Back to Journals » The Application of Clinical Genetics » Volume 17
Optical Genome Mapping Identifies a Novel Unbalanced Translocation Between Chromosomes 4q and 6q Leading to Feeding Difficulties and Hypotonia in a Neonate: A Case Report
Authors Wang Y, Bi S, Shi X, Dai L
Received 21 February 2024
Accepted for publication 14 May 2024
Published 27 May 2024 Volume 2024:17 Pages 63—69
DOI https://doi.org/10.2147/TACG.S465244
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Ying Wang, Shaohua Bi, Xiaoqing Shi, Liying Dai
Division of Neonatology, Anhui Provincial Children’s Hospital, Hefei, Anhui, 230051, People’s Republic of China
Correspondence: Liying Dai, Division of Neonatology, Anhui Province Children’s Hospital, No. 39, Wangjiang East Road, Baohe District, Hefei, Anhui, 230051, People’s Republic of China, Email [email protected]
Abstract: Optical Genome Mapping (OGM) technology has garnered growing interest for the identification of chromosomal structural variations (SVs), particularly complex ones that are implicated in genetic diseases in humans. In this study, we performed genetic diagnostics on a neonatal patient who presented with feeding difficulties, hypotonia, and an atrial septal defect. We utilized a combination of trio-whole exome sequencing and OGM for our analysis. The results revealed an unbalanced translocation between maternal chromosomes 4 and 6 in the proband, ogm[GRch38]t(4:6)(q35.2;q25.3), resulting in a 2.8 Mb deletion at the 4q35 terminal and a 10.2 Mb duplication at the 6q25 terminal. In summary, this study highlights how OGM, in conjunction with other genetic approaches, can unveil the genetic etiology of complex clinical syndromes. Neonatal patients often exhibit low specific phenotypes, underlining the significance of SV detection.
Keywords: optical genome mapping, unbalanced translocation, copy number variations, neonatal diseases
Introduction
Structural variations (SVs) in chromosomes encompass alterations in gene number, position, or sequence, stemming from physical, chemical, and genetic factors. SVs affect genome segments with ≥50 base pairs (bp) and include copy number variations (CNVs), rearrangements, and mobile element insertions.1,2 Next Generation Sequencing (NGS) technology is widely used for identifying single nucleotide variants (SNVs) or small insertions and deletions (Indels). Nonetheless, there exist certain technical limitations in detecting SVs, primarily stemming from the standard read lengths of NGS, which are typically confined to 100–150 bp. This limitation hinders the ability to span repetitive elements or provide contextual information.3,4 Optical genome mapping (OGM) is an innovative technology for detecting extremely long single DNA molecules (>150 kb). OGM uses specific recognition sequences, such as CTTAAG, to label DNA molecules and employs software-assisted whole-genome assembly to accurately identify various SV classes, including aneuploidy, deletions, translocations, inversions, and complex rearrangements.5
The unbalanced translocation between chromosome 4q and 6q is a rare SV. Due to a lack of necessary case reports, the correlation between this SV and its phenotype remains incompletely elucidated. Isolated deletions/duplications at the 4q or 6q terminal regions have been sporadically documented, and significant phenotypic heterogeneity has been observed in affected individuals. The most common manifestations include developmental delay and intellectual disability (ID); some patients also exhibit abnormal behavior and special facial features.6–17 Due to the limited number of reported cases of these SVs and the corresponding phenotypic heterogeneity, there remains controversy regarding the pathogenicity of this variant.
In this study, we outline the genetic diagnostic process for a Chinese family with ID, focusing on the proband, a neonatal patient displaying symptoms such as hypotonia, feeding intolerance, and an atrial septal defect. Notably, his mother had an ID. By employing OGM, we identified an unbalanced translocation between maternal chromosomes 4 and 6 in the proband, resulting in a deletion at the 4q terminal and a duplication at the 6q terminal. This study aims to elucidate the identification of novel unbalanced translocations in a neonatal patient with complex phenotypes and underscores the contribution of SV to the identification of the hereditary etiology.
Materials and Methods
Ethical Compliance
This study adhered to the principles of the Declaration of Helsinki and was approved by the Ethics Committee of Anhui Children’s Hospital. After obtaining written informed consent for diagnostic tests and research studies, 5 mL of peripheral blood samples were collected from the proband and his parents.
