Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 4
Optimal management of interstitial lung disease associated with dermatomyositis/polymyositis: lessons from the Japanese experience
Authors Kurasawa K, Arai S
Received 30 May 2014
Accepted for publication 30 July 2014
Published 21 November 2014 Volume 2014:4 Pages 93—107
DOI https://doi.org/10.2147/ODRR.S50355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Lise Aagaard
Kazuhiro Kurasawa,1,2 Satoko Arai2
1Center of Rheumatic Diseases, Dokkyo Medical University, Mibu, Tochigi, Japan; 2Department of Clinical Immunology, Dokkyo Medical University, Mibu, Tochigi, Japan
Abstract: Interstitial lung disease (ILD) is a serious complication in dermatomyositis (DM) and polymyositis (PM). In Japan, patients with DM/PM develop acute life-threatening ILD with high frequency. Physicians in Japan have shown the following: refractory acute/subacute (A/S)-ILD is not a rare complication in DM and amyopathic DM (ADM); anti-anti-melanoma differentiation-associated gene 5 (anti-MDA5) antibody (Ab) is closely related to A/S-ILD with poor outcomes in DM/ADM; and poor prognostic factors in A/S-ILD in DM/PM are ADM, DM with low creatine kinase elevation, positivity for anti-MDA5 Ab, serum ferritin elevation, and consolidation with ground-glass opacities on high-resolution computed tomography. There are subtypes in DM/PM-ILD: refractory DM/ADM A/S-ILD positive for anti-MDA5 Ab with poor prognosis; DM A/S-ILD with glucocorticoid (GC) resistance; PM A/S-ILD with GC sensitivity; chronic ILD positive for anti-aminoacyl-tRNA-synthetases (anti-ARS) Abs with GC responsiveness; and C-ILD negative for anti-ARS Abs. For patients with A/S-ILD with poor prognosis, initial combination therapy with cyclosporine and cyclophosphamide in addition to GC has been developed, which rescues 50%–80% of the patients, although elucidation of the efficacy of the combination therapy is required. A/S-ILD with potentially fatal outcomes is found worldwide, not only in Japan. Clinicians caring for patients with DM/PM should be cautious when dealing with A/S-ILD and treat the patients based on clinical subtypes.
Keywords: interstitial lung disease, dermatomyositis, polymyositis, management, cyclosporine, cyclophosphamide
Introduction
Dermatomyositis (DM) and polymyositis (PM) are systemic autoimmune diseases characterized by skeletal muscle inflammation and skin lesions.1,2 DM/PM affects many organs other than muscle and skin. The lung is one of the most commonly affected organs in DM/PM. Interstitial lung disease (ILD) is the most frequent pulmonary complication of DM/PM and determines the prognosis of patients with myositis.3–5 The clinical features of DM/PM vary widely. ILD is often asymptomatic and remains unchanged or progresses slowly, while some patients develop acute-onset, life-threatening ILD. Responses of ILD to immunosuppressive therapies also vary; some patients are improved by glucocorticoid (GC) therapy alone, but there are cases in which interstitial pneumonia deteriorates despite aggressive immunosuppressive therapy. Controlling ILD is a key issue in the management of DM/PM. Unfortunately, we have had few controlled trials showing which strategies/therapies are effective for the management of ILD in DM/PM. This review focuses on prognostic factors of ILD in DM/PM and the management of ILD based on experience with ILD associated with DM/PM (DM/PM-ILD) in Japan, where acute life-threatening ILD is frequently seen.
DM, PM, and amyopathic DM
Inflammatory myopathy includes three diseases: DM, PM, and inclusion body myositis. DM is characterized by myositis and skin lesions such as a heliotropic (purple) rash and Gottron’s papules, whereas PM shows myositis but no skin inflammation. Amyopathic DM (ADM) is a subtype of DM characterized by hallmark biopsy-confirmed cutaneous manifestations of classic DM occurring for 6 months or longer with no clinical evidence of proximal muscle weakness and no serum muscle enzyme abnormalities.6,7 Hypomyopathic DM differs from ADM in that, although patients do not exhibit any clinical evidence of muscle weakness, they may have subclinical evidence of muscle involvement on laboratory, electrophysiologic, and/or radiographic evaluation. Clinically amyopathic DM (CADM) encompasses both the amyopathic and hypomyopathic DM groups.
ILD associated with DM/PM
Epidemiology
ILD associated with DM/PM was first reported by Mills and Mathews.8 It used to be thought that ILD was not a common complication in DM/PM. However, it has become evident that ILD is common in DM/PM. The prevalence of ILD in DM/PM varies between 20%–65%, depending on detection methods. Using conventional radiographic examinations, the prevalence of lung diseases was 5%–9% in patients with DM/PM.9,10 High-resolution computed tomography (HRCT) examinations revealed that ILD was detected in 32%–78% of patients, including asymptomatic patients.11–13 Connors et al3 reviewed reports on the prevalence of ILD in DM/PM from 2002–2009 and reported that the prevalence was 37.4% overall (range, 21%–74%).
The frequency of ILD varies among subtypes of DM/PM. The frequency is high or similar in DM compared to PM.13,14 The prevalence of ILD is particularly high in patients with ADM. Moreover, patients positive for autoantibodies for anti-melanoma differentiation-associated gene 5 (MDA5) antibody (Ab)15 or anti-aminoacyl- tRNA-synthetases (ARS) Ab have a high incidence of ILD.16 In contrast, ILD is less common in patients with DM/PM associated with malignancies.17
Clinical features
Onset and progression
ILD has many clinical features. The onset of ILD is variable. The majority of patients are diagnosed as having ILD at the same time or after DM/PM is diagnosed. However, in 10%–30% of cases, ILD precedes muscle or skin manifestations.13,18–21 Particularly in patients positive for anti-ARS Abs, it is not uncommon that ILD precedes myositis or exists without muscle symptoms, and these patients are diagnosed as having idiopathic interstitial pneumonia (IIP).
As for the onset and progression of ILD, two types of ILD are identified: acute/subacute (A/S-ILD), sometimes termed rapidly progressive interstitial pneumonia; and chronic/asymptomatic ILD (C-ILD).19,20,22 Some investigators have classified three types of ILD: acute lung disease; lung disease with a chronic progressive course; and asymptomatic ILD with abnormalities evident on pulmonary function tests and HRCT scans.21 In this review, A/S-ILD and C-ILD are used.
A/S-ILD is rapidly progressive and can lead to respiratory failure requiring mechanical ventilation; some cases may be part of Hamman-Rich syndrome. Usually, A/S-ILD is defined as rapidly progressive ILD within 3 months from the onset of symptoms; progression of ILD is defined by 1) symptomatic exacerbation (dyspnea on exertion), 2) an increase in parenchymal abnormalities on HRCT scan, and 3) physiological change including a 10% decrease in vital capacity or a 1.33 kPa decrease in arterial oxygen tension (PaO2), referring to the international consensus statement on idiopathic pulmonary fibrosis of the American Thoracic Society and the European Respiratory Society.23,24 C-ILD is an asymptomatic nonrapidly progressive ILD or slowly progressive ILD over 3 months.
The prevalence of A/S-ILD is variable. Reports from Japan showed that 20%–34% of DM patients developed A/S-ILD, including 30%–40% DM-ILD, and 18% of PM patients developed A/S-ILD,14,15,25 while a recent analysis in our institute showed that 83% of DM/PM-ILD (92% of DM-ILD, 72% of PM-ILD) was A/S-ILD.26 A/S-ILD occurs with a higher frequency in DM, particularly in ADM, compared to PM.24,27,28 In countries other than Japan, the prevalence of A/S-ILD in DM/PM-ILD has been reported to be 17%–33%; the prevalence of asymptomatic ILD was up to 30%, and the majority of DM/PM with ILD has been considered to be chronic, slowly progressive.20–22 However, A/S-ILD is not uncommon in Japan.
Autoantibodies
Recent studies have shown a strong association between autoantibodies and clinical subsets of DM/PM.4,29 Some autoantibodies are positively associated with ILD in DM/PM, and other autoantibodies are negatively correlated with ILD. These autoantibody profiles provide important information about the clinical features, response to therapy, and prognosis. Anti-MDA5 Ab (another name for anti-CADM-140 Ab) and anti-ARS Ab are strongly associated with ILD in DM/PM.
