loaded mPEG-b-PCL micelle. However, the RAPA concentrations after standalone RAPA and combined FEN/RAPA injections were similar in the livers, spleens, kidneys, hearts, lungs, and muscles (Figure 8B).
 |
Figure 8 Biodistribution of (A) fenbendazole (FEN) and (B) rapamycin (RAPA) in each tissue 8 h after IV injection. (p < 0.05) (n = 3). |
Discussion
We previously performed FEN-loaded Soluplus® micellar studies.23 Based on these studies, we hypothesized that if a micellar formulation was made by encapsulating FEN with other anticancer drugs, it would be more effective than a single formulation on cancer cells. FEN is an antimitotic agent similar to vinblastine and paclitaxel, and RAPA is a drug that inhibits mTOR signaling in cancer cells. We predicted that these two drugs would exhibit synergistic effects by simultaneously targeting multiple key pathways with different mechanisms of action. However, both FEN and RAPA have low bioavailability as they are poorly soluble.68,69 To overcome this deficiency, the bioavailability of both agents could be increased by processing them into injectable formulations. To this end, we adopted polymeric micelles as carriers to co-encapsulate FEN and RAPA. Polymeric micelles are relatively more stable and comparatively less likely to get degraded in the blood than other nanocarriers.70 Moreover, their small size enables them to accumulate in tumors via enhanced permeability and retention (EPR).71
To evaluate the potential synergy between FEN and RAPA, the CI was calculated at various molar ratios of these drugs. When the FEN:RAPA molar ratio was 1:1, the CI was < 1.14. Therefore, true synergism could not be confirmed. However, other FEN:RAPA molar ratios demonstrated synergy of efficacy. Based on the solubilities of both drugs in the polymers and the pharmacokinetic RAPA dose with anticancer efficacy, FEN:RAPA molar ratios of 1:1 and 1:2 were selected. We encapsulated these drug combinations in various polymers and selected the mPEG-b-PCL micelle as the carrier for FEN:RAPA at a molar ratio of 1:2 because its particles were consistent in size and had high EE and DL capacity. We measured and compared the sizes of the FEN/RAPA-loaded mPEG-b-PCL micelles at 4°C, 25°C, and 37°C for 2 wk and found that they remained at < 50 nm for 7 d at 4°C. At the other temperatures, however, the particle sizes significantly changed within 1 d. Therefore, it is recommended to store the micelles at 4°C to maintain their stability.
Cytotoxicity evaluations based on the MTT assay showed that the IC50 value of free RAPA was substantially lower than the IC50 value of RAPA-loaded mPEG-b-PCL micelle. The free FEN and free FEN/RAPA also had lower IC50 values than the FEN- and FEN/RAPA-loaded mPEG-b-PCL micelles, respectively. Several situations can be considered as the reason why free drugs are more potent than micelle formulation in in vitro condition. First, it may take some time for FEN and RAPA to be released from the micelle and uptake into the cancer cell.49,72 Second, the process of endocytosis of micelles into cancer cells may take some time.73,74 In any case, nanoparticles have many advantages in in vivo situations, so if actually administered in the body, the results may be inconsistent with in vitro. In the cytotoxicity evaluation by clonogenic assay, the free FEN at 209 nM was 1.6x more effective at inhibiting colony formation than the FEN-loaded mPEG-b-PCL micelle at the same concentration. Both the free RAPA and the RAPA-loaded mPEG-b-PCL micelle were nearly equally effective at inhibiting colony formation at the same concentrations. However, FEN/RAPA-loaded mPEG-b-PCL micelle at 0.8 nM was 1.5x more effective at inhibiting colony formation than free FEN/RAPA at the same concentration. The IC50 value of the free RAPA and the IC50 value of the RAPA-loaded mPEG-b-PCL micelle significant differed according to the MTT assay results. Nevertheless, there was no significant difference between free RAPA and RAPA-loaded mPEG-b-PCL micelles in terms of colony inhibition efficacy according to the clonogenic assay.