Whole Exome Sequencing (WES)
Genomic DNA from the samples collected from the proband and his parents was extracted using the TIANamp Blood DNA Kit (#DP348-03, TIANamp Blood DNA Kit, Beijing, China) following the manufacturer’s instructions. DNA concentration and purity were assessed using the OneDrop™ OD-1000 spectrophotometer (RockGene, Shanghai, China), ensuring an OD 260/280 ratio between 1.6 and 2.0 and a total quantity of >1 μg. Fragmentation, end-repair, and adapter ligation were performed using the Hieff NGS® OnePot DNA Library Prep Kit for Illumina® (WeHealth BioMedical, Shanghai, China) to prepare pre-libraries. DNA libraries were constructed using the IDT xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, IA, USA). Sequencing was conducted on the NovaSeq 6000 sequencing platform (Illumina, San Diego, CA, USA) in PE150 mode. Subsequently, the sequencing data was converted into FastQ files using the bc2fastq software. Base sequences from the FastQ files were aligned to the GRCh38/hg20 reference genome using the BWA v0.7.12 software, resulting in BAM files. SNVs and Indels in the samples were analyzed using the GATK v4.0.5.2 software. CNVs were analyzed using the CNVkit software. Identified SNVs and Indels were formatted according to the Human Genome Variation Society format and annotated against various databases, including ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home), DECIPHER (https://www.deciphergenomics.org/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), and GenomeAD (https://gnomad.broadinstitute.org/) using the snpEff v4.2 annotation software. Variants were classified based on the guidelines of the American College of Medical Genetics and Genomics.18
OGM
Genomic DNA was extracted from the proband’s blood sample using the Bionano Prep™ Blood and Cell Culture DNA Isolation Kit (Bionano Genomics, San Diego, USA). The extracted DNA was fluorescently labeled using the DLE-1 enzyme (Bionano Genomics), sourced from the DLS DNA Labeling Kit (Bionano Genomics). The labeled DNA was loaded onto the Saphyr Chip G2.2 (Bionano, San Diego, CA, USA), and molecules >150 kb were collected, targeting a total data yield of 1300 Gb. Subsequently, the labeled DNA was loaded onto the Bionano Genomics Saphyr® system for linearization and visualization. Each sample yielded a minimum of 320 GB of data, followed by automated whole-genome de novo assembly.19 Genome assembly and variant calling were performed using Bionano Access version 1.7 (https://bionanogenomics.com/support/software-downloads/). SV data was identified by referencing the human reference genome (GRCh38/hg38). SV detection was performed using the Solve v.3.2.1 software (Bionano Genomics). Independent analyses were conducted for the CNV and SV algorithms, used for chromosome abnormalities >100 Kb and >500 bp, respectively.
Results
Clinical Phenotype
The patient was a newborn male, the first born, delivered via cesarean section due to fetal malposition at 40 weeks and 5 days gestation. He weighed 3100 g at birth and had Apgar scores of 8 at 1 and 5 min, with no history of birth asphyxia. The newborn was admitted to Anhui Children’s Hospital in August 2023, 2 days after birth, due to aspiration pneumonia.
Upon admission, a physical examination revealed erythematous skin, a flat and soft anterior fontanelle, and smooth oral mucosa without a cleft palate. Milk digestion disorders manifesting as feeding intolerance were observed after breastfeeding, leading to abdominal distension and vomiting. The limbs were relatively short and exhibited reduced muscle tone, and hip joint abduction was observed. Transient-evoked otoacoustic emission technology (MEDSEN Accuscreen full-function hearing screener, GN Otometrics, Denmark) was used to detect the newborn’s hearing, and the results showed abnormal hearing in the left ear (the otoacoustic emission meter screen displayed REFER).
Radiological examination primarily identified multiple atrial septal defects on cardiac ultrasonography. Laboratory tests showed inflammatory features, such as a slight increase in C-reactive protein, with no other substantial abnormalities (see Supplemental Table 1 for relevant laboratory examination). The child’s father (23-year-old) was in good health, while the mother (21-year-old) had mild intellectual disabilities (indicated by the Wechsler Intelligence Scale test results from another hospital). She was able to perform daily living activities and some household chores however, had mild social communication difficulties, and only maintained brief interactions with her family.
