Back to Archived Journals » Hypoxia » Volume 2
One more stem cell niche: how the sensitivity of chronic myeloid leukemia cells to imatinib mesylate is modulated within a "hypoxic" environment
Authors Rovida E, Marzi I, Cipolleschi MG, Sbarba P ![]()
Received 21 October 2013
Accepted for publication 10 December 2013
Published 21 January 2014 Volume 2014:2 Pages 1—10
DOI https://doi.org/10.2147/HP.S51812
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Elisabetta Rovida, Ilaria Marzi, Maria Grazia Cipolleschi, Persio Dello Sbarba
Dipartimento di Scienze Biomediche Sperimentali e Cliniche, Università degli Studi di Firenze, Florence, Italy; Istituto Toscano Tumori, Florence, Italy
Abstract: This is a review (by no means comprehensive) of how the stem cell niche evolved from an abstract concept to a complex system, implemented with a number of experimental data at the cellular and molecular levels, including metabolic cues, on which we focused in particular. The concept was introduced in 1978 to model bone marrow sites suited to host hematopoietic stem cells (HSCs) and favor their self-renewal, while restraining clonal expansion and commitment to differentiation. Studies of the effects of low oxygen tension on HSC maintenance in vitro led us to hypothesize niches were located within bone marrow areas where oxygen tension is lower than elsewhere. We named these areas hypoxic stem cell niches, although a low oxygen tension is to be considered physiological for the environment where HSCs are maintained. HSCs were later shown to have the option of cycling in low oxygen, which steers this cycling to the maintenance of stem cell potential. Cell subsets capable of withstanding incubation in very low oxygen were also detected within leukemia cell populations, including chronic myeloid leukemia (CML). The oncogenetic Bcr/Abl protein is completely suppressed in these subsets, whereas Bcr/Abl messenger ribonucleic acid is not, indicating that CML cells resistant to low oxygen are independent of Bcr/Abl for persistence in culture but remain genetically leukemic. Accordingly, leukemia stem cells of CML selected in low oxygen are refractory to the Bcr/Abl inhibitor imatinib mesylate. Bcr/Abl protein suppression turned out to be actually determined when glucose shortage complicated the effects of low oxygen, indicating that ischemia-like conditions are the driving force of leukemia stem cell refractoriness to imatinib mesylate. These studies pointed to “ischemic” stem cell niches as a novel scenario for the maintenance of minimal residual disease of CML. A possible functional relationship of the “ischemic” with the “hypoxic” stem cell niche is discussed.
Keywords: hypoxia, ischemia, chronic myeloid leukemia, stem cell niche, leukemia stem cell, drug resistance
More than one stem cell niche: places where stem cell maintenance is modulated
The stem cell niches
The environment where hematopoietic stem cells (HSCs) are maintained was conceptually modeled and defined “stem cell niche” by Ray Schofield in 1978.1 The predicted function of stem cell niche was to maintain stem cell potential by preventing HSCs from commitment to clonal expansion and differentiation, so that if HSCs proliferate, they do as stem cells, ie, without losing stem cell potential (self-renewal). The model was in keeping with the findings that HSCs and hematopoietic progenitor cells (HPCs) are selectively compartmentalized, rather than randomly distributed, within bone marrow (BM), relatively less mature progenitors being preferentially located close to the bone surface and more mature progenitors near the central sinus.2–7 Transplanted hematopoietic cells were shown to behave likewise in the course of homing into BM.8
Hints about the actual association of HSCs to defined bone/BM areas were also published as early as 1978.9 However, the intercellular relationships leading to the definition of the so-called endosteal (or paraendosteal) niches were characterized much later.10–18 An alternative niche scenario emerged with the finding that, in BM, HSCs can also interact with the sinusoidal endothelium.19 Tissue areas characterized by such an occurrence were named vascular (or perivascular, or perisinusoidal) niches.19,20 It has been estimated that about 60% of BM HSCs are located around blood vessels.21
The relationship between endosteal and vascular niches represents a critical aspect of HSC maintenance and clonal expansion.21–24 Accumulated evidence supports the model that HSCs generate HPCs that migrate, as they expand clonally and differentiate, to blood vessels at the center of the BM cavity,8 where their transendothelial migration is driven together with that of mature cells, resulting in the release into blood circulation. Accordingly, the vascular niche seems to be the site where actively dividing HPCs are located.15,17,19 However, it has been shown that blood sinuses may be close to the bone surface25 and that HSCs may be adjacent to the endosteum as well as to endothelial cells in both BM and the spleen.19 This suggests that HSCs, rather than being compartmentalized in different niches, may be subjected to antagonistic stimuli acting within a continuum niche space. Properties and regulative role of different niches and the relative distribution of HSCs and HPCs are summarized in Figure 1. Many features of the niche environment at the tissue, cellular, intracellular, and molecular level have been characterized. To deepen these issues is beyond the scope of this paper, so we refer to some (among a number of them) excellent and extensive reviews,17,26–29 as well as to many references therein.
