Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
Opinions and Attitudes of Pulmonologists About Augmentation Therapy in Patients with Alpha-1 Antitrypsin Deficiency. A Survey of the EARCO Group
Authors Greulich T, Albert A, Cassel W, Boeselt T, Peychev E, Klemmer A, Ferreira F, Clarenbach C ![]() , Torres-Duran ML
, Torres-Duran ML ![]() , Turner AM
, Turner AM ![]() , Miravitlles M
, Miravitlles M ![]()
Received 24 October 2021
Accepted for publication 20 December 2021
Published 5 January 2022 Volume 2022:17 Pages 53—64
DOI https://doi.org/10.2147/COPD.S346051
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Timm Greulich, 1,* Anna Albert, 1,* Werner Cassel, 2 Tobias Boeselt, 1 Erika Peychev, 1 Andreas Klemmer, 1 Francini Ferreira, 3 Christian Clarenbach, 4 Maria L Torres-Duran, 5, 6 Alice M Turner, 7 Marc Miravitlles 8
1University Medical Centre Giessen and Marburg, Philipps-University, Department of Medicine, Pulmonary and Critical Care Medicine, Member of the German Centre for Lung Research (DZL), Marburg, Germany; 2University Medical Centre Giessen and Marburg, Philipps-University, Department of Medicine, Pulmonary and Critical Care Medicine, Sleep Disorders Centre, Member of the German Centre for Lung Research (DZL), Marburg, Germany; 3Bioclever, Barcelona, Spain; 4Division of Pulmonology, University Hospital Zurich, Zurich, Switzerland; 5Pneumology Department, Hospital Álvaro Cunqueiro, AS Vigo, Spain; 6Instituto de Investigación Sanitaria Galicia Sur (IISGS), Vigo, Spain; 7Respiratory Medicine, University Hospitals Birmingham NHS Foundation Trust, Institute of Applied Health Research, University of Birmingham, Birmingham, UK; 8Pneumology Department, Hospital Universitari Vall d´Hebron/Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Barcelona Hospital Campus, CIBER de Enfermedades Respiratorias (CIBERES), Barcelona, Spain
*These authors contributed equally to this work
Correspondence: Marc Miravitlles
Pneumology Department, Vall d´Hebron Barcelona Hospital Campus, P. Vall d’Hebron 119-129, Barcelona, 08035, Spain
Tel/Fax +34 932746083
Email [email protected]
Background: Augmentation therapy (AT) is the only specific treatment licensed for patients with alpha-1 antitrypsin deficiency (AATD) associated lung disease. Since patients with severe AATD may have a very different prognosis and AT requires intravenous infusions for life, the decision to initiate AT may be challenging.
Methods: This survey was conducted on 63 experts in AATD from 13 European countries about their opinions and attitudes regarding AT. Participants were asked to rank the importance of 11 identified factors related with the prescription of AT. In addition, each participant was asked to respond to the indication of AT for 30 out of 500 hypothetical cases developed with the combinations of the 11 factors. Each case was evaluated by 3 experts to check the concordance.
Results: The variables that scored higher on preferences for initiating AT were AAT genotype (score 8.6 from a Likert scale 0– 10 (SD: 1.7)), AATD serum level (8.2 (SD:2.4)) and FEV1 (%) decline (7.9 (SD:2.4)). Among the 500 different cases, there was an agreement in indication of AT among the 3 experts in 291 (58.2%). Regarding the variables associated with AT, it was indicated to 81.9% of Pi*ZZ, 52.4% of Pi*SZ and 9.8% of Pi*MZ (p < 0.0001). For Pi*ZZ patients, multivariate analysis identified younger age, reduced FEV1 (%), higher FEV1 decline and worse emphysema as significantly associated with prescription (AUC = 0.8114); for Pi*SZ variables were younger age, worse FEV1 (%) and worse emphysema (AUC = 0.7414); and for Pi*MZ younger age, worse DLCO (%), higher DLCO decline and dyspnea (AUC = 0.8387).
Conclusion: There is a high variability in the criteria for prescription of AT among European experts. Most cases were recommended AT according to guidelines, but a significant number of patients with genotype Pi*SZ and almost 10% Pi*MZ were recommended to initiate AT despite the lack of evidence of efficacy in these genotypes.
Keywords: augmentation therapy, survey, EARCO, Europe
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a common but under-recognized genetic condition that affects approximately 1 in 2000 to 1 in 5000 individuals and predisposes to early-onset emphysema and liver disease.1 Alpha-1 antitrypsin (AAT) is mainly produced in the liver, and its main function is to protect the lung against the proteolytic damage, especially from neutrophil elastase.2
It is estimated that approximately 1 in 850 cases of chronic obstructive pulmonary disease (COPD) in Europe is associated with AATD, with large variations between countries.3 The only specific treatment available for lung disease associated with AATD is the intravenous administration of AAT obtained from blood donors, which is called augmentation therapy (AT).4 This treatment was approved in the United States and Europe in the late 80s based on its biochemical efficacy restoring the antielastase protection in the lungs4,5 and only recently demonstrated its clinical efficacy in reducing the rate of decline of lung density in patients with AATD associated emphysema.6 National and international guidelines have established criteria for initiation of AT based on the diagnosis of severe AATD and the demonstration of lung function impairment;7–9 however, it is recognized that clinical evolution of lung disease in AATD is difficult to predict on an individual basis.10 Some patients may have an accelerated decline in lung function and lung density despite quitting smoking and adequate COPD treatment,11,12 while non-index cases, identified by population or family screening, may have the same life expectancy than the general population.13 Therefore, a personalized approach to treatment according to the characteristics of the patients has been suggested.14
Since AT requires weekly intravenous infusions for life, usually in hospital facilities, the decision to initiate AT must be based on a case-by-case careful evaluation of risks and cost-benefit analysis in experienced reference centers.15 In the current study, we aimed to investigate the criteria used by European experts to indicate initiation of AT beyond the recommendations of the current guidelines.