Anti-MDA5 Ab (anti-CADM-140 Ab)
Sato et al30 found a new autoantibody that reacts with a 140 kD protein by immunoprecipitation and immunoblotting. This antibody was only found in sera from ADM and was named anti-CADM-140 Ab. Importantly, anti-CADM-140 Ab was strongly associated with A/S-ILD; 50% of patients with the antibody developed A/S-ILD. Later, MDA5 was shown to be an antigen recognized by anti-CADM-140 Ab, and the antibody was called anti-MDA5 Ab.31,32 MDA, also known as IFIH1, is one of the retinoic acid-inducible gene-I-like receptors, intracellular sensors of viral RNAs, and its expression is upregulated by interferon (IFN)-β, which might suggest an association of A/S-ILD with anti-MDA5 Ab and virus infection.
Further studies in Japan have shown that anti-MDA5 Ab is detected exclusively in patients with DM and ADM. The antibody was detected in 9%–26% of cases of DM with muscle symptoms and in 50%–85% of ADM cases.25,32–34
Importantly, there is a strong association of anti-MDA5 Ab and A/S-ILD with a poor prognosis. Nakashima et al32 showed that patients with the antibody showed a typical DM rash (100% of patients), fever (69%), ILD (92%), and A/S-ILD (54%). Despite intensive treatment, six of 12 patients with ILD died of respiratory failure within 6 months. Gono et al15 reported that 71% of ILD cases positive for anti-MDA5 Ab were A/S-ILD, 57% of DM-ILD cases with the Ab were ADM, and 36% of the patients died of respiratory failure. Koga et al25 also reported similar results; 94% and 71% of patients positive for anti-MDA5 Ab developed ILD and A/S-ILD, respectively, and 41% of them died of respiratory failure.
In countries other than Japan, Fiorentino et al35 reported that anti-MDA5 Ab was detected in 13% of DM cases in the USA, and 50% of patients with the antibody had ADM. They also reported that these patients had a characteristic cutaneous phenotype consisting of skin ulceration, tender palmar papules, or both, and that 25% of them had ILD, while A/S-ILD developed in only 7%. Labrador-Horrillo et al36 reported that anti-MDA5 Ab was detected in 11% of DM cases including ADM and 54% of ADM cases in the Mediterranean area; 57% of patients with the antibody developed A/S-ILD, and patients with the antibody had a poor prognosis. Reports from China showed that anti-MDA5 Ab was positive in 9%–50% of DM cases with muscle symptoms, 38%–100% of ADM cases, and 0% of PM cases; ILD and A/S-ILD were found in 100% and 38%–60% of patients positive for the antibody, respectively.37,38 Chen et al38 conducted a meta-analysis of the clinical features of anti-MDA5 Ab-positive patients and showed a higher frequency of ADM in Japanese than in non-Japanese patients, and that existence of the antibody is a risk factor for A/S-ILD (sensitivity 77%, specificity 86%). In contrast, Hall et al39 in the USA reported that most patients with anti-MDA5 Ab showed arthritis, a clinical myopathy, and ILD, which typically resolved with treatment; the MDA5 phenotype was a clinical mimic of the antisynthetase syndrome and was not associated with rapidly progressive ILD.
Taken together, these reports indicate that anti-MDA5 Ab is found exclusively in DM, especially in ADM, and has a strong association with ILD worldwide. In East Asia, particularly in Japan, the antibody is strongly associated with A/S-ILD with a poor prognosis and severe cutaneous lesions, but the certainty of this association is controversial in Western countries. However, it is certain that there is an association between anti-MDA5 Ab and A/S-ILD in some populations with DM/PM.
It has been shown that increased ferritin levels are strongly associated with the severity and activity of A/C-ILD in DM.40 Moreover, patients with anti-MDA5 Ab frequently show high ferritin levels,15,32 and the serum ferritin level correlates with a poor prognosis in A/S-ILD with anti-MDA5 Ab.15,25 Serum ferritin levels have been reported to also be correlated with interleukin-18 and IFN-α levels.41,42 Moreover, anti-MDA Ab levels are correlated with ILD activity and serum ferritin levels.41 In A/S-ILD with anti-MDA5 Ab, immunological mechanisms inducing macrophage activation resulting in increased ferritin levels and type 1 IFN and inflammatory cytokine production might play important roles in the development of the disease.
Anti-ARS Abs
Anti-ARS Abs are other autoantibodies strongly associated with ILD in DM/PM. Eight antibodies specific for different synthetases have been identified: anti-Jo-1 (histidyl), anti-PL-7, (threonyl), anti-PL-12 (alanyl),20 anti-EJ (glycyl), anti-OJ (isoleucyl), anti-KS (asparaginyl), anti-Zo (phenylalanyl), and anti- HA (tyrosyl) antibodies.43 Approximately 30%–40% of patients with myositis, regardless of their ILD status, are positive for one of the anti-ARS Abs.44 Anti-Jo-1 Ab is the most common and is found in approximately 20% of patients with myositis. The frequency of other non-Jo-1 Abs is between 1%–5%.4,45 Patients with anti-ARS Abs, despite the specificity of the Ab, show similar clinical features (ILD, myositis, arthritis, fever, Raynaud’s phenomenon, and mechanic’s hands), and this condition is termed “anti-synthetase syndrome.”4,46 ILD is the most common organ involvement in patients with anti-ARS Abs; more than 75% of patients with anti-ARS Abs develop ILD.4,47 Notably, 20%–30% of patients with anti-ARS Abs have ILD but no signs or symptoms of DM/PM, including myositis and skin lesions, or their ILD precedes the myositis.48 These patients are indistinguishable from IIP in clinical features, and the majority of them have a non-Jo-1 Ab.49 In fact, Fischer et al50 reported that 24% of IIP patients without Jo-1 Ab and antinuclear antibodies have non-Jo-1 Abs.
The majority of ILD cases positive for anti-ARS Abs develop gradually, but some show acute onset. In contrast to MDA5 Ab, ARS Ab positive-ILD generally responds well to GC, although a number of cases show recurrence after withdrawing or reducing doses of GC.19,51,52
The clinical features of ILD positive for anti-ARS Abs in Japan are similar to those reported in other countries. Yoshifuji et al52 analyzed 74 DM/PM cases and found that 28% were positive for anti-ARS Abs, 95% of the patients with antibodies had ILD, and ILD with anti-ARS Abs preceded myositis in 50% of cases. Moreover, they showed that ILD positive for anti-ARS Abs responded well to GC compared to ILD negative for the Abs, and only one patient with the Ab died of respiratory failure, although recurrences of ILD occurred in 40% of patients with the Abs.
Taken together, patients positive for ARS Abs have similar clinical features worldwide: chronic progression, good response to GC, and a fair prognosis.
Prognosis of ILD in DM/PM and prognostic factors
ILD determines the prognosis of DM/PM patients. Thus, controlling ILD is the key issue to avoid poor outcomes and to maintain patients’ quality of life. As described above, ILD in DM/PM has large heterogeneity. For ILD with a good response to standard therapy, intensive immunosuppressive therapy might hurt rather than benefit patients. In contrast, in patients with ILD refractory to standard therapy, standard therapy or delay of intensive therapy might cause poor outcomes, including death. Identification of patients with poor prognostic factors and selection of proper therapy are required in the management of ILD in DM/PM.
It is notable that the prognosis, the survival rate of patients, and the prognostic factors are changing with the introduction of new therapies and therapeutic strategies. For example, we introduced combination therapy with GC and cyclosporine (CsA) for GC-resistant A/S-ILD in the late 1990s, which dramatically improved the prognosis (Figure1). There remained, however, patients who did not respond to the combination therapy. Poor prognostic factors in the era of GC therapy (factors predicting GC resistance) are not the same as those in the era of combination therapy (factors predicting unresponsiveness to the combination therapy). When interpreting reports on prognosis and prognostic factors, it is important to pay attention to which therapies were used and the times when the data were collected.