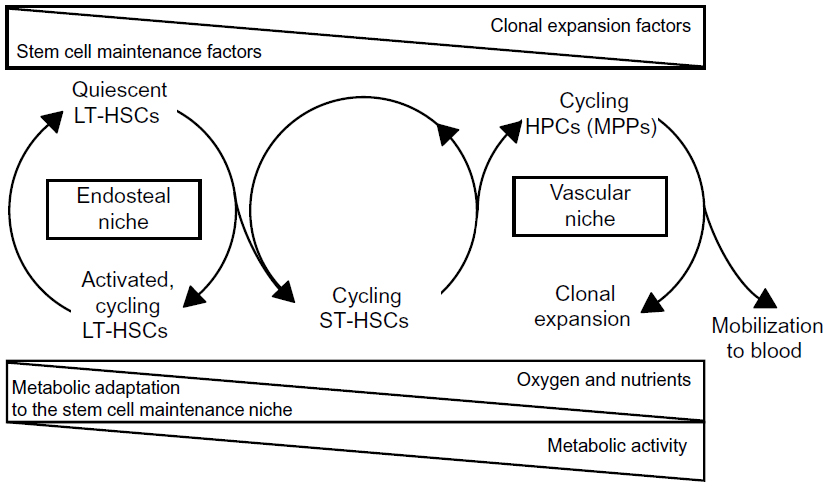 | Figure 1 Niche properties and distribution of hematopoietic stem and progenitor cell subsets. Top triangles: decrease or increase of environmental concentrations of stem/progenitor cell-extrinsic factors regulating the balance between stem cell maintenance and clonal expansion. Bottom triangles: nutrient supply to the different niche environments and metabolism in stem/progenitor cell subsets. |
The “hypoxic” stem cell niche
The first information about the effects of reduced oxygen tension on hematopoiesis came from the observation that the clonal growth of HSCs/HPCs is enhanced in an incubation atmosphere at ~7% O2 when compared to air (19%).30 One of the few later studies31–36 in the field showed that HSCs/HPCs residing within the compact bone, to the contrary of those located in paraendosteal regions and BM cavity, are resistant to ionizing radiations and sensitive to misonidazole, an enhancer of cytotoxicity in hypoxic cells.32
In the early 1990s, our laboratory was the first to address directly the effects of incubation in atmosphere at very low oxygen concentration (1%) on HSC maintenance.37 We found that 1) the overall murine hematopoiesis is inhibited at 1% oxygen, 2) the resistance of immature cells to 1% oxygen is higher the higher their level within the hematopoietic hierarchy, 3) short-term repopulating HSCs are maintained at 1% oxygen better than in air, so that 4) at 1% oxygen, HSCs are selected and enriched within the hematopoietic cell population.37–39 The effects of such stem cell-selective oxygen concentrations (1%–1.5%) were confirmed for human cells40–43 and extended to include long-term repopulating HSCs.44
On the basis of the in vitro data summarized above, we put forward the hypothesis of the existence in vivo of hypoxic HSC niches, where very low oxygen tensions favor maintenance and self-renewal of HSCs while restraining their clonal expansion, thereby protecting stem cell potential from exhaustion.37 A point crucial to this hypothesis is that oxygen tensions in vivo corresponding to those obtained in vitro using a 1%–1.5% oxygen-incubation atmosphere represent a physiological feature of stem cell niche, implying that an environment that is hypoxic for the bulk of hematopoietic cells is actually normoxic for HSCs. This issue is reviewed elsewhere.45–47 Accordingly, from here on, the “hypoxic” conditions suitable for HSC maintenance will be referred to as “low oxygen”, and the term “hypoxic” maintained only in conjunction with niche (hypoxic niche). Our hypothesis was strongly confirmed and enriched with experimental data, so that a clear relationship was established between the BM sites where oxygen tension is lowest and those where HSC maintenance and self-renewal are ensured (endosteal niches).17,21,48–56
HSC cycling at low oxygen
Low oxygen tension may ensure not only the best conditions for HSC maintenance but also their expansion with respect to time zero, provided an appropriate combination of stem cell-active factors is added to cultures.38 A corollary of these results is that the regulative role of tissue oxygen tension on hematopoiesis is integrated with those of other environmental factors, such as cytokines. The finding that the stem cell compartment can expand in low oxygen prompted us to verify whether stem cells can cycle in low oxygen, by treating cultures with 5-fluorouracil (5FU), a cell cycle-specific antiblastic drug. The maintenance of stem cell potential, assessed as described,57 in cultures incubated in low oxygen was highly sensitive to 5FU, which determined the suppression of two-thirds of this potential, while a third was 5FU-resistant. The most straightforward explanation of these results is that two-thirds of HSCs were cycling at the time of treatment, and that, in low oxygen, HSCs have the option between cycling and quiescence.57 These in vitro data support the compatibility of the “hypoxic” HSC niche environment in vivo with HSC cycling.56,58
Experiments carried out by PKH26 labeling and flow cytometry showed that, in low oxygen, stem cell potential is markedly enhanced in cells that have undergone one replication cycle, while it is rapidly lost when cycling is sustained beyond the first cycle. In air-incubated cultures, on the other hand, this loss occurs independently of the number of cycles accomplished.59 Thus, incubation in low oxygen allows but also restrains HSC cycling, resulting in optimal conditions to steer cycling in favor of the maintenance of stem cell potential. A reasonable comment on these findings is that HSC self-renewal is ensured immediately after their rescue from quiescence to cycling in low oxygen. In this perspective, low oxygen within the stem cell niche appears a factor driving dynamic stem cell maintenance, rather than a condition enforcing static stem cell maintenance via induction of quiescence. Thus, a location of stem cell niche in tissue areas at low oxygen tension contributes to define a scenario where the regulation of maintenance and cycling of HSCs relies not only on their direct contact with the niche structure or on the availability of soluble cytokines but also on the overall nature of the environment where the niche is located.56,58,60