Method
Study Design
Investigation about variability of prescription of AT and associated factors was identified as a research priority by the European Alpha-1 antitrypsin Research Collaboration (EARCO), see www.earco.org.16 Therefore, a cross-sectional, survey was conducted between April and December 2018 among European AATD expert pulmonologists identified from the EARCO network.17 The study involved the remittance of a survey to a group of previously selected experts, with the goal of identifying the most important factors influencing the decision to initiate AT in patients with AATD.
The study was carried out according to the principles of the Declaration of Helsinki and the prevailing norms for performing investigation in humans. Data confidentiality was ensured according to the Law of Data Protection 2016/679. The study was approved by the Phillips University of Giessen and Marburg (Germany) ethics committee on December 12th, 2017, no written informed consent was considered necessary.
Development of the Questionnaire
A nonsystematic literature review was conducted to identify variables associated with the prescription of AT for AATD, including those described in guidelines for management of patients with AATD7–9 and the outcomes described in clinical trials and observational studies.6,10–13 A questionnaire was designed that included the most relevant variables found, as well as additional contributions based on the experience of the scientific committee. The final version of the survey consisted of 11 items comprising age, AAT serum level, AAT genotype (PiZZ, PiMZ and PiSZ), forced expiratory volume in one second in percent predicted (FEV1 (%)), diffusion capacity of the lungs for CO in percent predicted (DLCO (%)), FEV1 (mL) decline, DLCO (%) decline, severity of emphysema on computed tomography (CT) scan, smoking history, frequency of exacerbations in the previous year and degree of dyspnea according to the modified Medical Research Council (mMRC) dyspnea scale.
Two aspects were evaluated in the questionnaire: 1) the degree of importance given by each expert to the different variables selected for the initiation of AT, and 2) their decision as to whether to initiate AT or not in 500 hypothetical cases constructed with different combinations of the 11 selected variables. The construction of the hypothetical cases was developed considering the biological plausibility of variables that were closely related; as an example, a patient with AAT serum levels < 25 mg/dL could only have a Pi*ZZ genotype, and conversely, a patient with AAT > 80 mg/dL could only have a Pi*MZ genotype (Table 1).
 |
Table 1 Stratification of Variables Analysed in the Study |
Questionnaire Administration
All the selected experts received an email with an invitation letter and the questionnaire. Email reminders were sent after 4 weeks. Participation in the study was voluntary and experts participated anonymously. The respondent’s sociodemographic data, such as age, sex, years of work experience and number of patients with AATD attended yearly, were also collected. The experts were required to respond to the importance of the different variables for the decision to initiate AT according to a 10-point, Likert-type ordinal scale (where 1 = not important and 10 = very important).
Finally, the 500 different fictitious cases were distributed in 50 batches of 10 cases. Every participant responded to three rounds of 1 batch of 10 cases; therefore, every participant responded to 30 different cases and each case was evaluated by 3 different experts in order to test concordance of decisions. Thirteen participants in the first round did not respond to rounds 2 and 3 and were replaced by 13 new experts, for a total of 63 participants.
Statistical Analysis
For the continuous variables, the description of the mean, median and standard deviation (SD) is presented and percentages for categorical variables.
A descriptive analysis of the variables associated with the decision of AT is presented indicating the frequency of yes/no observed in each clinical case and the concordance between the respondents. Afterwards, logistic regression was adjusted combining the three response rounds (ie, 500 cases evaluated by 3 participants, resulting in 1500 evaluations) with respect to initiation of AT. This analysis was performed in 3 steps: Step 1: univariate models were estimated to assess the possible association between the independent variables and the outcome, separately. Step 2: a logistic model, called “full model” for the present analysis, including all variables of step 1 with a p value <0.1 was adjusted. Step 3: several models were adjusted from the full model, excluding those variables that were less associated with the outcome in the presence of others, in order to reach the best model to explain the outcome. The final model was then evaluated by some metrics such as receiver operating (ROC) curve and its area under the curve (AUC). The significance level used was <0.05, unless otherwise specified. All statistical analyses were performed using the statistical package SAS® system version 9.4.
Results
Participants
The 63 experts participating in the study belonged to 13 countries, the most represented countries were Germany with 18, Spain 14, Portugal 9, and the United Kingdom 7. Up to 66.7% were male, 46% treated more than 20 patients with AATD and 40% between 5 and 20 patients, and 42 (66.7%) had between 5 and 20 years of experience treating AATD patients.
Variables Considered Important for Prescription of Augmentation
The variables that scored higher on preferences for initiating augmentation therapy were AAT genotype (score 8.6 (SD: 1.7)), AATD serum level (8.2 (SD:2.4)) and FEV1 (%) decline (7.9 (SD:2.4)). Complete results are shown in Figure 1.