In the in vivo pharmacokinetic study, when both the FEN-loaded mPEG-b-PCL micelle and the FEN solution were administered at 5 mg/kg, FEN in the FEN-loaded mPEG-b-PCL micelles exhibited 1.6x higher AUC0–2h, 1.6x higher Co than the FEN solution. The FEN-loaded mPEG-b-PCL micelle maintained high FEN concentrations in the plasma for a longer period than the FEN solution. Compared with the FEN solution, the relative bioavailability of FEN-loaded mPEG-b-PCL micelles to FEN was 162%. When 30 mg/kg RAPA solution and RAPA-loaded mPEG-b-PCL micelle were administered, RAPA in the RAPA-loaded mPEG-b-PCL micelles displayed 1.3x higher AUC0–8h and 1.6x higher Co than the RAPA solution. Compared with the RAPA solution, the relative bioavailability of RAPA-loaded mPEG-b-PCL micelles to RAPA was 132%. After IV injection of the FEN/RAPA solution and FEN/RAPA-loaded mPEG-b-PCL micelles, in terms of FEN, the AUC0–2h and Co of the FEN/RAPA-loaded mPEG-b-PCL micelle were higher than those of the FEN/RAPA solution. Similarly, in terms of RAPA, the AUC0–8h and Co of the FEN/RAPA-loaded mPEG-b-PCL micelle were higher than those of the FEN/RAPA solution. Compared with the FEN/RAPA solution, the relative bioavailability of FEN/RAPA-loaded mPEG-b-PCL micelles to FEN was 125% and the relative bioavailability of FEN/RAPA-loaded mPEG-b-PCL micelles to RAPA was 121%. Therefore, the FEN/RAPA-loaded mPEG-b-PCL micelle was predicted to apparently have superior bioavailability to the FEN/RAPA solution.75 It is believed that the performance will be improved if the drug stays in the body for a long time and exhibits an increased EPR effect by controlling the release rate of the drug through the modification of the micelles.
In the biodistribution study, RAPA showed similar distributions in the livers, spleens, kidneys, hearts, lungs, and muscles regardless of solution or micelle formulations. In contrast, in FEN, solution accumulated at a higher concentration in the lungs than the micelle after both the single and combination treatments.
Conclusion
A combination of two drugs with different modes of action might have greater anticancer efficacy than single drug treatments as there could be a synergy of efficacy between both agents. Here, we evaluated whether there is synergy between FEN and RAPA as each of these drugs has demonstrated anticancer efficacy. As they are poorly soluble, however, we loaded them into a polymeric micelle to increase their bioavailability. FEN and RAPA at 1:2 a ratio and the drug combination at a 1:2 ratio encapsulated in mPEG-b-PCL micelles both showed synergism. We evaluated the physicochemical properties of micelles according to the various polymers and established the optimal formulation consisted of mPEG-b-PCL micelles containing FEN and RAPA at a molar ratio of 1:2. We assessed the in vitro cytotoxicity of FEN, RAPA, and FEN/RAPA combinations against cancer cells. In the in vivo studies, we intravenously injected these formulations into rats and calculated the pharmacokinetic parameters of FEN and RAPA such as AUC0–2h, AUC0–8h, Co, and Vd. The micelle formulations showed relatively greater bioavailability than the drugs dissolved with solubilizing agents. We believe that the findings of the present study lay an empirical foundation for ongoing research into the clinical administration of FEN/RAPA polymeric micelles for anticancer therapy.