Besides those on HSC cycling, hypoxia has other effects, eg, on gene expression and metabolic adaptation. A summary of these effects would exceed the limits of this review, so we refer to others for an extensive description and discussion of the topic.52–56,61–63
One more stem cell niche: a place where leukemia stem cells are drug-insensitive
“Hypoxia” and drug resistance of leukemia cells
The first attempt to establish the effects of low oxygen tension in the incubation atmosphere on (acute lymphoblastic) leukemia cells was published in 1978.64 However, in this case, like in that of normal hematopoiesis,30 the oxygen concentration used (~7%) was much higher than what is nowadays commonly defined “hypoxia”, according to either the threshold for degradation of the hypoxia-inducible factors (≥2%) or for pimonidazole staining (≤1.3%). In our laboratory, instead, a stem cell-selective oxygen concentration (see above) was used to investigate leukemia stem cells (LSCs) of chronic myeloid leukemia (CML), and in particular their sensitivity to the tyrosine-kinase inhibitor (TKi) imatinib mesylate (IM) (Gleevec®; Novartis, East Hanover, NJ, USA), the prototype TKi used for CML therapy. In these studies, the effects of an oxygen concentration as low as 0.1% on the maintenance of stem cell potential were evaluated as described,57,65,66 together with those on the expression of the oncogenetic Bcr/Abl fusion protein responsible for the pathogenesis of CML and main target of IM. Total cell number in cultures of several different CML cell populations (from stabilized lines or primary explants) was 1-log reduced with respect to time zero as a consequence of incubation in low oxygen. In the remaining cells, Bcr/Abl protein (as detected by immunoblotting) was completely suppressed, whereas Bcr/Abl messenger ribonucleic acid (mRNA) was not, indicating that CML cells capable of withstanding low oxygen are independent of Bcr/Abl for persistence in culture but remain genetically leukemic. Indeed, when these cells are transferred to growth-permissive incubation conditions in air, cultures are repopulated with Bcr/Abl protein-expressing cells. Thus, CML cell adaptation to low oxygen is paralleled by reversible Bcr/Abl protein suppression.67–69 Whether this suppression is driven via posttranscriptional, translational, and/or posttranslational mechanisms is currently under investigation in our laboratory.
When the stem cell potential of Bcr/Abl protein-negative cells was measured following transfer to growth-permissive conditions, this potential was totally or largely maintained, although it was exploited according to a different kinetics of repopulation of secondary cultures incubated in air. The maintenance of stem cell potential was also found completely insensitive to the treatment of “hypoxic” cultures with IM, apparently because its molecular target was suppressed. Such refractoriness to IM seems to indicate that Bcr/Abl protein suppression in hypoxia is actually complete (and that Western blotting does not overestimate Bcr/Abl protein suppression). Full sensitivity to IM, on the other hand, is rescued in the course of clonal expansion of LSCs following their transfer from low oxygen to air.67,68
The results summarized in this section were the first to link IM resistance to the suppression of Bcr/Abl protein, and this suppression in turn to the capacity of a subset of CML cells to survive in the “hypoxic” stem cell niches ensuring HSC maintenance. The question of the metabolic adaptation to low oxygen of this “LSC-like” subset of CML cells, such as that of HSCs, is beyond the scope of this review; we address elsewhere some points relevant to this issue.68,70
Another way to the IM resistance of CML cells
The Bcr/Abl-dependent or -independent mechanisms traditionally believed to determine resistance to IM are: 1) BCR/ABL mutations affecting IM binding to the Bcr/Abl protein; 2) BCR/ABL amplification or increased transcription, resulting in an increased Bcr/Abl protein expression level,71 a view that has been challenged;72,73 3) mutations not involving BCR/ABL and determining Bcr/Abl-independent survival and proliferation74,75 (mutation-driven loss of oncogene addiction); 4) enhanced activity of drug exporters in LSCs; and 5) LSC quiescence. Our studies66–69 led to the emergence of mechanism 6: Bcr/Abl protein suppression enforced in LSCs within the “hypoxic” stem cell niches and acting independently of whether LSCs are cycling or quiescent (see next paragraph). Thus, mechanism 6 relies on the primary resistance (more appropriately referred to as refractoriness) of LSCs to IM due to the lack of its molecular target, a property which characterizes LSCs in relation to their capacity to home stem cell niches. It is worth stressing here the “simplicity” of mechanism 6, consisting in the fact that to explain IM resistance, it does not need to postulate secondary mutations occurring in a CML cell subset. Mechanism 6 rather hints at a phenotypical adaptation of LSCs, which is indeed fully reversible when microenvironmental changes allow reexpression of Bcr/Abl protein. A marked heterogeneity of phenotype among genetically identical cells has been shown in many situations,76 implying that leukemia cell populations, including cell lines, represent a continuum of phenotypes with different survival, growth, and differentiation properties.