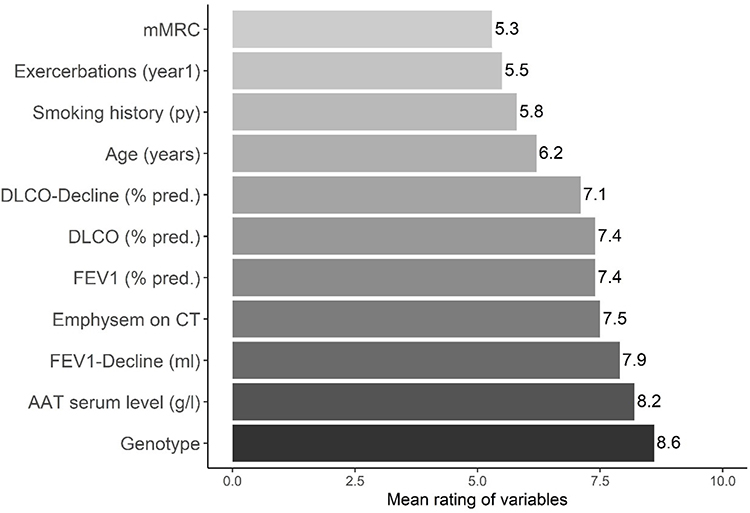 |
Figure 1 Mean scores of variables associated with prescription of augmentation therapy (range 0–10). Abbreviations: Py, pack-years; AAT, alpha-1 antitrypsin; g, grams; l, liter; FEV1, forced expiratory volume in one second; DLCO, diffusion capacity of the lung for carbon monoxide; CT, computed tomography; mMRC, modified Medical Research Council; mL, milliliter; y, year. |
Clinical Cases: Agreement Among Responders
Among the 500 different cases, there was an agreement among the 3 experts in 291 (58.2%), of which 156 (53.6%) corresponded to 3 negative and 135 (46.4%) to 3 positive responses.
Among the concordant negative all had moderate or mild deficiency, except 8 with a severe deficiency, 5 of them with normal lung function and 3 with combinations of Pi*SZ and moderate or absent emphysema. Among the concordant positive all had severe deficiency except 5 with moderate deficiency age younger than 45 years, FEV1 (%) <50% and severe emphysema.
Clinical Cases. Univariate Analysis of Variables Associated with Augmentation
Out of the 11 variables selected for the study, only mMRC degree of dyspnea was not significantly associated with the prescription of AT. However, for some of the variables, despite being significant, differences were very small. For example, AT would be prescribed in 43.1% of cases with no exacerbations, in 50.4% of patients with one and 51.3% of patients with 2 or more exacerbations the previous year (p = 0.0229). In contrast, there was a clear gradient in variables such as AAT genotype (with AT being prescribed to 81.9% of Pi*ZZ, 52.4% of Pi*SZ and 9.8% of Pi*MZ; p < 0.0001), AAT serum levels or emphysema on CT scan (Table 2).
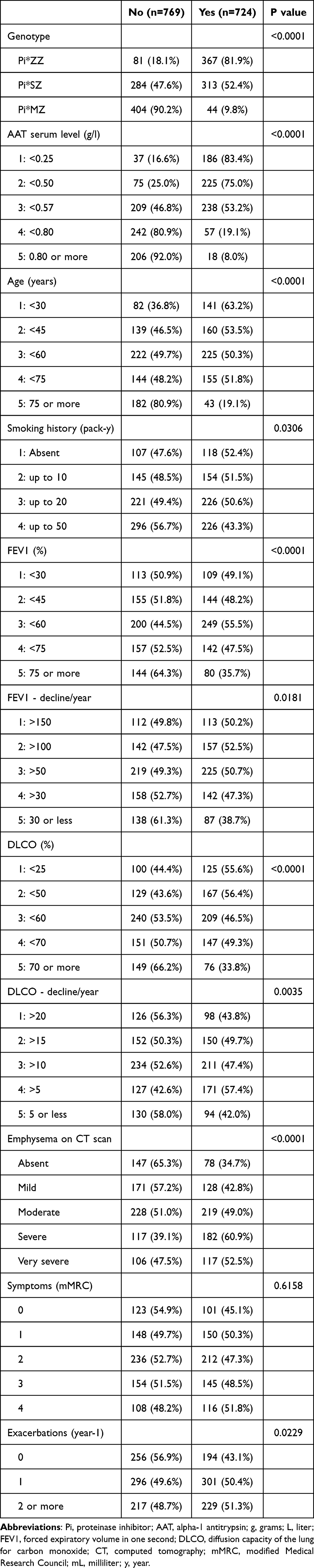 |
Table 2 Frequencies of No/Yes Within the Categories of Each Variable for the 1,500 Combinations of Case-Participants |
Clinical Cases. Logistic Regression for All Combinations of Cases-participants Included in the Analysis
The logistic regression analysis included the 1500 combinations of cases-participants and results are presented in Table 3.