WES and OGM Results
WES sequencing yielded 86.6 million reads for the proband, with an average sequencing depth of 131.74× and ≥30× coverage of 98.4%. However, WES did not identify SNP/INDELS genes related to the proband’s phenotype. The CNVkit software suggested that he may carry a copy number deletion variant of approximately 1.91 Mb in the 4q35.2q35.2 segment (deemed of uncertain significance according to guidelines) and a copy number duplication variant of approximately 10.64 Mb in the 6q25.3q27 region (deemed likely pathogenic). The mother had CNVs of the same length in the above two segments. The data control for the OGM test is included in Supplemental Table 2. The findings indicate an unbalanced translocation variation between chromosomes 4 and 6 of the proband, ogm[GRch38]t(4:6)(q35.2;q25.3). This SV results in a copy number deletion of approximately 2.8 Mb in the 4q35.2q35.3 segment and a copy number duplication of approximately 10.2 Mb in the 6q25.3q27 segment (Table 1). This variant resulted in the formation of a derivative chromosome 4, der(4)(4pter→4q35.2::6q25.3→6qter) (Figure 1).
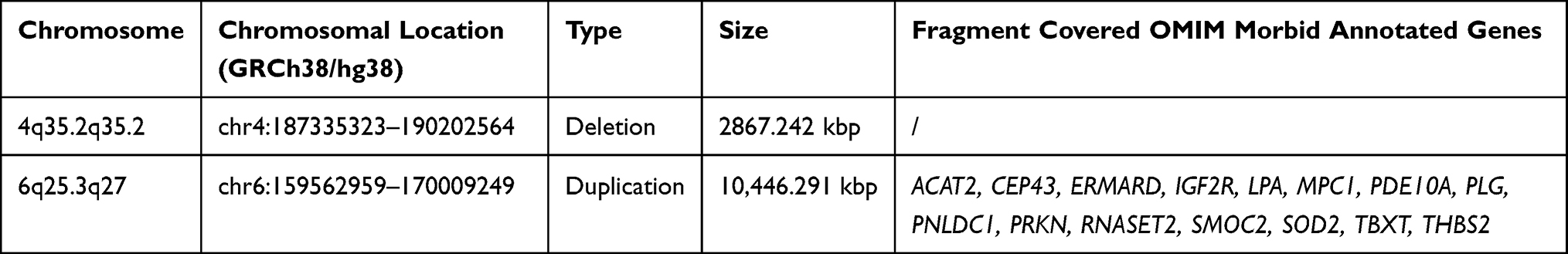 |
Table 1 Copy Number Variations Detected by Whole Genome Optical Mapping |
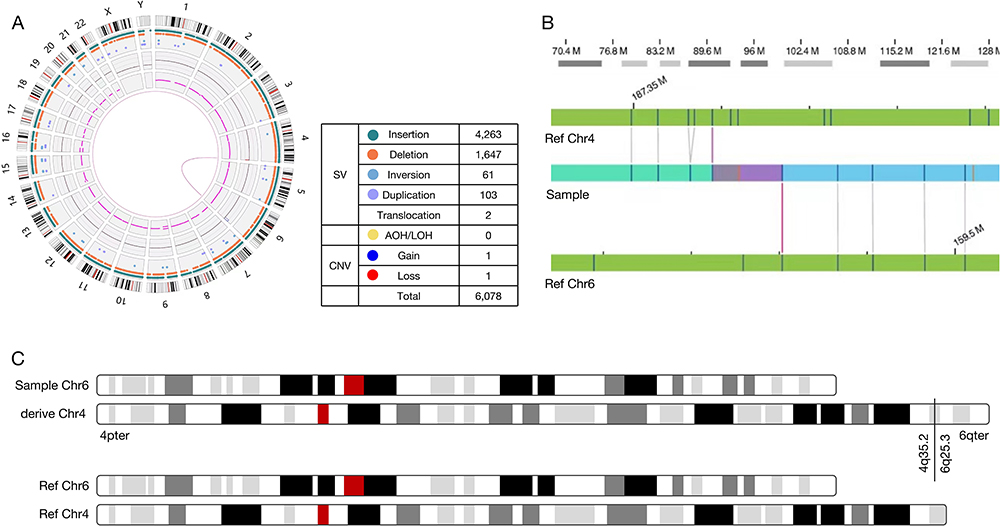 |
Figure 1 The proband’s OGM test results. (A) Circos plot, the pink line connecting Chr4 and Chr6 represents the unbalanced translocation between them; (B) The genome map view shows an unbalanced translocation, ogm[GRch38]t(4:6)(q35.2;q25.3); (C) Schematic diagram of the chromosomes of this sample. |
Discussion
In this study, we conducted genetic testing on a neonatal patient presenting with hypotonia, feeding intolerance, and an atrial septal defect. OGM results revealed an unbalanced translocation between the 4q and the 6q terminal in the proband, leading to the formation of a derivative chromosome 4, der(4)(4pter→4q35.2::6q25.3→6qter). While breakpoint validation was not conducted, based on the OGM data, this variant did not appear to involve pathogenic genes listed in the Online Mendelian Inheritance in Man (OMIM) database (https://www.omim.org/), nor did it result in the formation of fusion genes. WES detected no SNVs or Indels associated with the proband’s phenotype. However, it indicated the presence of a genetic microdeletion in the 4q35.2q35.2 region and a microduplication in the 6q25.3q27 region inherited from the proband’s mother.