How LSCs of CML behave within the “hypoxic” niche
CML cells capable of withstanding low oxygen apparently lose their growth advantage over normal hematopoiesis as a consequence of the forced suppression of Bcr/Abl-dependent signaling. However, there is no reason to suppose that under these conditions, LSCs are also deprived of the physiological properties of HSCs, such as the capacity to survive and cycle in low oxygen. Indeed, Bcr/Abl protein-negative LSCs were found to be in significant part sensitive to 5FU (Giuntoli S, Tanturli M, Dello Sbarba P, unpublished data, 2010). Thus, in low oxygen, LSCs of CML are likely to return to an HSC-like phenotype, losing the oncogene addiction (ie, the dependence on oncogene-conferred survival and proliferation signals) of the bulk of the leukemia population and possibly rescuing the dependence on physiological signals generated in the microenvironment.77,78 A crucial issue is that, given the unstable genotype of CML cells, LSC cycling in low oxygen sustains not only dynamic stem cell maintenance but also neoplastic progression, as transmission of mutations to progeny requires cell cycling. In this context, progression may well include the acquisition of secondary mutation/s, listed as case mechanism 1 in the section “Another way to the IM resistance of CML cells.” Thus, cycling of LSC in low oxygen, being like that of HSCs most probably coupled to self-renewal but not clonal expansion,60 results in the long-term maintenance of subclinical yet progressing disease. We define such a scenario as dynamic maintenance of minimal residual disease (MRD). We do not refer here, of course, to MRD that remains detectable forever in the absence of treatment as well as of relapse, and therefore actually corresponds to the clinical, if not biological, cure of disease. Rather, dynamic MRD describes the maintenance of an unstable equilibrium that is eventually unbalanced to lead to relapse.76 On this basis, one can argue that cycling of Bcr/Abl protein-negative LSCs in low oxygen explains better than LSC quiescence the combination of neoplastic progression with the maintenance of refractoriness to TKi.
All these factors led to the important conclusion that the refractoriness to TKi exhibited by LSCs in low oxygen is the effect of an adaptive phenotypical modulation that is reversibly enforced under environmental metabolic pressure in potentially all LSCs (and not in a minor subset of them), rather than a genetically blocked property that is stably transmitted to a fraction of the progeny as a permanent character. This is in keeping with the chiaroscuro model proposed for the control of normal hematopoiesis,79 based on a reversible transition between the stem and progenitor cell phenotypes. The relevance of this model to the interpretation of our experimental data is discussed elsewhere.66,68 However, it is worth pointing out here that our view of an environment-driven bouncing between the LSC and the leukemia progenitor cell (LPC) phenotypes is compatible with the findings of equal or higher Bcr/Abl expression in primary LSCs when compared to LPCs, as well as of the TKi resistance of CML progenitors shown to be Bcr/Abl-positive by fluorescence in situ hybridization or polymerase chain reaction.72,73,80–83 We believe indeed these progenitors to be, by and large, Bcr/Abl mRNA-positive/Bcr/Abl protein-negative, and the reversible chiaroscuro modulation of Bcr/Abl protein expression to occur only under metabolic pressure in selected tissue areas. This represents a simple answer to the question whether TKis inhibit Bcr/Abl kinase activity in CML cells responsible for MRD.84,85 In conclusion, if the only feature that differentiates LSCs of CML from HSCs is actually the presence of BCR/ABL gene, not Bcr/Abl protein function, then efforts to enhance inhibition of Bcr/Abl protein enzymatic activity via the development of more potent inhibitors appear useless if one wants to target LSCs.
The “ischemic” leukemia/cancer stem cell niche
The mechanisms underlying the fluctuations of Bcr/Abl protein expression observed in low oxygen were deepened by varying time-zero cell density and glucose concentration in CML cell cultures.68 We found that in low oxygen, although cell growth is markedly inhibited in comparison to air, the cell population is nevertheless capable of expanding significantly with respect to time zero, in keeping with the aforementioned capacity of normal hematopoietic and leukemic cells to cycle in hypoxia.57,59,65 On the other hand, glucose availability emerged as the crucial condition modulating the size of the CML cell population in low oxygen as well as the level of Bcr/Abl protein expression. It turned out, indeed, that culture expansion is possible in low oxygen until glucose is no longer available in culture medium. This is a frequent occurrence, due to the enhanced glucose-consumption rate (the Pasteur effect) typical of cell metabolism in low oxygen. After the onset of glucose shortage, the number of viable cells decreases, leading to the selection of a relatively small subset of cells capable of withstanding oxygen/glucose shortage but not of expanding under Bcr/Abl signaling. Thus, it is glucose shortage that ultimately drives Bcr/Abl protein suppression in low oxygen and the consequent acquisition of LSC refractoriness to IM.
On the basis of all this, we hypothesized66,68 that “ischemic” stem cell niches are established where glucose gets close to exhaustion within the core of the “hypoxic” niches. Such an occurrence is very likely to characterize the hyperplastic BM of CML patients. Indeed, tissue conditions resembling ischemia rather than hypoxia, where glucose supply in addition to that of oxygen is insufficient for long enough to affect cell viability, have been shown in neoplasias.86,87 On the other hand, the existence of “ischemic” stem cell niches appears also compatible with normal BM. In this respect, extremely relevant information came from Winkler et al88 and Lévesque and Winkler.89 They showed that blood perfusion rather than oxygen diffusion controls the hierarchical distribution of HSCs/HPCs in normal BM. With modern technologies, Lévesque, Winkler, and coworkers elaborated on a number of classical studies of blood and nutrient supply to BM.90–94 They hypothesized that the niches hosting the most primitive HSCs reside in tissue areas at very low blood perfusion. This would imply very low concentrations of blood-borne nutrients and oxygen, as well as the lowest dilution (by blood flow) of locally secreted soluble factors supporting HSC maintenance within the niche.87,88 On this basis, CML cells selectively maintained within the hypoxic/ischemic niches where HSCs reside may well correspond to the LSCs responsible for MRD.66,68
The “ischemic” versus the “hypoxic” LSC niche and cancer stem cell models
Figure 2 depicts the relationship of the “ischemic” to the “hypoxic” stem cell niche in CML. The “ischemic” niche would represent the site of Bcr/Abl-independent self-renewal as well as of the maintenance of dynamic, IM-resistant MRD. Moving from these sites to neighboring areas where oxygen tension is still low but glucose concentration is higher (the “hypoxic” niche), LSCs would undergo the rescue of Bcr/Abl protein expression and thereby an enhanced Bcr/Abl-stimulated self-renewal. Eventually, clonal expansion would be triggered in better-oxygenated tissue areas (the vascular niche), from which relapse of disease would be driven.