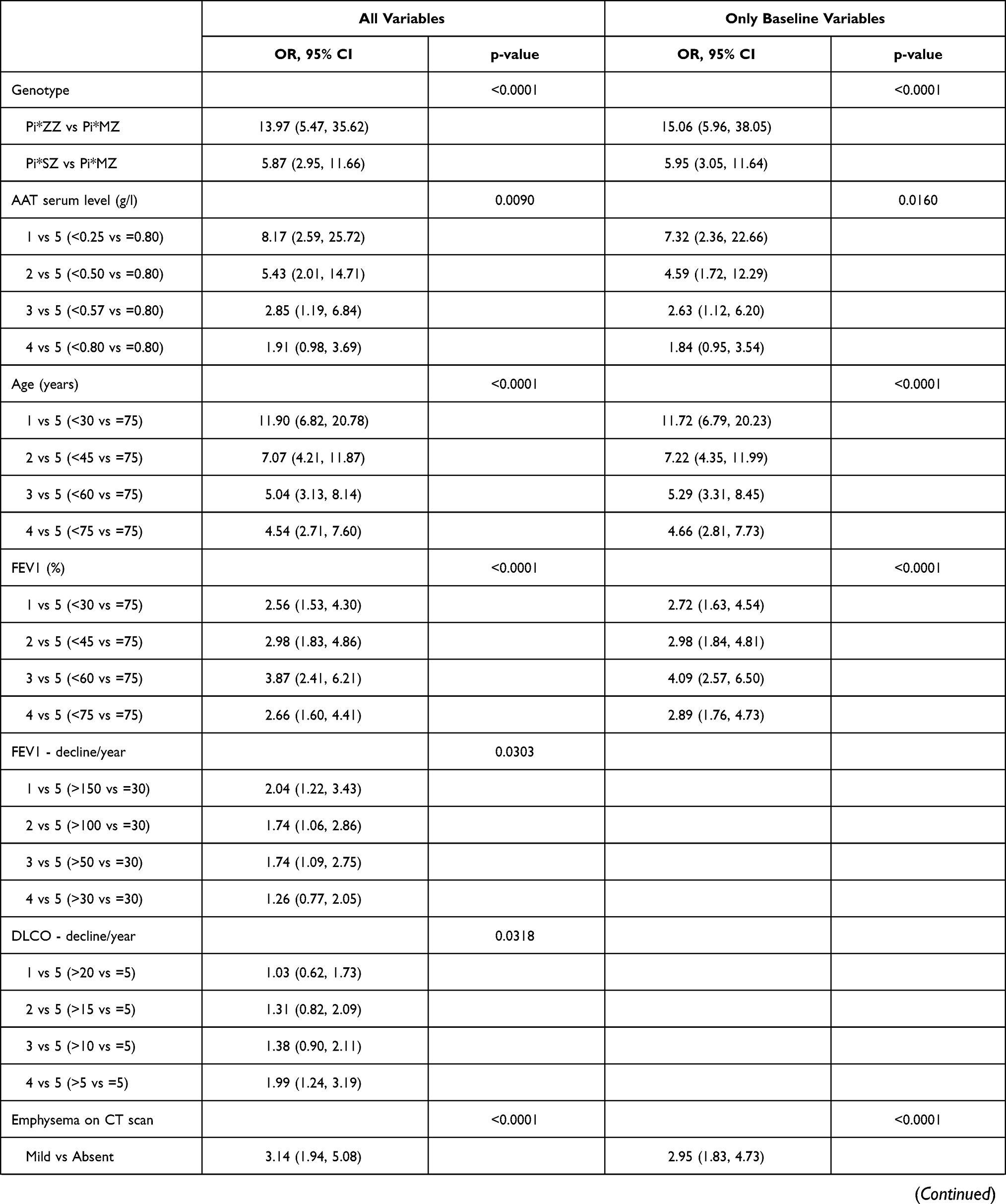 |
Table 3 Logistic Regression Analysis for the 1,500 Combinations of Case-Participants |
The first analysis included the 11 variables of the study. Seven of them were significant and the strongest gradient in odds ratios (ORs) was observed for AAT genotype, AAT serum levels and age. Results did not change significantly when the model was repeated including only baseline variables, excluding FEV1 (%) and DLCO (%) decline (Table 3).
Clinical Cases. Logistic Regression According to Different AAT Genotypes
Since genotype had the strongest influence in prescription of augmentation, we explored which factors were significantly associated with prescription in the three different genotype groups. When patients were grouped by genotypes, AAT serum levels were no longer significant for any of the genotypes. For Pi*ZZ patients, DLCO (%) and rate of decline in DLCO (%) were not significantly associated with the prescription of augmentation. For Pi*SZ patients only age, FEV1 (%) and emphysema on CT scan were significant, and finally for Pi*MZ age, DLCO (%), DLCO (%) decline and mMRC dyspnea degree were significant (Table 4). The AUC for the different models were 0.811 for the Pi*ZZ, 0.741 for the Pi*SZ and 0.838 for the Pi*MZ (Figure 2A–C).