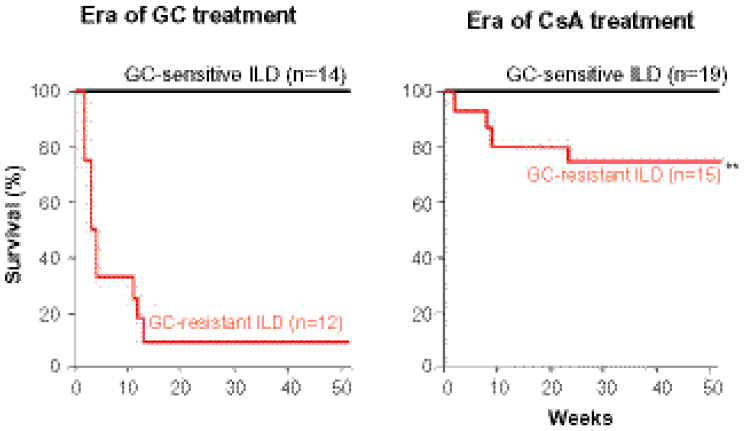 | Figure 1 Cyclosporine therapy improved prognosis of glucocorticoid-resistant ILD associated with DM/PM. |
In addition, of course, it is important to know which subjects are enrolled for analysis. As described in the above section, the predominant clinical features of ILD in DM/PM differ from one country to another. In Japan, A/S-ILD is predominant and includes many cases positive for anti-MDA5 Ab. Therefore, studies on prognosis and prognostic factors in Japan focus mainly on short-term outcomes of A/S-ILD.
Impact of ILD on the prognosis of DM/PM
In Japan, ILD is a major cause of death in DM/PM. Nawata et al14 showed that A/S-ILD is found in approximately 40% of DM/PM cases, and 42% of A/S-ILD patients died within 6 months in the era of GC therapy. Kameda et al53 reported that 75% of A-S/ILD cases died of respiratory failure within 3 months. Koga et al25 reported that 21% of DM patients had A/S-ILD, and that 62% of DM A/S-ILD cases died of respiratory failure.
Consistent with studies from Japan, ILD has been shown to be a factor associated with poor outcomes worldwide. Kang et al22 reported that ILD was observed in 40.3% of Korean DM/PM patients and was associated with poor survival. Won Huh et al20 in China demonstrated that ILD, particularly A/S-ILD, is a poor prognostic factor in DM/PM. Marie et al19 in France showed that the survival rate of patients with ILD was lower than that of those without ILD in the long-term when cancer-associated patients were excluded from the analysis, suggesting that the impact of ILD on prognosis might be larger in East Asian people than in Caucasian populations.
Predictive factors
The prognostic factors in DM/PM-ILD are summarized in Figure 2.
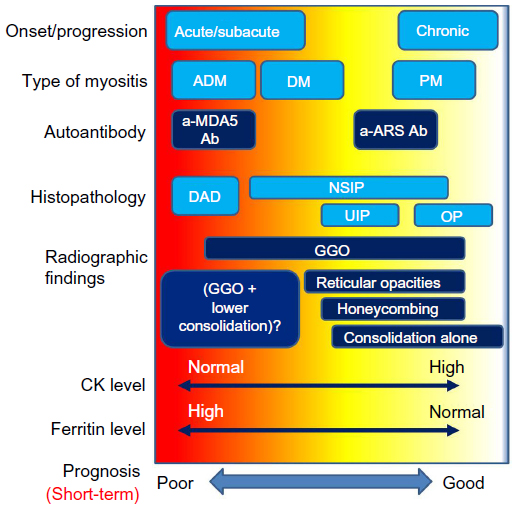 | Figure 2 Prognostic factors (short-term) in ILD associated with DM/PM. |
DM, CADM (DM with low creatine kinase elevation) is a poor prognostic factor
It has been reported that DM-ILD, particularly ADM-ILD, has a poor prognosis compared to PM-ILD. Nawata et al14 reported that the survival rate of ILD (most cases were A/S-ILD) in DM (including ADM) was significantly poorer than that in PM in the era when GC alone was used (6-month survival: DM 50% versus PM 91%). Similarly, Fujisawa et al27 reported that DM-ILD was refractory to GC, resulting in a poorer prognosis compared with PM-ILD (2-year survival: PM-ILD 87% versus DM-ILD 56%). Hayashi et al13 also showed that PM-ILD responded well to GC, but 38% of patients with DM-ILD did not and died despite additional intensive immunosuppressive therapy. The poor prognosis of DM-ILD compared to PM-ILD was supported by other studies from Japan.53,54 However, DM-ILD did not show a poor prognosis in Europe.55,56 The difference might be caused by exclusion or inclusion of ADM closely related to fatal ILD.
The prognosis of CADM, DM with no/low creatine kinase (CK) elevation, is much poorer than that of DM with CK elevation (classical DM). Nawata et al14 reported that 82% of DM-ILD without CK elevation was GC-resistant, while only 22% of DM cases with CK elevation and 0% of PM cases were refractory to GC. Mukae et al28 reported that the mortality rate was significantly higher for patients with CADM-ILD (45%) than for patients with classic DM-ILD (6%). Yamasaki et al54 concluded through analysis of 197 DM/PM cases that survival was lower in CADM and DM cases, mainly due to fatal ILD, than in PM cases. Supporting these findings, reports from China showed that ADM-ILD was a poor prognostic factor in DM/PM-ILD.38,57,58 The poor prognosis of DM, particularly of ADM, is associated with high frequencies of anti-MDA5 Ab in DM, especially ADM; the antibody is closely related to fatal, rapidly progressive ILD.15,25,30,32
A/S-ILD is a poor prognostic factor
ILD with acute presentation/ rapid progression is a poor prognostic factor that has been reported worldwide.19,20,22 In Japan, Gono et al40 reported that only DM patients with A/S-ILD died within 6 months after starting therapy, but not those without (6-month survival: DM A/S-ILD (+) 62.7% versus DM A/S-ILD (-) 100%). We observed similar results (1-year survival rate: DM A/S-ILD 79%, PM A/S-ILD 100%, DM/PM C-ILD 100%, DM/PM-ILD (-) 95%).26 Suda et al24 and Mukae et al28 independently reported that, among ADM cases, a poor outcome was associated with A/S-ILD, but not with C-ILD. Moreover, among cases positive for anti-MDA5 Ab, A/S-ILD but not C-ILD was associated with a poor prognosis.15,25,34 These findings indicate that the presence of A/S-ILD is a poor prognostic factor.
Anti-MDA5 Ab is a poor prognostic factor, but ARS Ab is not
Since the discovery of anti-MDA5 Ab, a strong relationship between the antibody and A/S-ILD with a poor prognosis has been shown and confirmed by many investigators worldwide. In Japan, 50%–71% of patients with anti-MDA5 Ab developed A/S-ILD, and their mortality rate was 36%–46%, which was significantly higher than that of those without the Ab.15,25,32 Studies done in countries other than Japan also demonstrated a high prevalence of A/S-ILD (22%–60%) and shorter survival time in anti-MDA5 Ab-positive patients, although the frequencies of A/S-ILD with the Ab in Western countries seemed to be low compared to Japan.35–38
It is notable that anti-MDA5 Ab-positive ILD is not always A/S-ILD; there are cases of C-ILD. Importantly, A/S-ILD but not C-ILD is associated with a poor prognosis.15,25,34 In addition, titers of anti-MDA Ab are correlated with the activity and severity of the disease, and successful therapy reduces Ab titers, suggesting that anti-MDA5 Ab is not only a prognostic factor, but also a marker of disease activity.37,40
In contrast to anti-MDA5 Ab, anti-ARS Ab is associated with C-ILD rather than A/S-ILD, has fewer negative effects on long-term survival,16,19 and is associated with a good response to GC compared to anti-ARS Ab (-) cases.18,19,51,52 Moreover, among patients positive for anti-ARS Abs, no difference was found in the survival rate between A/S-ILD and C-ILD with proper immunosuppressive therapy.59
In addition, recurrence of ILD is frequent in ILD positive for anti-ARS Abs,52 but rarely in anti-MDA5 Ab-positive patients who survive A/S-ILD.25
Taken together, A/S-ILD, but not C-ILD, positive for anti-MDA5 Ab, indicates fatal ILD, while both A/S-ILD and C-ILD with anti-ARS Abs respond well to immunosuppressive therapy and could have a good prognosis.