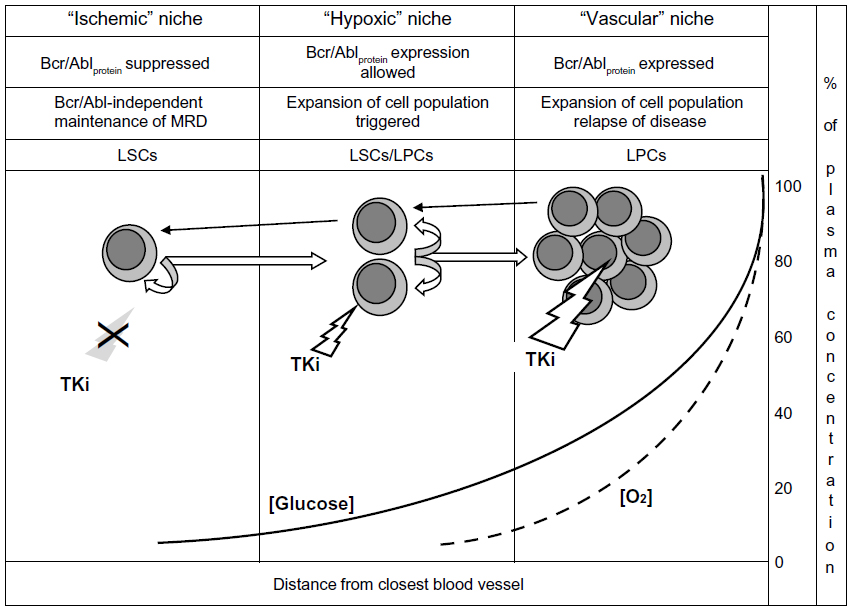 | Figure 2 Nutrient supply and niche homing of functional stem and progenitor cell subsets in chronic myeloid leukemia. Straight white arrows: hierarchical top-down phenotype shift (describing biology of disease from onset to clinical level). Curved arrows: self-renewal. Black arrows: environment-enforced bottom-up phenotype shift (according to the chiaroscuro model). Solid and dashed lines: decay of glucose concentration or oxygen tension, respectively (expressed as percentage of value within blood vessel), in function of distance from the closest source of blood supply and of the definition of individual niche types. White bolt: TKi effective. Grey bolt/X: TKi ineffective. Note the different sensitivity of cell subsets to TKi treatment in relation to Bcr/Abl protein expression. |
Two “alternative” models have been proposed to define the origin of cancer stem cells: the cancer stem cell in normal stem cell and the cancer stem cell in progenitor cell models.95 We propose that CML fits both models, the former describing Bcr/Abl protein-negative LSCs capable of Bcr/Abl-independent self-renewal and the latter self-renewing cells where Bcr/Abl protein expression is rescued (LSCs/LPCs in Figure 2). We believe that quiescent LSCs represent just a subset of cells exhibiting the LSC phenotype83 and that the LSCs/LPCs phenotype fits at best with that of the CML cells, characterized by a relatively low sensitivity to IM described in some studies.72,73,80–83,96 Figure 2 also shows that a stochastically determined minority of Bcr/Abl protein-expressing cells would always maintain the capacity to undergo the environment-enforced, bottom-up chiaroscuro shift, returning to the Bcr/Abl protein-negative LSC phenotype capable of sustaining MRD. All this is very well in keeping with the current view of CML as a stem cell-derived but progenitor cell-driven disease.97
Conclusion
The experimental data summarized in the previous paragraph indicated that, to ensure efficient maintenance of LSCs capable of sustaining MRD, the restrictions applied upon the leukemia-regeneration machinery by low oxygen tension are not sufficient unless glucose shortage occurs. Such a scenario made it necessary to introduce the “ischemic” stem cell niche concept. The proposed model suggests that Bcr/Abl protein is suppressed when and where glucose shortage occurs, which is facilitated in “hypoxia” due to the Pasteur effect. CML cell capacity to withstand “ischemia” implies the bypassing of requirements for Bcr/Abl signaling and, as far as LSCs are concerned, a sort of “reversion” to a normal stem cell phenotype, also resistant to oxygen and glucose shortage. On the other hand, in “hypoxic” but not “ischemic” niches, Bcr/Abl protein expression (or reexpression following suppression in ischemia) is allowed.
Our model allows, on one hand, to include the more immature CML cell compartments within the range of effectiveness of IM, as IM would affect LSC/LPC in the same way it affects all other leukemic cells, as long as they express Bcr/Abl protein (ie, within the vascular or the “hypoxic” stem cell niches). On the other hand, our model predicts that refractoriness to IM occurs in LSCs that are devoid of Bcr/Abl protein-dependent growth advantage over normal HSCs (ie, within the “ischemic” stem cell niches), independently of their cycling or quiescence. This scenario would confer upon LSCs the capacity to divide and even increase numerically under IM treatment, but not to outnumber HSCs,98 which is most probably crucial to the maintenance of disease at subclinical levels. The model explains well the observation that relapse of disease upon discontinuation of IM treatment often occurs in patients where there is no evidence of “molecular” resistance to IM (ie, due to changes of Bcr/Abl molecule), and who therefore typically respond well to the reintroduction of IM after relapse.99 Such an outcome would be impossible if a secondary mutation of IM binding to Bcr/Abl protein in a cell clone were the cause of relapse. On the contrary, the rescue of sensitivity to IM is obviously compatible with the rescue of expression of wild-type Bcr/Abl protein in CML cells that expand when disease is to return to a clinical phase.