The 6q25qter segment encompasses multiple genes associated with diseases documented in the OMIM database, including links to growth restriction, abnormal skull shape, facial anomalies, reduced muscle tone, seizures, and intellectual disabilities.16,17 Among these, the ERMARD gene (OMIM#615532) may be the key pathogenic gene in the 6q25qter segment that causes hypotonia and brain structural malformations. Conti et al have suggested that the ERMARD gene haploinsufficiency could impact the neuronal migration process.20 Clinical reports of microduplications in the 6q25qter region appear to be rarer than the 4q35.2 microdeletion, and no clear evidence exists of dosage-sensitive genes in this segment. However, data from the Decipher database (https://www.deciphergenomics.org/) (Patient ID: 400926, 501317, 345454, 331649) suggest that some patients with microduplications in the 6q26q27 region exhibit phenotypes similar to the those observed in this study, including cardiovascular morphological abnormalities, atrial septal defects, infantile feeding difficulties, hypotonia, and specific learning disabilities. Therefore, the possibility that the 6q25 microduplication may contribute to the pathogenic factors seen in this case cannot be ruled out.
Patients with 4q35.2 microdeletions have obvious phenotypic heterogeneity, mainly characterized by varying degrees of special facial features, ID, and attention deficit hyperactivity disorder (Table 2). Additionally, some parents of patients with 4q35.2 microdeletions did not show abnormal clinical characteristics, which may reflect the characteristics of incomplete penetrance.11 Although the neonatal patient in this study did not show signs of a neurodevelopmental disorder, his mother exhibited mild ID. Therefore, this cannot rule out the possibility of the proband having intellectual disabilities after birth or in childhood. Moreover, this condition may also be associated with the risk of other psychiatric diseases, such as learning disabilities or attention deficit hyperactivity disorders. The researchers in this study will continue to follow up on the clinical manifestations of this patient, paying particular attention to phenotypes related to neurodevelopmental disorders.