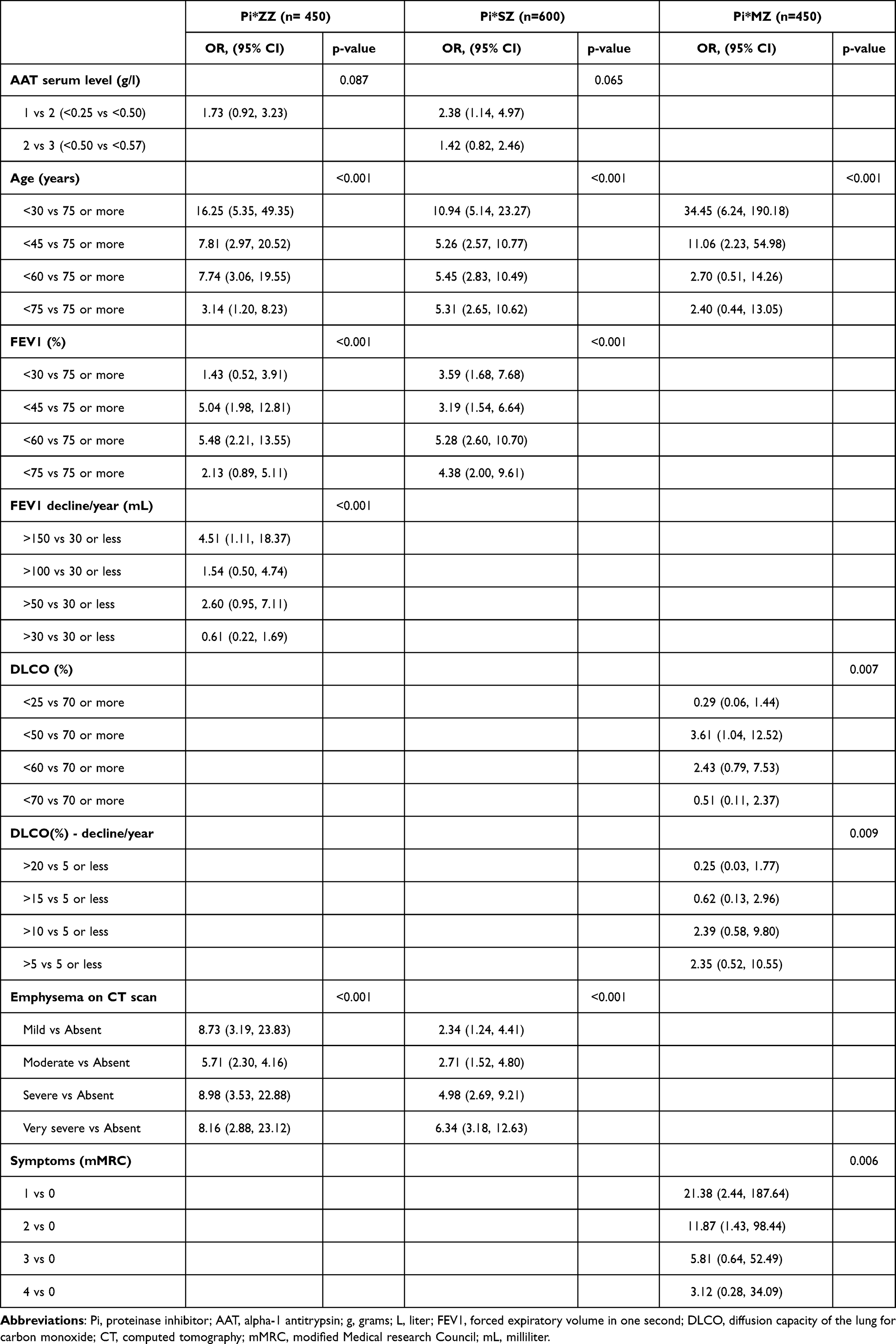 |
Table 4 Logistic Regression Analysis for the 1,500 Combinations of Case-Participants Divided by AAT Genotype |
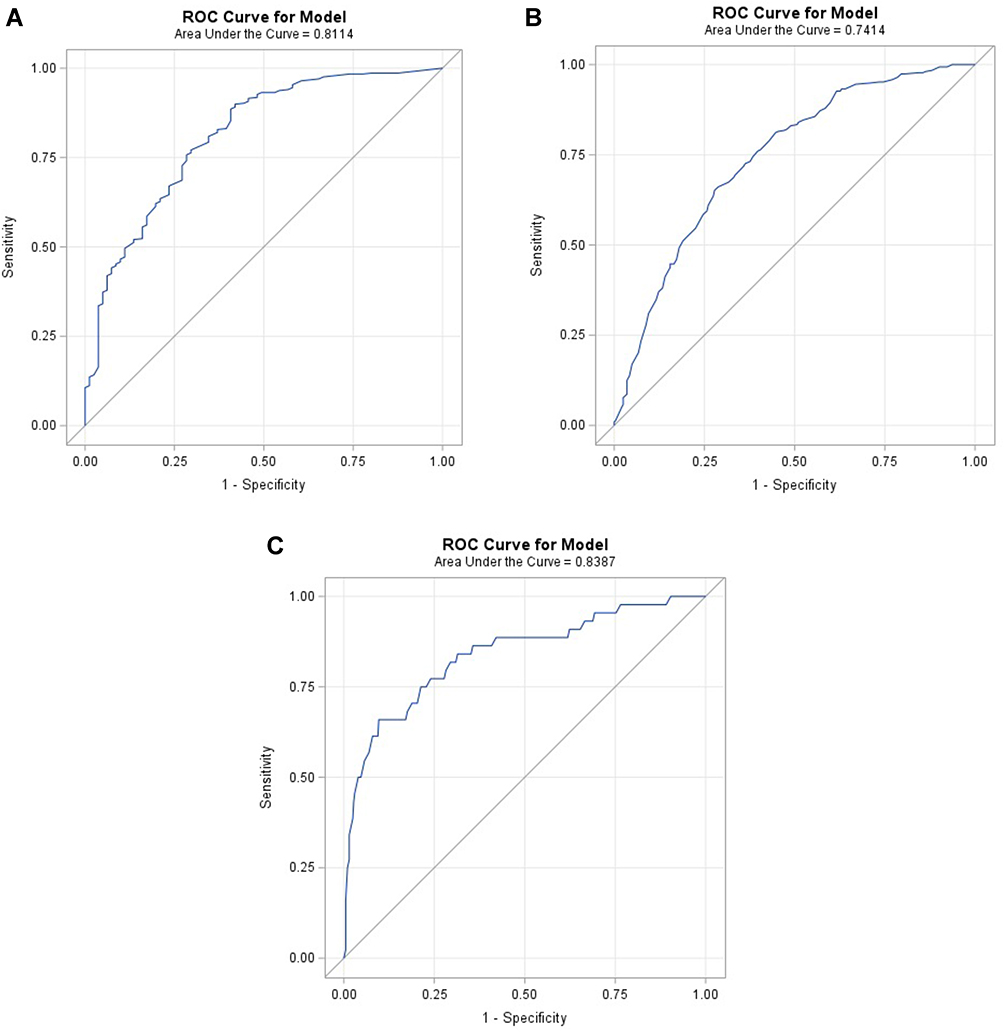 |
Figure 2 Receiver operating (ROC) curves and their area under the curve (AUC) for the indication of augmentation therapy for (A) Pi*ZZ, (B) Pi*SZ and (C) Pi*MZ. |
Discussion
Criteria for initiation of AT in clinical practice guidelines are broad and imprecise.7,9,18–20 Similarly the indications on the summary of product characteristics (SmPCs) of the available treatments for AT are also imprecise, with indications such as “evidence of progressive lung disease”21 or “clinical evidence of emphysema due to severe hereditary deficiency of AAT”22 but without a definition of progression or emphysema or even if severe AATD is restricted to Pi*ZZ or it also includes Pi*SZ. Moreover, due to the variable severity of the clinical manifestations of lung disease in patients with AATD10–12 and the different evolution and prognosis observed when patients quit smoking and initiate pharmacologic and non-pharmacologic treatments for COPD,23 the decision about if and when to start AT may be challenging and related to other demographic or clinical factors not included in the criteria for AT described in guidelines.