Abbreviations
ACN, acetonitrile; ANOVA, analysis of variance; AUC0–2h, area under the plasma concentration-time curve from 0 to 2 h; AUC0–8h, area under the plasma concentration-time curve from 0 to 8 h; Co, plasma concentration at time zero; CI, combination index; DL, drug loading; DLS, dynamic light scattering; DMA, dimethylacetamide; DMSO, dimethyl sulfoxide; DPBS, Dulbecco’s phosphate-buffered saline; DW, distilled water; EE, encapsulation efficiency; EPR, enhanced permeability and retention; EtOH, ethanol; FBS, fetal bovine serum; FEN, fenbendazole; HPLC, high-performance liquid chromatography; IC50, half maximal inhibitory concentration; MeOH, methanol; mTOR, mammalian target of rapamycin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PBS, phosphate-buffered saline; PCL, polycaprolactone; PEG, polyethylene glycol; PDI, polydispersity index; PVA, polyvinyl acetate; RAPA, rapamycin; RPMI, Roswell Park Memorial Institute; TEM, transmission electron microscopy; Vd, volume of distribution.
Data Sharing Statement
All the data contained in the article is available.
Ethics Approval
Animal experiments and experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Chungbuk National University (No. CBNUA-1502-21-01; approval date: February 10, 2021), and matters related to the welfare and treatment of laboratory animals were performed according to the guidelines of the Ministry of Food and Drug Safety (MFDS) in Republic of Korea.
Acknowledgments
The authors would like to thank Moon Sup Yoon and Yu Jin Lee for their technical assistance.
Author Contributions
Conceptualization, H.J.S., I.S.J., and D.H.S.; methodology, H.J.S. and M.J.J.; software, H.J.S. and M.J.J.; validation, H.J.S. and D.H.S.; formal analysis, H.J.S. and M.J.J.; investigation, H.J.S. and M.J.J.; resources, C.-W.P., J.-S.K., and D.H.S.; data curation, H.J.S.; interpretation, H.J.S., M.J.J., I.S.J., and D.H.S.; writing—original draft preparation, H.J.S. and M.J.J.; writing—review and editing, H.J.S., M.J.J., C.-W.P., J.-S.K., and D.H.S.; visualization, H.J.S. and M.J.J.; supervision, C.-W.P., J.-S.K., and D.H.S.; project administration, H.J.S., M.J.J., I.S.J., C.-W.P., J.-S.K., and D.H.S. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This research was funded by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, grant number NRF-2019R1C1C1009996. It was supported by a National Research Foundation of Korea Grant funded by the Korean government (MSIP) (No. MRC2017R1A5A2015541). These results were also supported by “Regional Innovation Strategy (RIS)” through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (MOE).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kim YS, Jin HO, Seo SK, et al. Sorafenib induces apoptotic cell death in human non-small cell lung cancer cells by down-regulating mammalian target of rapamycin (mTOR)-dependent survivin expression. Biochem Pharmacol. 2011;82(3):216–226. doi:10.1016/j.bcp.2011.04.011
2. Khan N, Afaq F, Khusro FH, Mustafa Adhami V, Suh Y, Mukhtar H. Dual inhibition of phosphatidylinositol 3-kinase/Akt and mammalian target of rapamycin signaling in human nonsmall cell lung cancer cells by a dietary flavonoid fisetin. Int J Cancer Res. 2012;130(7):1695–1705. doi:10.1002/ijc.26178
3. Miyake N, Chikumi H, Takata M, Nakamoto M, Igishi T, Shimizu E. Rapamycin induces p53-independent apoptosis through the mitochondrial pathway in non-small cell lung cancer cells. Oncol Rep. 2012;28(3):848–854. doi:10.3892/or.2012.1855
4. Cho H, Lai TC, Tomoda K, Kwon GS. Polymeric micelles for multi-drug delivery in cancer. Aaps Pharmscitech. 2015;16(1):10–20. doi:10.1208/s12249-014-0251-3
5. Tomoda K, Tam YT, Cho H, Buehler D, Kozak KR, Kwon GS. Triolimus: a multi‐drug loaded polymeric micelle containing paclitaxel, 17‐AAG, and rapamycin as a novel radiosensitizer. Macromol Biosci. 2017;17(1):1600194. doi:10.1002/mabi.201600194
6. Hasenstein JR, Shin H-C, Kasmerchak K, Buehler D, Kwon GS, Kozak KR. Antitumor activity of triolimus: a novel multidrug-loaded micelle containing paclitaxel, rapamycin, and 17-AAG. Mol Cancer Ther. 2012;11(10):2233–2242. doi:10.1158/1535-7163.MCT-11-0987
7. Koot D, Cromarty D. Anticancer efficacy and toxicokinetics of a novel paclitaxel-clofazimine nanoparticulate co-formulation. Drug Deliv Transl Res. 2015;5(3):257–267. doi:10.1007/s13346-015-0222-6
8. Batist G, Gelmon KA, Chi KN, et al. Safety, pharmacokinetics, and efficacy of CPX-1 liposome injection in patients with advanced solid tumors. Clini Cancer Res. 2009;15(2):692–700. doi:10.1158/1078-0432.CCR-08-0515
9. Villar D, Cray C, Zaias J, Altman NH. Biologic effects of fenbendazole in rats and mice: a review. J Am Assoc Lab Anim Sci. 2007;46(6):8–15.