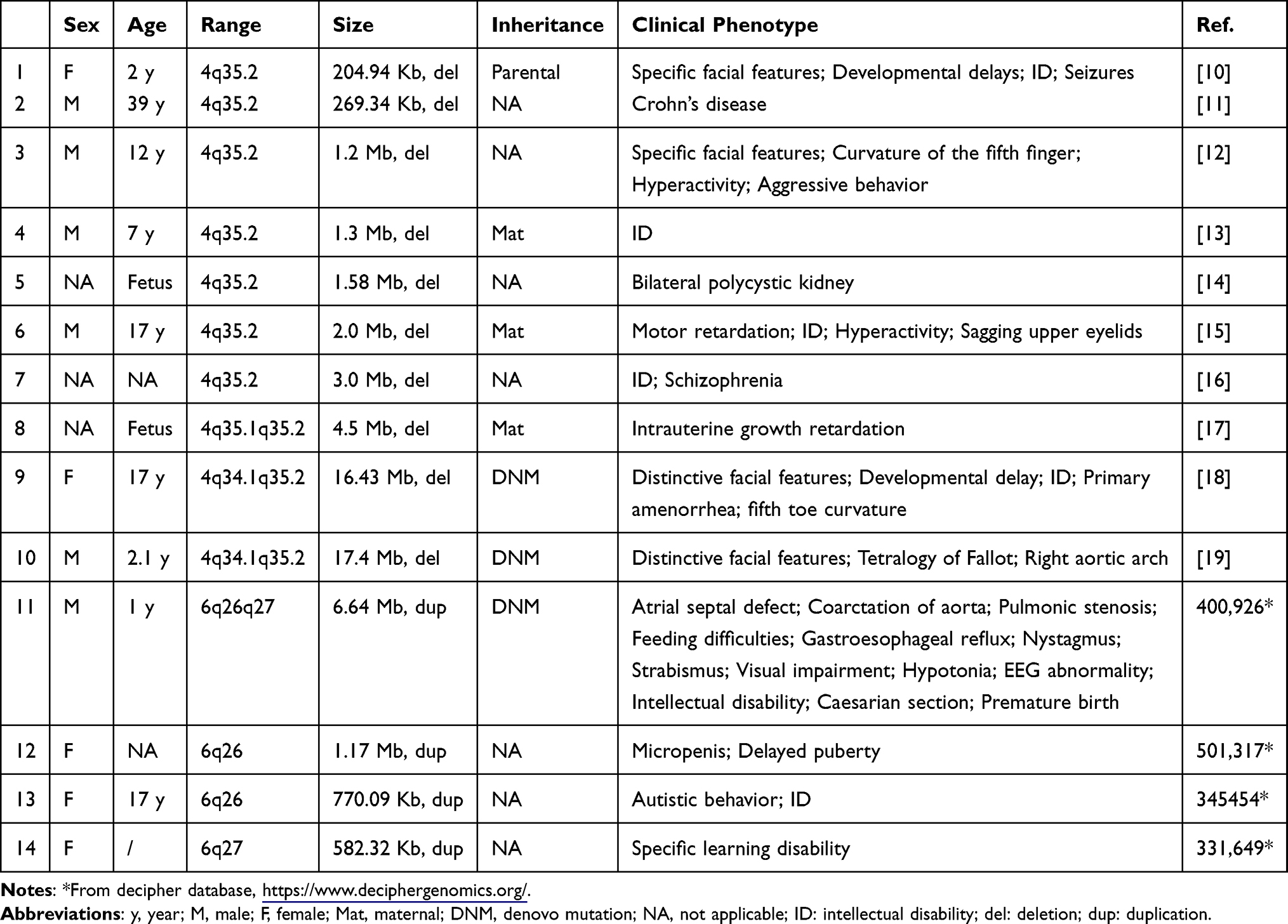 |
Table 2 Information on Patients with Reported 4q35 or 6q25qter Variants |
In this case, a combination of WES and OGM methods was employed. WES was primarily used for SNV and Indel screening, while OGM was utilized for SV detection. Currently, SV detection mainly relies on techniques such as chromosomal karyotyping, fluorescence in situ hybridization, and SNP arrays.21–23 Low-depth whole-genome sequencing (WGS) can be used to identify CNVs, while deep WGS can detect complex SVs and Indels.24,25 However, the short-read data reading principle of NGS-WES technology requires different algorithms to deal with different variant classes, may confuse the repeated elements of short-read mapping, and lacks accurate identification of the CNVs.26 Therefore, WES only provides directional pathological CNV results; for example, in this report, WES suggested a 6q25.3q27 duplication and a 4q35.2 microdeletion in the subjects, but did not accurately identify the breakpoints.