Histopathology and prognosis
Nonspecific interstitial pneumonia (NSIP) is a dominant histopathologic pattern in DM/PM-ILD, with a prevalence of approximately 80%, although some have diffuse alveolar damage (DAD), usual interstitial pneumonia (UIP), and organizing pneumonia (OP).3,18 The long-term prognosis of DM/PM-ILD has been reported to be similar to that of idiopathic NSIP.3 DAD has been reported in association with A/S-ILD and a poor prognosis in the short-term,27,60,61 while OP responds well to GC. Suda et al24 reported that DAD was 2/5 and NSIP was 3/5 in A/S-ILD in ADM, while NSIP was 2/3 and UIP was 1/3 in C-ILD in ADM.
Radiographic findings and prognosis
Radiological findings in DM/PM-ILD are similar to those seen in other ILDs. Recently, HRCT scans have been used for evaluation of DM/PM-ILD. HRCT findings of DM/PM-ILD (including A/S-ILD and C-ILD) were irregular linear opacities with lower lung predominance (63%–92%), consolidation (52%–53%), ground-glass opacities (GGOs) (43%–92%), micronodules (28%), and honeycombing (16%).18,62 Ikezoe et al62 reported that HRCT consolidation was associated with DAD or OP,62 but Arakawa et al63 showed that HRCT consolidation was frequently found in biopsy-proven NSIP in DM/PM.
A few studies have been performed on the correlation between radiological findings and prognosis in DM/PM-ILD. Hayashi et al13 categorized DM/PM-ILD into three groups based on HRCT findings: 1) consolidation dominant; 2) GGO/reticular opacity dominant without a chronic fibrosing process; and 3) GGO/reticular opacity dominant with a chronic fibrosing process. They reported that the disease was fatal in 0%, 83%, and 20%, of groups 1, 2, and 3, respectively. We analyzed 49 A/S-ILD cases and found that the existence of alveolar opacities with GGO and reticular opacities indicated a poor prognosis.26 Recently, Tanizawa et al64 reported that the characteristic HRCT findings of anti-MDA5 Ab ILD are lower consolidation or a GGO pattern, a random GGO pattern, and the absence of intralobular reticular opacities. They recently reported that the lower consolidation/GGO pattern (odds ratio =23.1) is an independent predictive factor for 90-day mortality in DM/PM A/S-ILD, as was the presence of anti-MDA5 Ab.65 These findings suggest that lower consolidation with GGO might be a poor prognostic sign in the short-term, although the number of examined cases was small and further studies are required.
Serum ferritin elevation is a poor prognostic factor
Gono et al40 found that serum ferritin levels were significantly elevated in DM A/S-ILD, and that the 6-month survival rate was significantly lower in the group with ferritin levels >1,500 ng/mL (29%) than in the group with ferritin levels <1,500 ng/mL (83%). The association between ferritin elevation and A/S-ILD with a poor prognosis was confirmed by other groups. Moreover, it has become evident that ferritin elevation is associated with positivity of anti-MDA5 Ab,15,25,32 and ferritin levels are correlated with the severity of ILD, anti-MDA5 Ab titers, and interleukin-18 levels.41 Thus, the serum ferritin level could be a predictive factor for prognosis and a biomarker of disease activity.
Increase in KL-6, but not KL-6 levels at one time point, is a poor prognostic factor
KL-6 is a biomarker for the presence and severity of ILD, reflecting damage to the alveolar–blood barrier and fibrotic changes.66 Elevation of serum KL-6 and surfactant protein D levels in interstitial pneumonia in DM/PM including C-ILD has been demonstrated, suggesting that KL-6 is a biomarker of extension of ILD.67 We recently examined whether KL-6 levels predict the prognosis of A/S-ILD and found that initial KL-6 levels were within the normal range in approximately 30% of A/S-ILD cases, and KL-6 levels failed to predict outcomes.26 However, the increase in KL-6 levels during the first 4 weeks after treatment correlated with a poor prognosis; an increase ratio in KL-6 levels >1.70 within the 4 weeks was a poor prognostic factor. Thus, measuring KL-6 levels in A/S-ILD is useful for monitoring ILD activity.
Other prognostic factors
Prognostic factors other than those described above have been reported. Older age, symptomatic ILD, lower values of vital capacity and diffusing capacity for carbon monoxide, a pattern of UIP on HRCT scan and lung biopsy, and neutrophil alveolitis were reported to be poor prognostic factors of DM/PM-ILD including C-ILD.19,21,22 Activation of T cells, particularly CD8 T cells, in bronchoalveolar lavage fluid predicted a poor response to GC in A/S-ILD.68 Cardiac involvement and fever (>38°C) have also been reported to be associated with a poor prognosis in A/S-ILD.32,58
Clinical subtypes of ILD in DM/PM from the clinical perspective
Based on the clinical features and prognosis of DM/PM-ILD as described above, the existence of subgroups is suggested (Table 1). In clinical practice, it is critical to identify patients whose ILD progresses rapidly, is resistant to standard immunosuppressive therapy, and has poor outcomes.
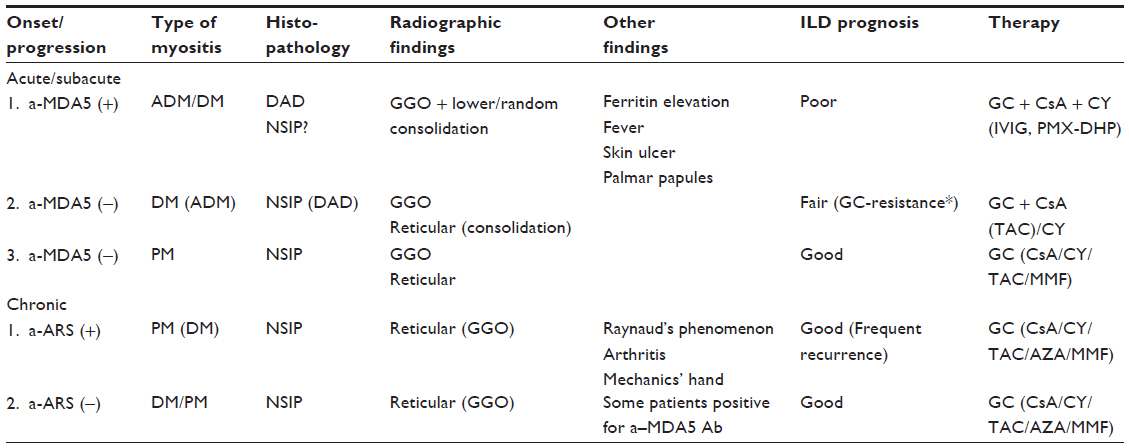 | Table 1 Subtypes of ILD associated with DM/PM |
We propose the subtypes of DM/PM as follows. DM/PM-ILD is divided into two types, A/S-ILD and C-ILD. The former is further subdivided into three subgroups: group 1, anti-MDA5 Ab-positive, characterized by ADM (DM), lower consolidation with GGO, serum ferritin elevation, and refractory ILD; group 2, DM negative for anti-MDA5 Ab; and group 3, PM. C-ILD is further subdivided into two groups: group 1, C-ILD positive for anti-ARS Ab, which is characterized by slowly progressive ILD, good response to GC, and frequent recurrence; and group 2, C-ILD negative for anti-ARS Abs.
Management of DM/PM ILD, particularly A/S-ILD, based on the Japanese experience
As described above, A/S-ILD, particularly with anti-MDA5 Ab, develops frequently in Japan. Before the discovery of anti-MDA5 Ab and serum ferritin elevation in fatal A/S-ILD, CADM (DM)-ILD had been recognized as ILD resistant to GC, and various intensive immunosuppressive therapies had been tried by many investigators. Unfortunately, there have been few controlled or randomized studies on A/S-ILD. However, accumulating small retrospective studies and case reports suggest the effectiveness of intensive immunosuppressive therapy through comparison with historical outcomes in the era of GC, although there remain some cases with no response to this therapy.
We now describe immunosuppressive therapy used for A/S-ILD in Japan, particularly using calcineurin inhibitors and cyclophosphamide (CY) in addition to GC.