10. Pritchett KR, Johnston NA. A review of treatments for the eradication of pinworm infections from laboratory rodent colonies. Contemp Top Lab Anim Sci. 2002;41(2):36–46.
11. Dawson PJ, Gutteridge WE, Gull K. A comparison of the interaction of anthelmintic benzimidazoles with tubulin isolated from mammalian tissue and the parasitic nematode Ascaridia galli. Biochem Pharmacol. 1984;33(7):1069–1074. doi:10.1016/0006-2952(84)90515-X
12. Lacey E. The role of the cytoskeletal protein, tubulin, in the mode of action and mechanism of drug resistance to benzimidazoles. Int J Parasitol. 1988;18(7):885–936. doi:10.1016/0020-7519(88)90175-0
13. Hayes RH, Oehme FW, Leipold H. Toxicity investigation of fenbendazole, an anthelmintic of swine. Am J Vet Res. 1983;44(6):1108–1111.
14. Hayes RH, Oehme FW, Leipold H. Safety of fenbendazole in swine. Am J Vet Res. 1983;44(6):1112–1116.
15. Schwartz RD, Donoghue AR, Baggs RB, Clark T, Partington C. Evaluation of the safety of fenbendazole in cats. Am J Vet Res. 2000;61(3):330–332. doi:10.2460/ajvr.2000.61.330
16. Muser RK, Paul JW. Safety of fenbendazole use in cattle. Mod Vet Pract. 1984;65(5):371–374.
17. Chu SW, Badar S, Morris DL, Pourgholami MH. Potent inhibition of tubulin polymerisation and proliferation of paclitaxel-resistant 1A9PTX22 human ovarian cancer cells by albendazole. Anticancer Res. 2009;29(10):3791–3796.
18. Dogra N, Kumar A, Mukhopadhyay T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci Rep. 2018;8(1):11926. doi:10.1038/s41598-018-30158-6
19. Aycock-Williams A, Pham L, Liang M, et al. Effects of fenbendazole and vitamin E succinate on the growth and survival of prostate cancer cells. J Cancer Res Exp Oncol. 2011;3:115–21.
20. Han Y, Joo H-G. Involvement of reactive oxygen species in the anti-cancer activity of fenbendazole, a benzimidazole anthelmintic. Korean J Vet Res. 2020;60(2):79–83. doi:10.14405/kjvr.2020.60.2.79
21. Dogra N, Mukhopadhyay T. Impairment of the ubiquitin-proteasome pathway by methyl N-(6-phenylsulfanyl-1H-benzimidazol-2-yl)carbamate leads to a potent cytotoxic effect in tumor cells: a novel antiproliferative agent with a potential therapeutic implication. J Biol Chem. 2012;287(36):30625–30640. doi:10.1074/jbc.M111.324228
22. Melian ME, Paredes A, Munguía B, et al. Nanocrystals of novel valerolactam-fenbendazole hybrid with improved in vitro dissolution performance. AAPS PharmSciTech. 2020;21(7):1–15. doi:10.1208/s12249-020-01777-y
23. Jin IS, Jo MJ, Park C-W, Chung YB, Kim J-S, Shin DH. Physicochemical, pharmacokinetic, and toxicity evaluation of soluplus® polymeric micelles encapsulating fenbendazole. Pharmaceutics. 2020;12(10):1000. doi:10.3390/pharmaceutics12101000
24. Vezina C, Kudelski A, Sehgal S. Rapamycin (AY-22, 989), a new antifungal antibiotic I. taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 1975;28(10):721–726. doi:10.7164/antibiotics.28.721