OGM produces high-resolution fluorescence imaging by producing multiple specific enzyme digestion labeling sites across the genome, which has a lower error rate than high-throughput sequencing technology that requires amplification steps.27,28 Numerous research findings indicate that OGM, which leverages sequencing technology, is quicker and offers distinct advantages in identifying chromosomal deletions, duplications, inversions, translocations, and assessing local structural variations.5,27 For example, Zhang et al reported on the case of a 5-year-old girl with skeletal malformations, ID, and congenital cranial dysinnervation disorder. Only 6q25.3 microduplication and 2q37.1 microdeletion were identified through WES. OGM determined that these variants were inherited from a balanced translocation between 2q and 6q carried by her healthy father.28 Similarly, Xie et al describe an unbalanced translocation between 5p and 6p in a proband and her mother using WES combined with OGM. At the same time, OGM also revealed that the proband’s grandmother had a balanced translocation that caused multiple malformations in the subsequent two generations.29 These case reports have shown that OGM has technical advantages in identifying SVs. However, the high cost of OGM currently limits its widespread application. As the technology develops, it can become a reliable and practical diagnostic method for detecting genomic SVs. Additionally, due to the generation of large amounts of SV data from OGM, there is an urgent need to establish specialized databases to collect and correlate patient clinical phenotypes with SV data.
In summary, we employed a combination of OGM and WES technologies to identify a new unbalanced translocation in a neonatal individual presenting with hypotonia, feeding challenges, and an atrial septal defect. However, this study has certain limitations. First, there is a lack of revalidation of the SV by other detection methods or breakpoint verification through qPCR. Secondly, the SV analysis of the patient’s maternal family members should be expanded, which may enrich more comprehensive family information. For non-specific phenotypes in newborns, such as hypotonia or feeding difficulties, or a family history with ID, it is advisable to employ a comprehensive testing approach that thoroughly examines various types of genetic variants. OGM is gaining clinical recognition for its capability to identify SVs, and this report aims to underscore the importance of prioritizing SV detection in clinical practice.
Data Sharing Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Ethics
Informed consent for publishing this case was obtained from the patient’ s parents. The study was conducted according to the guidelines of the World Medical Association (Declaration of Helsinki) and approved by the Ethics Committee of Anhui Provincial Children’s Hospital.
Patient Consent for Publication
The parents of the patient provided written informed consent regarding the publication of the medical data and images of the case.
Acknowledgment
We would like to express our gratitude the neonatal patient and his family in this study. We also want to acknowledge the healthcare professionals involved in the care of the patient. We are thankful for support and cooperation throughout the study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12(5):363–376. doi:10.1038/nrg2958
2. Morin SJ, Eccles J, Iturriaga A, et al. Translocations, inversions and other chromosome rearrangements. Fertil Steril. 2017;107(1):19–26. doi:10.1016/j.fertnstert.2016.10.013
3. Caspar SM, Dubacher N, Kopps AM, et al. Clinical sequencing: from raw data to diagnosis with lifetime value. Clin Genet. 2018;93(3):508–519. doi:10.1111/cge.13190
4. Bhattacharya S, Barseghyan H, Délot EC, et al. nanotatoR: a tool for enhanced annotation of genomic structural variants. BMC Genomics. 2021;22(1):10. doi:10.1186/s12864-020-07182-w
5. Mantere T, Neveling K, Pebrel-Richard C, et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am J Hum Genet. 2021;108(8):1409–1422. doi:10.1016/j.ajhg.2021.05.012
6. Karaman Mercan T, Altiok Clark O, Erkal O, et al. Coexistence of a Homozygous Chromosome 4q35.2 Deletion and Hidden IQSEC2 Pathogenic Variant in a Child with Intellectual Disability. Cytogenet Genome Res. 2021;161:153–159. doi:10.1159/000515368