Glucocorticoids
GC is the mainstay of therapy for DM/PM including myositis and ILD, although no evidence-based data on efficacy has yet been obtained. Approximately 50% of ILD in DM/PM-ILD (mainly C-ILD) responded well to initial GC therapy.19,69,70
Patients who were young, with PM rather than DM, with OP with an NSIP pattern rather than a UIP pattern on HRCT, or positive for anti-ARS Abs, showed a high response rate to GC alone.16,18,19,52,60
In Japan, Fujiwawa et al27 reported that GC alone was effective in six patients (37.5%) with PM-ILD, but in only one (8.3%) case with DM-ILD. Nawata et al14 reported that GC alone was effective in 68% of A/S-ILD (DM 50%, PM 91%) cases. However, ILD with normal CK levels was resistant to GC alone, and its prognosis was poor compared with ILD with CK elevation (1-year survival: normal CK 31% versus elevated CK 89%). Taken together, GC alone is effective against C-ILD, particularly with PM, but not strong enough to control A/S-ILD, particularly with DM and ADM.
Calcineurin inhibitors
Cyclosporine
Cyclosporine (CsA) is a carnelian inhibitor that is widely used as an immunosuppressant with GC for A/S-ILD in Japan. The effectiveness of CsA in GC-refractory cases was first reported by Gruhn and Diaz-Buxo71 Since then, there have been accumulating case reports and retrospective or open-label studies analyzing the efficacy of CsA in DM/PM-ILD, although no controlled study has been conducted. As shown in Figure 1 from our own experience, CsA dramatically improved the prognosis of GC-resistant ILD, most of which were DM/ADM A/S-ILD. Nagasaka et al72 performed a multicenter, retrospective analysis of 38 cases with DM A/S-ILD. They reported that the 5-year survival rate was 32% in patients receiving GC alone for more than 2 weeks as initial therapy, and nonsurvivors died within 9 months, while the survival rate was 69% in those who received CsA within 2 weeks of starting GC therapy.
The survival rate in response to the combination with CsA and GC ranged from 42%–73% (as we found and show in Figure 1).53,72–74 The difference in survival rates could be caused by many factors, including characteristics of patients (DM/ ADM or PM, kinds of autoantibodies, etc) and therapeutic strategies (initial combination therapy versus step-up therapy).
We would emphasize the importance of maintaining the CsA concentration in the management of A/S-ILD. The calcineurin blocking effect correlates with CsA concentration. A low CsA concentration or delay in achieving proper CsA concentrations would allow ILD to progress and result in poor outcomes. Indeed, we had several cases showing deterioration of ILD in association with low CsA concentrations.14 Kotani et al75 reported that GC–CsA combination therapy keeping the 2-hour postdose blood concentration (C2) >1,000 ng/mL is effective; the 1-year survival rate was 89%. Their group also showed that monitoring trough and C2 levels is important to obtain clinical benefits and avoid adverse effects of CsA, and once daily preprandial administration was beneficial in DM A/S-ILD.76
In our institute, to achieve rapid therapeutic concentrations to control A/S-ILD, CsA is administered intravenously, which is not commonly used for the treatment for ILD in DM/PM. CsA is administered intravenously for 12 hours every day.14 Initially, CsA is given at 200 mg/day, and the doses are adjusted to keep concentrations at 200–250 ng/mL just before CsA administration. When ILD is controlled and respiratory conditions become stable, CsA administration is switched from intravenous to oral.
In addition to A/S-ILD, CsA has been used in patients with C-ILD, including a small number of cases refractory to other therapies, and it has shown beneficial effects.72,77,78 CsA is an effective immunosuppressant for DM/PM-ILD and widely used as an initial agent in combination with GC for DM A/S-ILD in Japan.
Tacrolimus
Tacrolimus (TAC) is another calcineurin inhibitor. The efficacy of TAC for refractory DM/PM-ILD including A/S-ILD and C-ILD has been reported to be as good as CsA.78–81 There was a study showing that acute respiratory distress syndrome secondary to antisynthetase syndrome was reversible with TAC.82 Interestingly, TAC was effective against DM A/S-ILD refractory to combination therapy with GC and CsA.80 In addition, Oddis et al79 and Wilkes et al81 reported that TAC improved pulmonary function, including forced vital capacity and diffusing capacity of the lung for carbon monoxide, and showed a GC-sparing effect in ILD (mainly C-ILD) with anti-ARS Ab.
The effectiveness of calcineurin inhibitors, T cell-specific inhibitors suggests the important role of T cells in the development of ILD in DM/PM. Yamadori et al83 demonstrated CD8 T cell infiltration in DM/PM-ILD using surgical biopsy samples. We previously reported that T cells, particularly Th1 cells, were activated in bronchoalveolar lavage fluid from A/S-ILD, and that CD8 T cell activation was observed in GC-resistant ILD, but not in GC-sensitive ILD.68 However, the existence of ILD refractory to combination therapy with GC and calcineurin inhibitors suggests that important mechanisms other than T cells are involved in ILD in DM/PM. Clarifying these mechanisms could lead to a new therapy for DM/PM-ILD refractory to GC and calcineurin inhibitors.
Cyclophosphamide
Cyclophosphamide, oral or intravenous pulse (IVCY), is commonly used for patients with systemic lupus erythematosus or vasculitis. The effectiveness of CY has been reported in refractory DM/PM-ILD, including A/S-ILD.84,85 Yamasaki et al85 reported that IVCY (300–800 mg/m2 at least six times every 4 weeks) improved symptoms, pulmonary function tests, and HRCT findings in patients with DM/PM, including A/S-ILD. Interestingly, there was a patient who did not respond to IVCY but improved with CsA, suggesting that CY and CsA block different pathways in the development of ILD.
Other agents
Azathioprine
Azathioprine is commonly used as a second-line agent for DM/PM-ILD in Western countries and is often effective in the treatment of NSIP due to DM or PM.18,19 It is not known if azathioprine is effective in patients with A/S-ILD.
Mycophenolate mofetil
Mycophenolate mofetil is an inhibitor of lymphocyte proliferation widely used in systemic lupus erythematosus. Case reports and case series have shown that the agent is effective for NSIP associated with DM and PM, and it stabilized or improved pulmonary function.86–88
Intravenous immunoglobulin
Intravenous immunoglobulin has been used as a second-line agent for muscular symptoms of refractory DM and PM. Its usefulness in DM/PM- ILD is uncertain, although Suzuki et al89 reported its use in five patients with severe ILD.
Rituximab
Rituximab is an anti-CD20 antibody widely used for B cell lymphoma and rheumatoid arthritis, which depletes B cells in vivo. Limited data suggest a benefit from rituximab in patients with progressive ILD in patients with PM or DM, the majority of whom were ILD with anti-ARS Abs.90–93 Sem et al93 reported eleven refractory ILD cases in anti-synthetase syndrome patients treated with rituximab and showed that the agent stabilized and/or improved the ILD in seven of eleven patients. It is not clear whether rituximab is effective for ILD cases positive for anti-MDA5 Ab. Further studies are required to elucidate the efficacy of B cell depletion therapy on DM/PM-ILD.
Polymyxin-B direct hemoperfusion
Polymyxin-B direct hemoperfusion, originally developed for the removal of endotoxin, has been demonstrated to be effective for treating various types of acute respiratory failure. A case of the effective use of polymyxin-B direct hemoperfusion in refractory A/S-ILD positive for anti-MDA5 Ab has been reported.94
Treatment principles in DM/PM-ILD
The goal of management of DM/PM-ILD is survival in A/S-ILD and maintenance of pulmonary function for daily life in C-ILD. As described above, DM/PM-ILD is heterogeneous and contains several clinical subsets. Management of ILD involves assessment of ILD, determining which subtypes of ILD the patient belongs to, and treating the patient based on the subtype, severity of ILD, and other factors such as organ involvement other than the lung. In the management of A/S-ILD, delaying effective treatment causes poor outcomes. Thus, intensive therapy should be considered as initial treatment, particularly in DM with anti-MDA5 Ab or in ADM. In addition, tight monitoring is required in A/S-ILD. If clinical effects are not observed during therapy, second-line or salvage therapies should be considered.
In C-ILD, initial intensive therapy is not usually required. In management of C-ILD, a balance between therapeutic effects and adverse effects is critical. In several cases, particularly asymptomatic patients, treatment for ILD is not necessary. In patients with symptomatic C-ILD, GC is used as initial therapy for ILD, and when the effects are insufficient or a decrease in GC dose is required, additional immunosuppressive therapy is considered. In many cases, therapy for myositis also improves C-ILD.