One of the most relevant conclusions we can draw from all this is at the translational level. The data that led us to put forward the “ischemic” stem cell niche model also definitely led us to predict that even the next generations of Bcr/Abl-active TKis will fail to suppress MRD. This prediction is already progressing towards confirmation, as second-generation Bcr/Abl inhibitors do not seem to ensure CML suppression, although they exhibit an enhanced action on the bulk of the CML cell population.100
Acknowledgments
This research was supported by the following sources of funding: Istituto Toscano Tumori (ITT); Istituto Superiore di Sanità (ISS) – national program “Stem Cells” (grant #CS64); Ministero della Salute (grant RF-TOS-2008-1163728); Regione Toscana – Programma per la Ricerca in Materia di Salute; Associazione Italiana per la Ricerca sul Cancro (AIRC; grants IG5220 and IG13466); Federazione Italiana per la Ricerca sul Cancro (FIRC); Associazione Italiana per la Lotta contro le Leucemie e i Linfomi (AIL; Sezione di Prato); Fondazione Oretta Bartolomei-Corsi; Fondazione Cassa di Risparmio di Volterra. The important intellectual contribution of Massimo Olivotto (professor emeritus of our department) is also acknowledged.
Disclosure
The authors report no conflicts of interest in this work.
References
Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7–25. | |
Lord BI, Hendry JH. The distribution of haemopoietic colony-forming units in the mouse femur, and its modification by x rays. Br J Radiol. 1972;45:110–115. | |
Lord BI, Testa NG, Hendry JH. The relative spatial distributions of CFU-S and CFU-C in the normal mouse femur. Blood. 1975;46:65–72. | |
Shackney SE, Ford SS, Wittig AB. Kinetic-microarchitectural correlations in the bone marrow of the mouse. Cell Tissue Kinet. 1975;8: 505–516. | |
Mason TM, Lord BI, Hendry JH. The development of spatial distributions of CFU-S and in-vitro CFU-C in femora of mice of different ages. Br J Haematol. 1989;73:455–461. | |
Lord BI. The architecture of bone marrow cell populations. Int J Cell Cloning. 1990;8:317–331. | |
Haylock DN, Williams B, Johnston HM, et al. Hemopoietic stem cells with higher hemopoietic potential reside at the bone marrow endosteum. Stem Cells. 2007;25:1062–1069. | |
Nilsson SK, Johnston HM, Coverdale JA. Spatial localization of transplanted hemopoietic stem cells: inferences for the localization of stem cell niches. Blood. 2001;97:2293–2299. | |
Gong JK. Endosteal marrow: a rich source of hematopoietic stem cells. Science. 1978;199:1443–1445. | |
Taichman RS, Emerson SG. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med. 1994;179:1677–1682. | |
Taichman RS, Reilly MJ, Emerson SG. The hematopoietic microenvironment: osteoblasts and the hematopoietic microenvironment. Hematology. 2000;4:421–426. | |
Heissig B, Hattori K, Dias S, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–637. | |
Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425: 836–841. | |
Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. | |
Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. | |
Zhu J, Emerson SG. A new bone to pick: osteoblasts and the haematopoietic stem-cell niche. Bioessays. 2004;26:595–599. | |
Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116: 1195–1201. | |
Bianco P. Bone and the hematopoietic niche: a tale of two stem cells. Blood. 2011;117:5281–5288. | |
Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. | |
Kopp HG, Avecilla ST, Hooper AT, Rafii S. The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology (Bethesda). 2005;20:349–356. | |
Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. | |
Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. | |
Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. | |
Ellis SL, Grassinger J, Jones A, et al. The relationship between bone, hemopoietic stem cells, and vasculature. Blood. 2011;118: 1516–1524. | |
Bourke VA, Watchman CJ, Reith JD, Jorgensen ML, Dieudonnè A, Bolch WE. Spatial gradients of blood vessels and hematopoietic stem and progenitor cells within the marrow cavities of the human skeleton. Blood. 2009;114:4077–4080. | |
Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood. 2005;105: 2631–2639. | |
Urao N, Ushio-Fukai M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic Biol Med. 2013;54:26–39. | |
Nakamura-Ishizu A, Suda T. Hematopoietic stem cell niche: an interplay among a repertoire of multiple functional niches. Biochim Biophys Acta. 2013;1830:2404–2409. | |
Matsumoto A, Nakayama KI. Role of key regulators of the cell cycle in maintenance of hematopoietic stem cells. Biochim Biophys Acta. 2013;1830:2335–2344. | |
Bradley TR, Hodgson GS, Rosendaal M. The effect of oxygen tension on haemopoietic and fibroblast cell proliferation in vitro. J Cell Physiol. 1978;97:517–522. | |
Rich IN, Kubanek B. The effect of reduced oxygen tension on colony formation of erythropoietic cells in vitro. Br J Haematol. 1982;52: 579–588. | |
Allalunis MJ, Chapman JD, Turner AR. Identification of a hypoxic population of bone marrow cells. Int J Radiat Oncol Biol Phys. 1983;9: 227–232. | |