25. Sehgal S. Sirolimus: its discovery, biological properties, and mechanism of action.
26. Kahan BD, Gibbons S, Tejpal N, Stepkowski SM, Chou T-C. Synergistic interactions of cyclosporine and rapamycin to inhibit immune performances of normal human peripheral blood lymphocytes in vitro. Transplantation. 1991;51(1):232–239. doi:10.1097/00007890-199101000-00038
27. Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol. 1996;14(1):483–510. doi:10.1146/annurev.immunol.14.1.483
28. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729–734. doi:10.1038/nrc1974
29. Gulhati P, Cai Q, Li J, et al. Targeted inhibition of mammalian target of rapamycin signaling inhibits tumorigenesis of colorectal cancer. Clin Cancer Res. 2009;15(23):7207–7216. doi:10.1158/1078-0432.CCR-09-1249
30. Eng C, Sehgal S, Vézina C. Activity of rapamycin (AY-22, 989) against transplanted tumors. J Antibiot (Tokyo). 1984;37(10):1231–1237. doi:10.7164/antibiotics.37.1231
31. Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13(4):136–142. doi:10.1016/0167-5699(92)90111-J
32. Noh W-C, Mondesire WH, Peng J, et al. Determinants of rapamycin sensitivity in breast cancer cells. Clini Cancer Res. 2004;10(3):1013–1023. doi:10.1158/1078-0432.CCR-03-0043
33. Marinov M, Fischer B, Arcaro A. Targeting mTOR signaling in lung cancer. Crit Rev Oncol Hematol. 2007;63(2):172–182. doi:10.1016/j.critrevonc.2007.04.002
34. Dumont FJ, Su Q. Mechanism of action of the immunosuppressant rapamycin. Life Sci. 1995;58(5):373–395. doi:10.1016/0024-3205(95)02233-3
35. Wanner K, Hipp S, Oelsner M, et al. Mammalian target of rapamycin inhibition induces cell cycle arrest in diffuse large B cell lymphoma (DLBCL) cells and sensitises DLBCL cells to rituximab. Br J Haematol. 2006;134(5):475–484. doi:10.1111/j.1365-2141.2006.06210.x
36. Raught B, Gingras A-C, Sonenberg N. The target of rapamycin (TOR) proteins. Proc Natl Acad Sci U S A. 2001;98(13):7037–7044. doi:10.1073/pnas.121145898
37. Shafer A, Zhou C, Gehrig PA, Boggess JF, Bae‐Jump VL. Rapamycin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and induction of apoptosis. Int J Cancer Res. 2010;126(5):1144–1154. doi:10.1002/ijc.24837
38. Marimpietri D, Brignole C, Nico B, et al. Combined therapeutic effects of vinblastine and rapamycin on human neuroblastoma growth, apoptosis, and angiogenesis. Clini Cancer Res. 2007;13(13):3977–3988. doi:10.1158/1078-0432.CCR-06-2757
39. Zhou Q, Lui VWY, Lau CPY, et al. Sustained antitumor activity by co-targeting mTOR and the microtubule with temsirolimus/vinblastine combination in hepatocellular carcinoma. Biochem Pharmacol. 2012;83(9):1146–1158. doi:10.1016/j.bcp.2012.01.013
40. Simamora P, Alvarez JM, Yalkowsky SH. Solubilization of rapamycin. Int J Pharm. 2001;213(1–2):25–29. doi:10.1016/S0378-5173(00)00617-7
41. Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid− polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2(8):1696–1702. doi:10.1021/nn800275r
42. Shin DH, Tam YT, Kwon GS. Polymeric micelle nanocarriers in cancer research. Front Chem Sci Eng. 2016;10(3):348–359. doi:10.1007/s11705-016-1582-2