Assessment of poor risk factors for ILD and determination of subtypes
In A/S-ILD, there is a form of fatal, rapidly progressive ILD refractory to GC therapy. The first step is to determine whether the patient belongs to this type of ILD. When a patient shows ADM, positivity for anti-MDA5 Ab, low CK levels (<500 U), elevated ferritin levels, and GGO plus lower consolidation on HRCT, the ILD is potentially fatal and refractory. If a patient has PM or positivity for anti-ARS Ab, the patient has a low possibility for fatal progressive ILD.
Initial combination therapy for A/S-ILD
Initial combination therapy is superior to step-up therapy for A/S-ILD. When CsA was introduced for A/S-ILD, CsA was administered if GC was ineffective. However, Nagasaka et al72 reported that early CsA administration (within 2 weeks after GC therapy) improved the survival rate of DM A/S-ILD compared to early GC alone therapy (5-year survival rate: 69% early combination versus 39% GC alone). Similarly, Kotani et al75 reported that initial combination therapy with GC and CsA brought better survival than step-up treatment (add CsA/CY if GC was ineffective) in DM A/S-ILD (1-year survival: 89% initial combination versus 43% step-up). Takada et al95 also showed that the primary intensive approach, involving immunosuppressant with GC initially, was superior to the step-up approach, involving additional immunosuppressant if GC was ineffective (5-year survival rate: primary intensive 74% versus step-up 54%) in DM/PM-ILD. For A/S-ILD with poor prognostic factors, initial combination therapy should be considered.
Triple therapy for refractory A/S-ILD
For combination therapy, CsA or CY is used with GC. However, the efficacy of these combinations is not sufficient. Kameda et al53 reported that combination therapy with one immunosuppressant (CsA or CY) plus GC saved only three of 12 (25%) of DM A/S-ILD patients. They treated ten DM A/S-ILD cases with triple therapy (CsA [trough level >150–250 ng/mL], IVCY [10–30 mg/kg every 3–4 weeks], and high-dose GC), and 50% of the patients survived; the survival rate was high compared to that of combination therapy with one immunosuppressant. The efficacy of this triple therapy has not been evaluated in controlled studies, although several cases of A/S-ILD treated with triple therapy were reported.96,97 In our experience, triple therapy was effective, particularly for anti-MDA5 Ab-positive DM A/S-ILD that was frequently resistant to GC–CsA combination therapy.
We think that triple therapy should be considered for A/S-ILD with poor prognostic factors such as anti-MDA5 Ab positivity, although evaluation of its efficacy is necessary in the future.
Importance of monitoring and reconsideration of treatment
In patients with DM/PM-ILD, particularly A/S-ILD, clinical condition changes quickly; some patients develop respiratory failure requiring mechanical ventilation within a few days. Thus, tight monitoring of the clinical condition is required in the management of DM/PM-ILD. The monitoring of the clinical condition in DM/PM-ILD involves three kinds of monitoring: for disease activity, for respiratory condition, and for adverse events (AEs) including opportunistic infections that frequently occur and become fatal during intensive immunosuppressive therapy. During monitoring of disease activity in A/S-ILD, serum ferritin levels are a good marker, as well as the anti-MDA5 Ab titer, in addition to clinical signs such as skin rash.41 For monitoring the respiratory condition, symptoms such as dyspnea, cough, alveolar-arterial oxygen tension difference, and change in the extension of GGO/consolidation on HRCT are useful. In addition, the increase in KL-6 and surfactant protein D levels also provides useful information, including prognostic information.26 Monitoring for AEs is critical in the management of A/S-ILD, because not a small number of patients die of infection, mainly opportunistic infections such as pneumocystis pneumonia.25,65 Routine monitoring for pneumocystis pneumonia and fungal infections by measuring plasma β-D glucan levels and cytomegalovirus reactivation by examining cytomegalovirus antigenemia is widely performed in Japan, as well as prophylactic administration of trimethoprim-sulfamethoxazole for pneumocystis pneumonia. Since nephrotoxicity and hepatotoxicity due to CsA and hepatotoxicity due to CY are frequently found during therapy, monitoring for these adverse events is necessary.
When disease activity is not controlled and the respiratory condition is worsening, salvage therapy should be considered.
Algorithm for management in DM/PM-ILD
The algorithm for management of DM/PM-ILD in our institute is shown in Figure 3; it starts with assessment of poor risk factors and identification of subtypes.
 | Figure 3 Algorithm for management of DM/PM-ILD. |
Lessons learned from experience with ILD in Japan
A/S-ILD is found in Japan at a high frequency compared with other countries. Among patients positive for anti-MDA5, A/S-ILD also develops frequently. Rapid progression and resistance to treatment are common in A/S-ILD in Japan. Taken together, a characteristic clinical feature of DM/PM-ILD in Japan is A/S-ILD with strong inflammation leading to a potentially fatal course.
Why is A/S-ILD with strong inflammation frequently found in Japan? A pulmonary hyperimmune/inflammatory reaction has been suggested.98 Several agents that rarely cause ILD frequently induce fatal ILD in Japan. Leflunomide, a widely used disease-modifying antirheumatic drug, causes fatal A/S-ILD with a high frequency (1.2%) in Japan, but not in other countries, including Korea and China.99 Gefatinib also caused ILD in Japan with a high incidence compared to other countries.100 Most cases of bleomycin-induced ILD were Japanese.98 These findings suggest that Japanese has a background, although it is unclear what that background is, that causes pulmonary hyperimmune/inflammatory reactions, including drug-induced ILD. The high frequency of A/S-ILD in DM and ILD with a poor prognosis might reflect this background. Since it has been suggested that viral infection, IFN induction, and MDA5 expression are involved, the high incidence of A/S-ILD in DM/PM in Japan might be explained by pulmonary hyperimmune/inflammatory reactions among Japanese people that cause drug-induced ILD rarely found in other countries.
Physicians managing DM/PM-ILD in Japan have had to face refractory, fatal A/S-ILD. They first found that there were many A/S-ILD cases that had been thought to develop rarely, most of which were DM, especially without CK elevation and resistant to GC therapy. During the struggle to control A/S-ILD, combination therapy with CsA/CY was developed, although CsA/CY had rarely been used for ILD. The combination therapy improved the prognosis of A/S-ILD and has been widely used for A/S-ILD in DM in Japan, although confirmation of the efficacy through controlled studies has not been performed and is required. Moreover, a subgroup of A/S-ILD cases was identified that was characterized by DM (particularly ADM), anti-MDA5 Ab, refractory rapidly progressive interstitial pneumonia, and serum ferritin elevation.
Experience with DM/PM-ILD has demonstrated the importance of DM A/S-ILD in clinical practice, combination therapy with CsA and CY for A/S-ILD, and the subtype of DM associated with anti-MDA5 Ab and refractory ILD. Are these findings common worldwide? We believe these findings are not specific for Japan, but are universal, because many investigators report the existence of A/S-ILD worldwide and identify a clinical subset characterized by anti-MDA5 Ab and A/S-ILD globally.35–38 However, the effectiveness of combination therapy with CsA and CY has yet to be elucidated through experience and studies worldwide.
It is notable that there remains approximately 20%–50% of patients with A/S-ILD who fail to respond to initial intensive combination therapy with CsA and CY.25,26,53,65 Development of new treatments for these cases is required, which might be different from immunosuppressive or anti-inflammatory agents and could control common pathways of the end stage of acute ILD (including acute exacerbation of idiopathic pulmonary fibrosis) that result in the destruction of pulmonary structure.
Conclusion
From experience with DM/PM-ILD, we learned the following: refractory A/S-ILD is not a rare complication in DM and ADM, anti-MDA5 Ab is closely related with A/S-ILD and poor outcomes in DM/ADM, and initial combination therapy with CsA/CY should be considered when patients show poor prognostic factors such as ADM, DM with low CK elevation, positivity for anti-MDA5 Ab, serum ferritin elevation, or consolidation with GGO on HRCT. A/S-ILD with potentially fatal outcomes is found worldwide, not only in Japan. Clinicians caring for patients with DM/PM should be cautious when dealing with A/S-ILD and treat the patients based on clinical subtypes. Further studies are required to elucidate the efficacy of combination therapy with CsA/CY on A/S-ILD and to develop new therapies for A/S-ILD with poor outcomes despite intensive immunosuppressive therapy, including CsA/CY.
Disclosure
The authors report no conflicts of interest in this work.