The current survey conducted on experts in AATD from 13 European countries has shown a large variability in their approach to AT. When hypothetical cases were presented to groups of 3 randomly selected participants, their indication of AT was concordant in only 58% of cases. Although the AAT genotype and serum levels were the most important criteria selected, some participants agreed to prescribe AT to a minority of patients with a non-severe AATD if they suffered a severe lung disease at a very young age, despite not being supported by evidence. In addition, there was agreement not to prescribe AT to severe AATD patients with only mild impairment in lung function, despite the protective role of AT in the rate of decline of lung density.6 In contrast, mMRC dyspnea score, history of exacerbations and smoking history were the variables considered to have a very minor role in the decision to treat with AT.
In most European countries, AT is administered intravenously at weekly intervals in day care hospital facilities.24 This means that from initiation, potentially to the end of life, patients will be requested to attend the hospital weekly for some hours; moreover, treatment is costly and some patients, even with severe deficiency, may have a benign clinical course irrespective of AT, with a normal survival and very few symptoms.13 Taking these considerations into account, some experts might not initiate AT right after diagnosis even if at face value they fulfill criteria for AT. In these cases, other variables not included in guidelines’ recommendations may be considered in addition to the main criteria for treatment, ie, severe AATD and reduced lung function.7,9 Age may be one of these criteria; as an example, AT may not always be considered in an 80-year-old Pi*ZZ patient with a FEV1 (%) of 65% predicted, despite fulfilling criteria for AT. However, most physicians would probably start treatment immediately if the same patient was 38 years-old instead. The same could be valid for other variables such as CT scan; a 65-year-old Pi*ZZ with FEV1 (%) of 70% and severe emphysema on CT will probably be recommended to start AT as soon as possible, but the recommendation might be different if the CT showed no or minimal emphysema.
Since AATD is a rare disease, it is difficult for a practicing physician or a single clinical center to accumulate enough experience to make informed decisions about AT and this may lead to significant differences in management between countries and even centers in the same country.15 Consequently, the EU council25 and the ERS statement16 recommend that patients with AATD should be attended in expert reference centers. In this respect, in almost half of the 500 cases there were dissenting opinions about initiating AT among the three evaluators. Among the concordant cases, concordance on initiating therapy occurred in cases of severe AATD, and five cases with moderate deficiency but younger than 45 years with severe emphysema and FEV1 (%) <45% predicted. Where concordance occurred for not initiating AT, the majority of cases were nonsevere AATD, except eight with severe deficiency and with either preserved lung function or moderate or absent emphysema on CT scans. This suggests that concordance happens largely around areas that are clearly supported by trial evidence, or conversely not supported by either trial data or the product license.
Globally, AT was recommended in 52% of the Pi*SZ cases, compared to 82% of Pi*ZZ and 10% of Pi*MZ. For Pi*SZ individuals, the factors significantly associated with the decision to treat were younger age, more severely impaired FEV1 (%) and more severe emphysema on CT scan. These results are somewhat unexpected because, although Pi*SZ individuals are included among the indications described in the SMPCs of the available therapies21,22 and a few Pi*SZ patients have an AAT level <11 µM and have been included in RCTs, the evidence for treatment in Pi*SZ is very limited26,27 and the risk of lung disease associated with this genotype is considered to be more similar to Pi*MZ than to Pi*ZZ.28,29 More surprising is the indication of AT in almost 10% of cases with Pi*MZ genotypein this case, age <45 years, a DLCO (%) <50% and being symptomatic were the most relevant variables to decide the initiation of AT. It is of note that AT in Pi*MZ patients is off label, there is no evidence of efficacy of AT in Pi*MZ individuals30,31 and, therefore, it is not recommended by guidelines.7–9
Since the introduction of AT in the late 80s the same regimen of 60 mg/Kg weekly has been recommended for patients with severe AATD and lung disease based on biochemical efficacy restoring serum AAT levels above the protective threshold,4,5 irrespective of the severity and phenotype of the respiratory disease. Prospective data from registries have shown that evolution of AATD-associated lung disease may be very variable, and therefore some patients, in particular non-index cases, may do well without AT, while others with severe disease and frequent exacerbations may require higher doses of AAT than the standard regimen to protect the lungs from the action of proteases.12–14 In this context, it would be of great help to identify responders to AT, patients that share some characteristics that either increase the concentrations or activity of proteases or make them more susceptible to the effects of proteases; however, the largest placebo-controlled, randomized clinical trial of AT to date included only 90 patients per arm, which makes subgroup analysis of responders unfeasible.6 Some authors have proposed a personalized approach to AT considering variables such as age, rate of decline of lung function and CT imaging of the lungs;11,14,32 our results indicate that most experts consider these variables for the prescription of AT despite the lack of definitive evidence.