43. Kazunori K, Masayuki Y, Teruo O, Yasuhisa S, Yasuhisa S. Block copolymer micelles as vehicles for drug delivery. J Control Release. 1993;24(1–3):119–132. doi:10.1016/0168-3659(93)90172-2
44. Manjili HK, Ghasemi P, Malvandi H, Mousavi MS, Attari E, Danafar H. Pharmacokinetics and in vivo delivery of curcumin by copolymeric mPEG-PCL micelles. Eur J Pharm Biopharm. 2017;116:17–30. doi:10.1016/j.ejpb.2016.10.003
45. Sezgin Z, Yüksel N, Baykara T. Preparation and characterization of polymeric micelles for solubilization of poorly soluble anticancer drugs. Eur J Pharm Biopharm. 2006;64(3):261–268. doi:10.1016/j.ejpb.2006.06.003
46. Jo MJ, Jo YH, Lee YJ, et al. Physicochemical, pharmacokinetic, and toxicity evaluation of methoxy poly(ethylene glycol)-b-poly(d,l-lactide) polymeric micelles encapsulating alpinumisoflavone extracted from unripe cudrania tricuspidata fruit. Pharmaceutics. 2019;11(8):366. doi:10.3390/pharmaceutics11080366
47. Kwon GS, Okano T. Polymeric micelles as new drug carriers. Adv Drug Deliv Rev. 1996;21(2):107–116. doi:10.1016/S0169-409X(96)00401-2
48. Zamani M, Shirinzadeh A, Aghajanzadeh M, Andalib S, Danafar H. In vivo study of mPEG–PCL as a nanocarriers for anti-inflammatory drug delivery of simvastatin. Pharm Dev Technol. 2019;24(6):663–670. doi:10.1080/10837450.2018.1556689
49. Jo MJ, Lee YJ, Park C-W, et al. Evaluation of the physicochemical properties, pharmacokinetics, and in vitro anticancer effects of docetaxel and osthol encapsulated in methoxy poly (ethylene glycol)-b-poly (caprolactone) polymeric micelles. Int J Mol Sci. 2021;22(1):231. doi:10.3390/ijms22010231
50. Wei X, Gong C, Gou M, et al. Biodegradable poly (ε-caprolactone)–poly (ethylene glycol) copolymers as drug delivery system. Int J Pharm. 2009;381(1):1–18. doi:10.1016/j.ijpharm.2009.07.033
51. Danafar H, Sharafi A, Kheiri Manjili H, Andalib S. Sulforaphane delivery using mPEG–PCL co-polymer nanoparticles to breast cancer cells. Pharm Dev Technol. 2017;22(5):642–651. doi:10.3109/10837450.2016.1146296
52. Doddapaneni BS, Al-Fatease AM, Rao DA, Alani AW. Dual-drug loaded micelle for combinatorial therapy targeting HIF and mTOR signaling pathways for ovarian cancer treatment. J Control Release. 2019;307:272–281. doi:10.1016/j.jconrel.2019.06.036
53. Croy S, Kwon G. Polymeric micelles for drug delivery. Curr Pharm Des. 2006;12(36):4669–4684. doi:10.2174/138161206779026245
54. Dian L, Yu E, Chen X, et al. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res Lett. 2014;9(1):1–11. doi:10.1186/1556-276X-9-684
55. Zhao J, Xu Y, Wang C, et al. Soluplus/TPGS mixed micelles for dioscin delivery in cancer therapy. Drug Dev Ind Pharm. 2017;43(7):1197–1204. doi:10.1080/03639045.2017.1304956
56. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. doi:10.1158/0008-5472.CAN-09-1947
57. Das UK, Bordoloi R, Ganguly S. Freeze-drying technique and its wide application in biomedical and pharmaceutical sciences. Res J Chem Environ Sci. 2014;2(3):01–04.