References
Zong M, Lundberg IE. Pathogenesis, classification and treatment of inflammatory myopathies. Nat Rev Rheumatol. 2011;7(5):297–306. | |
Khan S, Christopher-Stine L. Polymyositis, dermatomyositis, and autoimmune necrotizing myopathy: clinical features. Rheum Dis Clin North Am. 2011;37(2):143–158. | |
Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest. 2010;138(6):1464–1474. | |
Labirua A, Lundberg IE. Interstitial lung disease and idiopathic inflammatory myopathies: progress and pitfalls. Curr Opin Rheumatol. 2010;22(6):633–638. | |
Mimori T, Nakashima R, Hosono Y. Interstitial lung disease in myositis: clinical subsets, biomarkers, and treatment. Curr Rheumatol Rep. 2012;14(3):264–274. | |
Sontheimer RD. Would a new name hasten the acceptance of amyopathic dermatomyositis (dermatomyositis siné myositis) as a distinctive subset within the idiopathic inflammatory dermatomyopathies spectrum of clinical illness? J Am Acad Dermatol. 2002;46(4):626–636. | |
Gerami P, Schope JM, McDonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54(4):597–613. | |
Mills ES, Mathews WH. Interstitial pneumonitis in dermatomyositis. J Am Med Assoc. 1956;160(17):1467–1470. | |
Frazier AR, Miller RD. Interstitial pneumonitis in association with polymyositis and dermatomyositis. Chest. 1974;65(4):403–407. | |
Salmeron G, Greenberg SD, Lidsky MD. Polymyositis and diffuse interstitial lung disease. A review of the pulmonary histopathologic findings. Arch Intern Med. 1981;141(8):1005–1010. | |
Schnabel A, Reuter M, Biederer J, Richter C, Gross WL. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum. 2003;32(5):273–284. | |
Fathi M, Vikgren J, Boijsen M, et al. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum. 2008;59(5):677–685. | |
Hayashi S, Tanaka M, Kobayashi H, et al. High-resolution computed tomography characterization of interstitial lung diseases in polymyositis/dermatomyositis. J Rheumatol. 2008;35(2):260–269. | |
Nawata Y, Kurasawa K, Takabayashi K, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol. 1999;26(7):1527–1533. | |
Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49(9):1713–1719. | |
Richards TJ, Eggebeen A, Gibson K, et al. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum. 2009;60(7):2183–2192. | |
Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol. 2001;144(4):825–831. | |
Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis- dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med. 2001;164(7):1182–1185. | |
Marie I, Hachulla E, Chérin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum. 2002;47(6):614–622. | |
Won Huh J, Soon Kim D, Keun Lee C, et al. Two distinct clinical types of interstitial lung disease associated with polymyositis-dermatomyositis. Respir Med. 2007;101(8):1761–1769. | |
Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF. Short-term and long-term outcomes of interstitial lung disease in polymyositis and dermatomyositis: a series of 107 patients. Arthritis Rheum. 2011;63(11):3439–3447. | |
Kang EH, Lee EB, Shin KC, et al. Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositis. Rheumatology (Oxford). 2005;44(10):1282–1286. | |
American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646–664. | |
Suda T, Fujisawa T, Enomoto N, et al. Interstitial lung diseases associated with amyopathic dermatomyositis. Eur Respir J. 2006;28(5):1005–1012. | |
Koga T, Fujikawa K, Horai Y, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology (Oxford). 2012;51(7):1278–1284. | |
Arai S, Kurasawa K, Maezawa R, Owada T, Okada H, Fukuda T. Marked increase in serum KL-6 and surfactant protein D levels during the first 4 weeks after treatment predicts poor prognosis in patients with active interstitial pneumonia associated with polymyositis/dermatomyositis. Mod Rheumatol. 2013;23(5):872–883. | |
Fujisawa T, Suda T, Nakamura Y, et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J Rheumatol. 2005;32(1):58–64. | |
Mukae H, Ishimoto H, Sakamoto N, et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest. 2009;136(5):1341–1347. | |
Casciola-Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol. 2012;24(6):602–608. | |
Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52(5):1571–1576. | |
Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60(7):2193–2200. | |
Nakashima R, Imura Y, Kobayashi S, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology (Oxford). 2010;49(3):433–440. | |
Hoshino K, Muro Y, Sugiura K, Tomita Y, Nakashima R, Mimori T. Anti-MDA5 and anti-TIF1-gamma antibodies have clinical significance for patients with dermatomyositis. Rheumatology (Oxford). 2010;49(9):1726–1733. | |
Hamaguchi Y, Kuwana M, Hoshino K, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147(4):391–398. | |
Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65(1):25–34. | |
Labrador-Horrillo M, Martinez MA, Selva-O’Callaghan A, et al. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J Immunol Res. 2014;2014:290797. | |
Cao H, Pan M, Kang Y, et al. Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody. Arthritis Care Res (Hoboken). 2012;64(10):1602–1610. | |
Chen Z, Cao M, Plana MN, et al. Utility of anti-melanoma differentiation-associated gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res (Hoboken). 2013;65(8):1316–1324. | |
Hall JC, Casciola-Rosen L, Samedy LA, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res (Hoboken). 2013; 65(8):1307–1315. | |
Gono T, Kawaguchi Y, Hara M, et al. Increased ferritin predicts development and severity of acute interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49(7):1354–1360. | |
Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology (Oxford). 2012;51(9):1563–1570. | |
Horai Y, Koga T, Fujikawa K, et al. Serum interferon-α is a useful biomarker in patients with anti-melanoma differentiation-associated gene 5 (MDA5) antibody-positive dermatomyositis. Mod Rheumatol. Epub April 9, 2014. | |
Targoff IN. Autoantibodies and their significance in myositis. Curr Rheumatol Rep. 2008;10(4):333–340. | |
Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity. 2006;39(3):217–221. | |
Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford). 2009;48(6):607–612. | |
Targoff IN. Autoantibodies in polymyositis. Rheum Dis Clin North Am. 1992;18(2):455–482. | |
Grau JM, Miró O, Pedrol E, et al. Interstitial lung disease related to dermatomyositis. Comparative study with patients without lung involvement. J Rheumatol. 1996;23(11):1921–1926. | |
Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum. 1996;26(1):459–467. | |
Ghirardello A, Bassi N, Palma L, et al. Autoantibodies in polymyositis and dermatomyositis. Curr Rheumatol Rep. 2013;15(6):335. | |
Fischer A, Swigris JJ, du Bois RM, et al. Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presenting with idiopathic interstitial pneumonia. Respir Med. 2009;103(11):1719–1724. | |
Mielnik P, Wiesik-Szewczyk E, Olesinska M, Chwalinska-Sadowska H, Zabek J. Clinical features and prognosis of patients with idiopathic inflammatory myopathies and anti-Jo-1 antibodies. Autoimmunity. 2006;39(3):243–247. | |
Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity. 2006;39(3):233–241. | |
Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32(9):1719–1726. | |
Yamasaki Y, Yamada H, Ohkubo M, et al. Longterm survival and associated risk factors in patients with adult-onset idiopathic inflammatory myopathies and amyopathic dermatomyositis: experience in a single institute in Japan. J Rheumatol. 2011;38(8):1636–1643. | |
Dankó K, Ponyi A, Constantin T, Borgulya G, Szegedi G. Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine (Baltimore). 2004;83(1):35–42. | |
Airio A, Kautiainen H, Hakala M. Prognosis and mortality of polymyositis and dermatomyositis patients. Clin Rheumatol. 2006;25(2):234–239. | |
Ye S, Chen XX, Lu XY, et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: a retrospective cohort study. Clin Rheumatol. 2007;26(10):1647–1654. | |