Conclusions
Decisions about initiating AT take into consideration several variables not included in the current recommendations described in guidelines. There is a considerable variability among European experts in the criteria used to decide the prescription of AT. Unfortunately, the limited sample size of the existing clinical trials does not permit the analysis of responders, or the evaluation of demographic and clinical characteristics associated with better response to AT.
Abbreviations
AAT, alpha-1 antitrypsin; AATD, alpha-1 antitrypsin deficiency; AT, augmentation therapy; AUC, area under the curve; COPD, chronic obstructive pulmonary disease; CT, computed tomography; DLCO, diffusing capacity of the lungs for carbon monoxide; EARCO, European Alpha-1 antitrypsin Research Collaboration; EU, European Union; FEV1, forced expiratory volume in one second; FVC, forced vital capacity; mMRC, Modified Medical Research Council; PI*, protease inhibitor; ROC, receiver-operating characteristics; SAS, statistical analysis system; SMPC, summary of product characteristics; SD, standard deviation.
Acknowledgment
The authors want to thank the participants in the survey: Stefano Aliberti, Robert Bals, Miriam Barrecheguren, Marcus Berkefeld, Ignacio Blanco, Martin Brutsche, Ana Bustamante, Myriam Calle, Francisco Casas, Silvia Castillo, Jan Chlumsky, Joanna Chorowstoska-Wynimko, Christian Clarenbach, Bebiana Conde, Luciano Corda, Angelo Corsico, Filipa Costa, Eric Derom, Paul Ellis, Sebastian Fähndrich, Joachim Ficker, Wolfgang Gleiber, Joana Gomes, José María Hernández, Nicholas Hopkinson, Wim Janssens, Joana Gomes, Verena Knipel, Heiko Knoop, Rembert Koczulla, Thomas Köhnlein, Beatriz Lara, Lourdes Lazaro, Paulo Lopes, José Luis López-Campos, Ravi Mahadeva, Gerry McElvany, Javier Michel, Alexandra Mineiro, Marc Miravitlles, Jean-Francois Mornex, Anna Neb, David Parr, Isabel Parra, Esther Rodriguez-González, Gabriela Santos, Christine Seebacher, Dirk Skowasch, Robert A. Stockley, Jan Stolk, Maria Sucena, Vitor Teixeira, Gabriel Thabut, Maria L. Torres, Alice M Turner, Claus F. Vogelmeier.
Funding
This study was supported by the European Respiratory Society EARCO Clinical Research Collaboration.
Disclosure
Timm Greulich reports grants from CSL-Behring, grants from Grifols, grants from Kamada, during the conduct of the study; personal fees from Astra Zeneca, personal fees from Berlin-Chemie, personal fees from Boehringer-Ingelheim, personal fees from Chiesi, personal fees from CSL-Behring, grants and personal fees from Grifols, personal fees from GSK, personal fees from Novartis, grants from German Centre for Lung Research (DZL), Marburg, Germany (Deutsches Zentrum für Lungenforschung), outside the submitted work. Christian Clarenbach received advisory fees from Roche, Novartis, Boehringer, GSK, Astra Zeneca, Sanofi, Vifor, OM Pharma, Grifols and Mundipharma. María L Torres-Duran has received speaker and consulting fees from CSL Behring and Grifols. Alice M Turner has received either grants or speaker fees from AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim, Chiesi, CSL Behring, Takeda, Vertex and Grifols Biotherapeutics. Marc Miravitlles has received speaker fees from AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, Menarini, Rovi, Bial, Sandoz, Zambon, CSL Behring, Grifols and Novartis, consulting fees from AstraZeneca, Atriva Therapeutics, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Bial, Gebro Pharma, CSL Behring, Laboratorios Esteve, Ferrer, Mereo Biopharma, Verona Pharma, Spin Therapeutics, ONO Pharma, pH Pharma, Palobiofarma SL, Takeda, Novartis, Sanofi and Grifols and research grants from Grifols. The authors report no other conflicts of interest in this work.
References
1. Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obst Pulm Dis. 2017;12:561–569. doi:10.2147/COPD.S125389
2. Strnad P, McElvaney NG, Lomas DA. Alpha1-antitrypsin deficiency. N Engl J Med. 2020;382(15):1443–1455. doi:10.1056/NEJMra1910234
3. Blanco I, Diego I, Bueno P, Pérez-Holanda S, Casas-Maldonado F, Miravitlles M. Prevalence of alpha-1 antitrypsin genotypes in patients with COPD in Europe: a systematic review. Eur Respir Rev. 2020;29(157):200014. doi:10.1183/16000617.0014-2020
4. Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987;316(17):1055–1062. doi:10.1056/NEJM198704233161704
5. Stockley RA, Bayley DL, Unsal I, Dowson LJ. The effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2002;165(11):1494–1498. doi:10.1164/rccm.2109013
6. Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360–368. doi:10.1016/S0140-6736(15)60860-1
7. American Thoracic Society. European Respiratory Society Statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168:820–899.
8. Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society Statement: diagnosis and treatment of pulmonary disease in alpha-1 antitrypsin deficiency. Eur Respir J. 2017;50:1700610. doi:10.1183/13993003.00610-2017
9. Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis. 2016;3(3):668–682.
10. Demeo DL, Sandhaus RA, Barker AF, et al. Determinants of airflow obstruction in severe alpha-1-antitrypsin deficiency. Thorax. 2007;62(9):806–813. doi:10.1136/thx.2006.075846
11. Stockley RA, Edgar RG, Pillai A, Turner AM. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? Int J Chron Obst Pulm Dis. 2016;11:1745–1756.