Lu L, Broxmeyer HE. Comparative influences of phytohemoagglutinin-stimulated leukocyte conditioned medium, hemin, prostaglandin E, and low oxygen tension on colony formation by erythroid progenitor cells in normal human bone marrow. Exp Hematol. 1985;13:989–993. | |
Allalunis-Turner MJ, Koch CJ, Chapman JD. Growth of murine bone marrow under various oxygen conditions in media buffered with HEPES. Int J Cell Cloning. 1987;5:315–321. | |
Ishikawa Y, Ito T. Kinetics of hemopoietic stem cells in a hypoxic cultures. Eur J Haematol. 1988;40:126–129. | |
Broxmeyer HE, Cooper S, Lu L, Miller ME, Langfeld CD, Ralph P. Enhanced stimulation of human bone marrow macrophage colony formation in vitro by recombinant human macrophage colony-stimulating factor in agarose medium and at low oxygen tension. Blood. 1990;76:323–329. | |
Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82: 2031–2037. | |
Ivanovic´ Z, Bartolozzi B, Bernabei PA, et al. Incubation of murine bone marrow cells in hypoxia ensures the maintenance of marrow-repopulating ability together with the expansion of committed progenitors. Br J Haematol. 2000;108:424–429. | |
Kovacevic´-Filipovic´ M, Petakov M, Hermitte F, et al. Interleukin-6 (IL-6) and low O2 tension (1%) synergize to improve the maintenance of hematopoietic stem cells (pre-CFC). J Cell Physiol. 2007;212:68–75. | |
Cipolleschi MG, D’Ippolito G, Bernabei PA, et al. Severe hypoxia enhances the formation of erythroid bursts from human cord blood cells and the maintenance of BFU-E in vitro. Exp Hematol. 1997;25:1187–1194. | |
Ivanovic´ Z, Dello Sbarba P, Trimoreau F, Faucher JL, Praloran V. Primitive human HPCs are better maintained and expanded in vitro at 1 percent oxygen than at 20 percent. Transfusion. 2000;40:1482–1488. | |
Ivanovic´ Z, Hermitte F, Brunet de la Grange P, et al. Simultaneous maintenance of human cord blood SCID-repopulating cells and expansion of committed progenitors at low O2 tension (3%). Stem Cells. 2004;22:716–724. | |
Hammoud M, Vlaski M, Duchez P, et al. Combination of low O2 tension and mesenchymal stromal cells during culture of cord blood CD34(+) cells improves the maintenance and proliferative capacity of hematopoietic stem cells. J Cell Physiol. 2012;227:2750–2758. | |
Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest. 2003;112:126–135. | |
Ivanovic´ Z. Hypoxia or in situ normoxia: the stem cell paradigm. J Cell Physiol. 2009;219:271–275. | |
Guitart AV, Hammoud M, Dello Sbarba P, Ivanovic´ Z, Praloran V. Slow-cycling/quiescence balance of hematopoietic stem cells is related to physiological gradient of oxygen. Exp Hematol. 2010;38:847–851. | |
Ivanovic´ Z. Respect the anaerobic nature of stem cells to exploit their potential in regenerative medicine. Regen Med. 2013;8:1–4. | |
Eliasson P, Karlsson R, Jönsson JI. Hypoxia expands primitive hematopoietic progenitor cells from mouse bone marrow during in-vitro culture and preserves the colony-forming ability. J Stem Cells. 2006;1: 247–257. | |
Levesque JP, Winkler IG, Hendy J, et al. Hematopoietic progenitor cell mobilization results in hypoxia with increased hypoxia-inducible transcription factor-1a and vascular endothelial growth factor A in bone marrow. Stem Cells. 2007;25:1954–1965. | |
Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104:5431–5436. | |
Kubota Y, Takubo K, Suda T. Bone marrow long label-retaining cells reside in the sinusoidal hypoxic niche. Biochem Biophys Res Commun. 2008;366:335–339. | |
Mohyeldin A, Garzón-Muvdi T, Quiñones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7:150–161. | |
Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol. 2010;10:201–209. | |
Eliasson P, Jönsson JI. The hematopoietic stem cell niche: low in oxygen but a nice place to be. J Cell Physiol. 2010;222:17–22. | |
Simsek T, Kocabas F, Zheng J, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. | |
Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310. | |
Cipolleschi MG, Rovida E, Ivanovic´ Z, Praloran V, Olivotto M, Dello Sbarba P. The expansion of murine bone marrow cells preincubated in hypoxia as an in vitro indicator of their marrow-repopulating ability. Leukemia. 2000;14:735–739. | |
Eliasson P, Rehn M, Hammar P, et al. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term-reconstituting hematopoietic stem cells during in vitro culture. Exp Hematol. 2010;38:301–310. | |
Ivanovic´ Z, Belloc F, Faucher JL, Cipolleschi MG, Praloran V, Dello Sbarba P. Hypoxia maintains and interleukin-3 reduces the pre-colony-forming cell potential of dividing CD34(+) murine bone marrow cells. Exp Hematol. 2002;30:67–73. | |
Hermitte F, Brunet de la Grange P, Belloc F, Praloran V, Ivanovic Z. Very low O2 concentration (0.1%) favors G0 return of dividing CD34þ cells. Stem Cells. 2006;24:65–73. | |
Warr MR, Passegué E. Metabolic makeover for HSCs. Cell Stem Cell. 2013;12:1–3. | |
Vacanti NM, Metallo CM. Exploring metabolic pathways that contribute to the stem cell phenotype. Biochim Biophys Acta. 2013;1830: 2361–2369. | |
Takubo K, Suda T. Roles of the hypoxia response system in hematopoietic and leukemic stem cells. Int J Hematol. 2012;95:478–483. | |