7. Riccardi F, Rivolta GF, Uliana V, et al. Cryptic 13q34 and 4q35.2 Deletions in an Italian Family. Cytogenet Genome Res. 2015;147:24–30. doi:10.1159/000442068
8. Youngs EL, Henkhaus RS, Hellings JA, et al. 12-year-old boy with a 4q35.2 microdeletion and involvement of MTNR1A, FAT1, and F11 genes. Clin Dysmorphol. 2012;21:93–96. doi:10.1097/MCD.0b013e32834e9216
9. Balikova I, Menten B, de Ravel T, et al. Subtelomeric imbalances in phenotypically normal individuals. Hum Mutat. 2007;28(10):958–967. doi:10.1002/humu.20537
10. Fu F, Chen F, Li R, et al. Prenatal diagnosis of fetal multicystic dysplastic kidney via high-resolution whole-genome array. Nephrol Dial Transplant. 2016;31(10):1693–1698. doi:10.1093/ndt/gfv465
11. Zhuang J, Liu S, Chen X, et al. Identification of a novel isolated 4q35.2 microdeletion in a Chinese pediatric patient using chromosomal microarray analysis: a case report and literature review. Mol Cytogenet. 2023;16:18. doi:10.1186/s13039-023-00651-3
12. Pickard BS, Hollox EJ, Malloy MP, et al. A 4q35.2 subtelomeric deletion identified in a screen of patients with co-morbid psychiatric illness and mental retardation. BMC Med Genet. 2004;5(1):21. doi:10.1186/1471-2350-5-21
13. Xiao G, Qiu X, Zhou Y, et al. Prenatal diagnosis of a 4.5-Mb deletion at chromosome 4q35.1q35.2: case report and literature review. Mol Cytogenet. 2021;14:53. doi:10.1186/s13039-021-00573-y
14. Rossi MR, DiMaio MS, Xiang B, et al. Clinical and genomic characterization of distal duplications and deletions of chromosome 4q: study of two cases and review of the literature. Am J Med Genet A. 2009;149A(12):2788–2794. doi:10.1002/ajmg.a.33088
15. Cuturilo G, Menten B, Krstic A, et al. 4q34.1-q35.2 deletion in a boy with phenotype resembling 22q11.2 deletion syndrome. Eur J Pediatr. 2011;170:1465–1470. doi:10.1007/s00431-011-1533-3
16. Nagamani SC, Erez A, Eng C, et al. Interstitial deletion of 6q25.2-q25.3: a novel microdeletion syndrome associated with microcephaly, developmental delay, dysmorphic features and hearing loss. Eur J Hum Genet. 2009;17:573–581. doi:10.1038/ejhg.2008.220
17. Michelson M, Ben-Sasson A, Vinkler C, et al. Delineation of the interstitial 6q25 microdeletion syndrome: refinement of the critical causative region. Am J Med Genet A. 2012;158A(6):1395–1399. doi:10.1002/ajmg.a.35361
18. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
19. Barseghyan H, Tang W, Wang RT, et al. Next-generation mapping: a novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017;9(1):90. doi:10.1186/s13073-017-0479-0
20. Conti V, Carabalona A, Pallesi-Pocachard E, et al. Periventricular heterotopia in 6q terminal deletion syndrome: role of the C6orf70 gene. Brain. 2013;136(Pt 11):3378–3394. doi:10.1093/brain/awt249
21. Mukherjee S, Sathanoori M, Ma Z, et al. Addition of chromosomal microarray and next generation sequencing to FISH and classical cytogenetics enhances genomic profiling of myeloid malignancies. Cancer Genet. 2017;216-217:128–141. doi:10.1016/j.cancergen.2017.07.010
22. Su D, Zhang D, Chen K, et al. High performance of targeted next generation sequencing on variance detection in clinical tumor specimens in comparison with current conventional methods. J Exp Clin Cancer Res. 2017;36(1):121. doi:10.1186/s13046-017-0591-4
23. Lu X, Shaw CA, Patel A, et al. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS One. 2007;2(3):e327. doi:10.1371/journal.pone.0000327
24. Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinf. 2009;10(1):80. doi:10.1186/1471-2105-10-80
25. Chiang C, Scott AJ, Davis JR, et al. The impact of structural variation on human gene expression. Nat Genet. 2017;49(5):692–699. doi:10.1038/ng.3834
26. Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nat Methods. 2011;8(1):61–65. doi:10.1038/nmeth.1527
27. Pan W, Lonardi S. Accurate detection of chimeric contigs via Bionano optical maps. Bioinformatics. 2019;35:1760–1762. doi:10.1093/bioinformatics/bty850
28. Zhang S, Cui Q, Yang S, et al. Exome and genome sequencing to unravel the precise breakpoints of partial trisomy 6q and partial Monosomy 2q. BMC Pediatr. 2023;23(1):586. doi:10.1186/s12887-023-04368-5
29. Xie M, Xue J, Zhang Y, et al. Combination of trio-based whole exome sequencing and optical genome mapping reveals a cryptic balanced translocation that causes unbalanced chromosomal rearrangements in a family with multiple anomalies. Front Genet. 2023;14:1248544. doi:10.3389/fgene.2023.1248544
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.