12. Esquinas C, Serreri S, Barrecheguren M, et al. Long-term evolution of lung function in individuals with alpha-1 antitrypsin deficiency from the Spanish registry (REDAAT). Int J Chron Obstruct Pulmon Dis. 2018;13:1001–1007. doi:10.2147/COPD.S155226
13. Tanash HA, Ekström M, Rönmark E, Lindberg A, Piitulainen E. Survival in individuals with severe alpha 1-antitrypsin deficiency (PiZZ) in comparison to a general population with known smoking habits. Eur Respir J. 2017;50(3):1700198. doi:10.1183/13993003.00198-2017
14. Stockley RA, Miravitlles M, Vogelmeier C. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149. doi:10.1186/1750-1172-8-149
15. Miravitlles M, Nuñez A, Torres-Durán M, et al. The importance of reference centers and registries for rare diseases: the example of Alpha-1 antitrypsin deficiency. COPD. 2020;17(4):346–354. doi:10.1080/15412555.2020.1795824
16. Barrecheguren M, O’Hara K, Wilkens M, et al. Research Priorities in alpha-1 antitrypsin deficiency: results of a patients’ and healthcare providers’ international survey from the EARCO Clinical Research Collaboration. ERJ Open Res. 2020;6(4):00523–2020. doi:10.1183/23120541.00523-2020
17. Miravitlles M, Chorostowska-Wynimko J, Ferrarotti I, et al. The European Alpha-1 Research Collaboration (EARCO): a new ERS Clinical Research Collaboration to promote research in alpha-1 antitrypsin deficiency. Eur Respir J. 2019;53(2):1900138. doi:10.1183/13993003.00138-2019
18. Casas F, Blanco I, Martínez MT, et al. Indications for active case searches and intravenous alpha-1 antitrypsin treatment for patients with alpha-1 antitrypsin deficiency chronic obstructive pulmonary disease: an update. Arch Bronconeumol. 2015;51:185–192. doi:10.1016/j.arbres.2014.05.008
19. Lopes AP, Mineiro MA, Costa F, et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018;24(Suppl 1):1–21. doi:10.1016/j.pulmoe.2018.09.004
20. Attaway A, Majumdar U, Sandhaus RA, Nowacki AS, Stoller JK. An analysis of the degree of concordance among international guidelines regarding alpha-1 antitrypsin deficiency. Int J Chron Obstruct Pulmon Dis. 2019;14:2089–2101. doi:10.2147/COPD.S208591
21. Respreeza. Summary of product characteristics (SmPC). Available from: https://ec.europa.eu/health/documents/community-register/2015/20150820132594/dec_132594_es.pdf.
22. Prolastin. Summary of product characteristics (SmPC). Available from: http://grifols.com/documents/10192/27754341/ft-prolastin-es-en/786cce6a-effb-41b6-867f-59d7a34ca086.
23. Stockley JA, Stockley RA, Sapey E. There is no fast track to identify fast decliners in Alpha-1 antitrypsin deficiency by spirometry: a longitudinal study of repeated measurements. Int J Chron Obstruct Pulmon Dis. 2021;16:835–840. doi:10.2147/COPD.S298585
24. Horváth I, Canotilho M, Chlumský J, et al. Diagnosis and management of α1-antitrypsin deficiency in Europe: an expert survey. ERJ Open Res. 2019;5(1):00171–2018. doi:10.1183/23120541.00171-2018
25. The Council of the European Union. Council recommendation of 8 June 2009 on an action in the field of rare diseases, 2009/C 151/02. Available from: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:C:2009:151:0007:0010:EN:PDF.
26. McElvaney GN, Sandhaus RA, Miravitlles M, et al. Clinical considerations in individuals with Alpha-1 Antitrypsin PI*SZ genotype. Eur Respir J. 2020;55(pii):1902410. doi:10.1183/13993003.02410-2019
27. Lara B, Miravitlles M. Spanish registry of patients with Alpha-1 antitrypsin deficiency; comparison of the characteristics of PISZ and PIZZ Individuals. COPD. 2015;12(Suppl 1):27–31. doi:10.3109/15412555.2015.1021912
28. Green CE, Vayalapra S, Hampson JA, Mukherjee D, Stockley RA, Turner AM. PiSZ alpha-1 antitrypsin deficiency (AATD): pulmonary phenotype and prognosis relative to PiZZ AATD and PiMM COPD. Thorax. 2015;70(10):939–945.
29. Franciosi AN, Carroll TP, McElvaney NG. SZ alpha-1 antitrypsin deficiency and pulmonary disease: more like MZ, not like ZZ. Thorax. 2021;76(3):298–301. doi:10.1136/thoraxjnl-2020-215250
30. Sandhaus RA, Turino G, Stocks J, et al. alpha1-Antitrypsin augmentation therapy for PI*MZ heterozygotes: a cautionary note. Chest. 2008;134(4):831–834. doi:10.1378/chest.08-0868
31. Barjaktarevic I, Miravitlles M. Alpha-1 antitrypsin (AAT) augmentation therapy in individuals with the PI*MZ genotype: a pro/con debate on a working hypothesis. BMC Pulm Med. 2021;21(1):99. doi:10.1186/s12890-021-01466-x
32. Parr DG, Lara B. Clinical utility of alpha-1 proteinase inhibitor in the management of adult patients with severe alpha-1 antitrypsin deficiency: a review of the current literature. Drug Des Devel Ther. 2017;11:2149–2162. doi:10.2147/DDDT.S105207
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.