Ji SY, Zeng FQ, Guo Q, et al. Predictive factors and unfavourable prognostic factors of interstitial lung disease in patients with polymyositis or dermatomyositis: a retrospective study. Chin Med J (Engl). 2010;123(5):517–522. | |
Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax. 2008;63(1):53–59. | |
Tazelaar HD, Viggiano RW, Pickersgill J, Colby TV. Interstitial lung disease in polymyositis and dermatomyositis. Clinical features and prognosis as correlated with histologic findings. Am Rev Respir Dis. 1990;141(3):727–733. | |
Ito M, Kaise S, Suzuki S, et al. Clinico-laboratory characteristics of patients with dermatomyositis accompanied by rapidly progressive interstitial lung disease. Clin Rheumatol. 1999;18(6):462–467. | |
Ikezoe J, Johkoh T, Kohno N, Takeuchi N, Ichikado K, Nakamura H. High-resolution CT findings of lung disease in patients with polymyositis and dermatomyositis. J Thorac Imaging. 1996;11(4):250–259. | |
Arakawa H, Yamada H, Kurihara Y, et al. Nonspecific interstitial pneumonia associated with polymyositis and dermatomyositis: serial high-resolution CT findings and functional correlation. Chest. 2003;123(4):1096–1103. | |
Tanizawa K, Handa T, Nakashima R, et al. HRCT features of interstitial lung disease in dermatomyositis with anti-CADM-140 antibody. Respir Med. 2011;105(9):1380–1387. | |
Tanizawa K, Handa T, Nakashima R, et al. The prognostic value of HRCT in myositis-associated interstitial lung disease. Respir Med. 2013;107(5):745–752. | |
Nukiwa T. The role of biomarkers in management of interstitiallung disease: implications of biomarkers derived from type II pneumocytes. In: du Bois RM, Richeldi L, editors. Interstitial Lung Diseases. European Respiratory Society Monograph. Lausanne, Switzerland: European Respiratory Society Journals Ltd; 2009:47–66. | |
Kubo M, Ihn H, Yamane K, et al. Serum KL-6 in adult patients with polymyositis and dermatomyositis. Rheumatology (Oxford). 2000;39(6):632–636. | |
Kurasawa K, Nawata Y, Takabayashi K, et al. Activation of pulmonary T cells in corticosteroid-resistant and -sensitive interstitial pneumonitis in dermatomyositis/polymyositis. Clin Exp Immunol. 2002;129(3):541–548. | |
Dickey BF, Myers AR. Pulmonary disease in polymyositis/dermatomyositis. Semin Arthritis Rheum. 1984;14(1):60–76. | |
Schwarz MI, Matthay RA, Sahn SA, Stanford RE, Marmorstein BL, Scheinhorn DJ. Interstitial lung disease in polymyositis and dermatomyositis: analysis of six cases and review of the literature. Medicine (Baltimore). 1976;55(1):89–104. | |
Gruhn WB, Diaz-Buxo JA. Cyclosporine treatment of steroid resistant interstitial pneumonitis associated with dermatomyositis/polymyositis. J Rheumatol. 1987;14(5):1045–1047. | |
Nagasaka K, Harigai M, Tateishi M, et al. Efficacy of combination treatment with cyclosporin A and corticosteroids for acute interstitial pneumonitis associated with dermatomyositis. Mod Rheumatol. 2003;13(3):231–238. | |
Kuroda H, Morinaga H, Satoh C, Miyata A, Sunami K. Clinical study of 10 cases of acute or subacute interstitial pneumonia associated with dermatomyositis. Mod Rheumatol. 2003;13(4):313–318. | |
Maeda K, Kimura R, Komuta K, Igarashi T. Cyclosporine treatment for polymyositis/dermatomyositis: is it possible to rescue the deteriorating cases with interstitial pneumonitis? Scand J Rheumatol. 1997;26(1):24–29. | |
Kotani T, Makino S, Takeuchi T, et al. Early intervention with corticosteroids and cyclosporin A and 2-hour postdose blood concentration monitoring improves the prognosis of acute/subacute interstitial pneumonia in dermatomyositis. J Rheumatol. 2008;35(2):254–259. | |
Nagai K, Takeuchi T, Kotani T, et al. Therapeutic drug monitoring of cyclosporine microemulsion in interstitial pneumonia with dermatomyositis. Mod Rheumatol. 2011;21(1):32–36. | |
Qushmaq KA, Chalmers A, Esdaile JM. Cyclosporin A in the treatment of refractory adult polymyositis/dermatomyositis: population based experience in 6 patients and literature review. J Rheumatol. 2000;27(12):2855–2859. | |
Cavagna L, Caporali R, Abdì-Alì L, Dore R, Meloni F, Montecucco C. Cyclosporine in anti-Jo1-positive patients with corticosteroid-refractory interstitial lung disease. J Rheumatol. 2013;40(4):484–492. | |
Oddis CV, Sciurba FC, Elmagd KA, Starzl TE. Tacrolimus in refractory polymyositis with interstitial lung disease. Lancet. 1999;353(9166):1762–1763. | |
Takada K, Nagasaka K, Miyasaka N. Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunity. 2005;38(5):383–392. | |
Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum. 2005;52(8):2439–2446. | |
Guglielmi S, Merz TM, Gugger M, Suter C, Nicod LP. Acute respiratory distress syndrome secondary to antisynthetase syndrome is reversible with tacrolimus. Eur Respir J. 2008;31(1):213–217. | |
Yamadori I, Fujita J, Kajitani H, et al. Lymphocyte subsets in lung tissues of interstitial pneumonia associated with untreated polymyositis/dermatomyositis. Rheumatol Int. 2001;21(3):89–93. | |
Mok CC, To CH, Szeto ML. Successful treatment of dermatomyositis-related rapidly progressive interstitial pneumonitis with sequential oral cyclophosphamide and azathioprine. Scand J Rheumatol. 2003;32(3):181–183. | |
Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford). 2007;46(1):124–130. | |
Swigris JJ, Olson AL, Fischer A, et al. Mycophenolate mofetil is safe, well tolerated, and preserves lung function in patients with connective tissue disease-related interstitial lung disease. Chest. 2006;130(1):30–36. | |
Saketkoo LA, Espinoza LR. Experience of mycophenolate mofetil in 10 patients with autoimmune-related interstitial lung disease demonstrates promising effects. Am J Med Sci. 2009;337(5):329–335. | |
Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken). 2010;62(10):1496–1501. | |
Suzuki Y, Hayakawa H, Miwa S, et al. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung. 2009;187(3):201–206. | |
Lambotte O, Kotb R, Maigne G, Blanc FX, Goujard C, Delfraissy JF. Efficacy of rituximab in refractory polymyositis. J Rheumatol. 2005; 32(7):1369–1370. | |
Brulhart L, Waldburger JM, Gabay C. Rituximab in the treatment of antisynthetase syndrome. Ann Rheum Dis. 2006;65(7):974–975. | |
Vandenbroucke E, Grutters JC, Altenburg J, Boersma WG, ter Borg EJ, van den Bosch JM. Rituximab in life threatening antisynthetase syndrome. Rheumatol Int. 2009;29(12):1499–1502. | |
Sem M, Molberg O, Lund MB, Gran JT. Rituximab treatment of the anti-synthetase syndrome: a retrospective case series. Rheumatology (Oxford). 2009;48(8):968–971. | |
Teruya A, Kawamura K, Ichikado K, Sato S, Yasuda Y, Yoshioka M. Successful polymyxin B hemoperfusion treatment associated with serial reduction of serum anti-CADM-140/MDA5 antibody levels in rapidly progressive interstitial lung disease with amyopathic dermatomyositis. Chest. 2013;144(6):1934–1936. | |
Takada K, Kishi J, Miyasaka N. Step-up versus primary intensive approach to the treatment of interstitial pneumonia associated with dermatomyositis/polymyositis: a retrospective study. Mod Rheumatol. 2007;17(2):123–130. | |
Tanaka F, Origuchi T, Migita K, et al. Successful combined therapy of cyclophosphamide and cyclosporine for acute exacerbated interstitial pneumonia associated with dermatomyositis. Intern Med. 2000;39(5):428–430. | |
Miyazaki E, Ando M, Muramatsu T, et al. Early assessment of rapidly progressive interstitial pneumonia associated with amyopathic dermatomyositis. Clin Rheumatol. 2007;26(3):436–439. | |
Azuma A, Kudoh S. High prevalence of drug-induced pneumonia in Japan. JMAJ. 2007;50(5):405–411. | |
Sawada T, Inokuma S, Sato T, et al; Study Committee for Leflunomide-induced Lung Injury, Japan College of Rheumatology. Leflunomide-induced interstitial lung disease: prevalence and risk factors in Japanese patients with rheumatoid arthritis. Rheumatology (Oxford). 2009;48(9):1069–1072. | |
Kudoh S, Kato H, Nishiwaki Y, et al; Japan Thoracic Radiology Group. Interstitial lung disease in Japanese patients with lung cancer: a cohort and nested case-control study. Am J Respir Crit Care Med. 2008;177(12):1348–1357. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.