Smith SD, Uyeki EM, Lowman JT. Colony formation in vitro by leukemic cells in acute lymphoblastic leukemia (ALL). Blood. 1978;52: 712–718. | |
Giuntoli S, Rovida E, Gozzini A, et al. Severe hypoxia defines heterogeneity and selects highly immature progenitors within clonal erythroleukaemia cells. Stem Cells. 2007;25:1119–1125. | |
Cipolleschi MG, Rovida E, Dello Sbarba P. The culture-repopulating ability assays and incubation in low oxygen: a simple way to test drugs on leukaemia stem or progenitor cells. Curr Pharm Des. 2013;19:5374–5383. | |
Giuntoli S, Rovida E, Barbetti V, Cipolleschi MG, Olivotto M, Dello Sbarba P. Hypoxia suppresses BCR/Abl and selects imatinib-insensitive progenitors within clonal CML population. Leukemia. 2006;20:1291–1293. | |
Giuntoli S, Tanturli M, Di Gesualdo F, Barbetti V, Rovida E, Dello Sbarba P. Glucose availability in hypoxia regulates the selection of chronic myeloid leukaemia progenitor subsets with different resistance to imatinib-mesylate. Haematologica. 2011;96;204–212. | |
Tanturli M, Giuntoli S, Barbetti V, Rovida E, Dello Sbarba P. Hypoxia selects bortezomib-resistant stem cells of chronic myeloid leukemia. PLoS One. 2011;6:e17008. | |
Olivotto M, Dello Sbarba P. Environmental restrictions within tumor ecosystems select for a convergent, hypoxia-resistant phenotype of cancer stem cells. Cell Cycle. 2008;7:176–187. | |
Barnes DJ, Palaiologou D, Panousopoulou E, et al. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65:8912–8919. | |
Modi H, McDonald T, Chu S, Yee JK, Forman SJ, Bhatia R. Role of BCR/ABL gene expression levels in determining the phenotype and imatinib sensitivity of transformed human hematopoietic cells. Blood. 2007;109:5411–5421. | |
Kumari A, Brendel C, Hochhaus A, Neubauer A, Burchert A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood. 2012;119:530–539. | |
Sharma SV, Gajowniczek P, Way IP, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10:425–435. | |
Felsher DW. Reversing cancer from inside and out: oncogene addiction, cellular senescence, and the angiogenic switch. Lymphat Res Biol. 2008;6:149–154. | |
Tang M, Foo J, Gönen M, Guilhot J, Mahon FX, Michor F. Selection pressure exerted by imatinib therapy leads to disparate outcomes of imatinib discontinuation trials. Haematologica. 2012;97:1553–1561. | |
Donato NJ, Wu JY, Stapley J, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–698. | |
Chu S, Holtz M, Gupta M, Bhatia R. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34 cells. Blood. 2004;103:3167–3174. | |
Quesenberry PJ, Colvin GA, Lambert JF. The chiaroscuro stem cell: a unified stem cell theory. Blood. 2002;100:4266–4271. | |
Bhatia R, Holtz M, Niu N, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101:4701–4707. | |
Jiang X, Zhao Y, Smith C, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–935. | |
Abe A, Minami Y, Hayakawa F, et al. Retention but significant reduction of BCR-ABL transcript in hematopoietic stem cells in chronic myelogenous leukemia after imatinib therapy. Int J Hematol. 2008;88:471–475. | |
Graham SM, Jørgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. | |
Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2010;121: 396–409. | |
Perl A, Carroll M. BCR-ABL kinase is dead; long live the CML stem cell. J Clin Invest. 2011;121:22–25. | |
Tannock IF. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br J Cancer. 1968;22:258–273. | |
Vaupel P, Schlenger K, Knoop C, Höckel M. Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res. 1991;51:3316–3322. | |
Winkler IG, Barbier V, Wadley R, Zannettino AC, Williams S, Lévesque JP. Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood. 2010;116: 375–385. | |
Lévesque JP, Winkler IG. Hierarchy of immature hematopoietic cells related to blood flow and niche. Curr Opin Hematol. 2011;18: 220–225. | |
Grant WC, Root WS. The relation of O2 in bone marrow blood to post-hemorrhagic erythropoiesis. Am J Physiol. 1947;150:618–627. | |
Branemark PI. Experimental investigation of microcirculation in bone marrow. Angiology. 1961;12:293–305. | |
Lichtman MA. The ultrastructure of the hemopoietic environment of the marrow: a review. Exp Hematol. 1981;9:391–410. | |
Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO2 distributions in the bone marrow hematopoietic compartment. I: Krogh’s model. Biophys J. 2001;81:675–684. | |
Adams GB. Stem cells hold their breath. Blood. 2010;116:307–308. | |
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. | |
Zhang H, Li S. Molecular mechanisms for survival regulation of chronic myeloid leukemia stem cells. Protein Cell. 2013;4:186–196. | |
Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer. 2008;8:341–350. | |
Tomasetti C. A new hypothesis: imatinib affects leukemic stem cells in the same way it affects all other leukemic cells. Blood Cancer. 2011;1:e19. | |
Mahon FX, Réa D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–1035. | |
Konig H, Holtz M, Modi H, et al. Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia. 2008;22:748–755. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.