58. Shukla S. Freeze drying process: a review. Int J Pharm Sci Res. 2011;2(12):3061.
59. Ma L, Geng H, Song J, Li J, Chen G, Li Q. Hierarchical self-assembly of polyhedral oligomeric silsesquioxane end-capped stimuli-responsive polymer: from single micelle to complex micelle. J Phys Chem B. 2011;115(36):10586–10591. doi:10.1021/jp203782g
60. Cui H, Chen Z, Wooley KL, Pochan DJ. Origins of toroidal micelle formation through charged triblock copolymer self-assembly. Soft Matter. 2009;5(6):1269–1278. doi:10.1039/B811619A
61. D’Souza S, Zhang H, Dou L. A review of in vitro drug release test methods for nano-sized dosage forms. Adv Pharm. 2014;6(2):2014. doi:10.3390/pharmaceutics6020298
62. Aslantürk ÖS. In vitro Cytotoxicity and Cell Viability Assays: Principles, Advantages, and Disadvantages. Vol. 2. InTech; 2018.
63. Twentyman PR, Luscombe M. A study of some variables in a tetrazolium dye (MTT) based assay for cell growth and chemosensitivity. Br J Cancer. 1987;56(3):279–285. doi:10.1038/bjc.1987.190
64. Franken NA, Rodermond HM, Stap J, Haveman J, Van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–2319. doi:10.1038/nprot.2006.339
65. Berger D, Henss H, Winterhalter B, Fiebig H. The clonogenic assay with human tumor xenografts: evaluation, predictive value and application for drug screening. Ann Oncol. 1990;1(5):333–341. doi:10.1093/oxfordjournals.annonc.a057770
66. Xia B, Heimbach T, He H, Lin T. Nilotinib preclinical pharmacokinetics and practical application toward clinical projections of oral absorption and systemic availability. Biopharm Drug Dispos. 2012;33(9):536–549. doi:10.1002/bdd.1821
67. Zhang RX, Wong HL, Xue HY, Eoh JY, Wu XY. Nanomedicine of synergistic drug combinations for cancer therapy–strategies and perspectives. J Control Release. 2016;240:489–503. doi:10.1016/j.jconrel.2016.06.012
68. Petersen MB, Friis C. Pharmacokinetics of fenbendazole following intravenous and oral administration to pigs. Am J Vet Res. 2000;61(5):573–576. doi:10.2460/ajvr.2000.61.573
69. MacDonald A, Scarola J, Burke JT, Zimmerman JJ. Clinical pharmacokinetics and therapeutic drug monitoring of sirolimus. Clin Ther. 2000;22:B101–B121. doi:10.1016/S0149-2918(00)89027-X
70. Aw MS, Kurian M, Losic D. Polymeric micelles for multidrug delivery and combination therapy. Chem Eur J. 2013;19(38):12586–12601. doi:10.1002/chem.201302097
71. Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Del Rev. 2011;63(3):131–135. doi:10.1016/j.addr.2010.03.011
72. Woo HN, Chung HK, Ju EJ, et al. Preclinical evaluation of injectable sirolimus formulated with polymeric nanoparticle for cancer therapy. Int J Nanomedicine. 2012;7:2197. doi:10.2147/IJN.S29480
73. Zeng X, Zhang Y, Nyström AM. Endocytic uptake and intracellular trafficking of bis-MPA-based hyperbranched copolymer micelles in breast cancer cells. Biomacromolecules. 2012;13(11):3814–3822. doi:10.1021/bm301281k
74. Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. doi:10.1016/j.jconrel.2010.01.036
75. Manjili HK, Malvandi H, Mousavi MS, Attari E, Danafar H. In vitro and in vivo delivery of artemisinin loaded PCL–PEG–PCL micelles and its pharmacokinetic study. Artif Cells Nanomed Biotechnol. 2018;46(5):926–936. doi:10.1080/21691401.2017.1347880
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.