Back to Archived Journals » Oncolytic Virotherapy » Volume 4
Oncolytic virotherapy using herpes simplex virus: how far have we come?
Authors Sokolowski N, Rizos H, Diefenbach R ![]()
Received 21 July 2015
Accepted for publication 16 October 2015
Published 25 November 2015 Volume 2015:4 Pages 207—219
DOI https://doi.org/10.2147/OV.S66086
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Faris Farassati
Nicolas AS Sokolowski,1 Helen Rizos,2 Russell J Diefenbach1
1Centre for Virus Research, Westmead Millennium Institute for Medical Research, The University of Sydney, 2Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University, NSW, Australia
Abstract: Oncolytic virotherapy exploits the properties of human viruses to naturally cause cytolysis of cancer cells. The human pathogen herpes simplex virus (HSV) has proven particularly amenable for use in oncolytic virotherapy. The relative safety of HSV coupled with extensive knowledge on how HSV interacts with the host has provided a platform for manipulating HSV to enhance the targeting and killing of human cancer cells. This has culminated in the approval of talimogene laherparepvec for the treatment of melanoma. This review focuses on the development of HSV as an oncolytic virus and where the field is likely to head in the future.
Keywords: herpes simplex virus, cancer, immunity, combination therapy, oncolysis
Introduction
Oncolytic virotherapy began with the observation that patients with cancer and a viral infection could, on the rare occasion, briefly enter cancer remission.1 Although such cases were noted as early as the mid-1800s (before viruses were known to exist), it was not until the 1950s and 1960s that an earnest effort was mounted to put this observation into practice. Clinical trials using different human viruses including hepatitis,2 Epstein–Barr,3 adenovirus,4 and rabies5 were undertaken with mixed results. None of the studies were able to overcome the risk that infecting humans with wild type virus posed. Attempts were also made to identify and adapt animal viruses for oncolytic virotherapy. Animal viruses that lacked pathogenicity in normal human tissue, yet still retained a propensity for replicating in human cancer cells, were identified in order to circumvent the side effects of human oncolytic viruses. However, the potential for these viruses to alter their tropism to normal human cells was an impediment that halted the majority of virotherapy research with animal viruses.6
Interest in oncolytic virotherapy was reborn in the 1990s with the advent of genetic engineering. Combined with the extensive knowledge that had been accumulated on viruses, genetic engineering allowed the generation of viruses with specific attenuations. The deletion of viral genes that, while essential for replication in normal tissue, were not required for replication in cancerous cells, allowed viruses to be retargeted toward cancer cells.
Herpes simplex virus (HSV)7 was identified as a highly attractive candidate for oncolytic virotherapy due to several characteristics including: 1) a naturally cytolytic life cycle with the ability to infect a broad range of cell types; 2) a highly prevalent human pathogen which in the vast majority of cases causes a self-limiting disease that can be treated with antivirals in life-threatening cases; 3) a very large genome, with many nonessential genes that can be replaced with foreign genes; and 4) an envelope with separate attachment and fusion glycoproteins which can be modified for improved cancer cell targeting.
The initial focus of oncolytic HSV (oHSV) virotherapy involved demonstrating the safety of oHSVs for the treatment of cancer. Subsequent research has focused on confirming that oHSV mutants can be effective in various cancers and enhanced by targeting areas such as host immunity, tumor microenvironment, and cancer-specific cell functions, either by insertion of human genes into the oHSV and/or combining with other therapies. This has culminated in the approval by the US Food and Drug Administration (FDA) of the oHSV talimogene laherparepvec (T-VEC) for the treatment of melanoma.8–10 This review highlights the development of oHSV for the treatment of cancers and potential improvements for the activity and use of oHSV. For a perspective, the reader should consult a number of recent reviews on the field of oncolytic virotherapy as a whole.11–13
Generation of oHSVs
Development of oHSVs involved initial deletion of a single viral gene and subsequently multiple viral gene deletions and modifications. Key examples of this development are discussed further. A summary of the viral genes modified in oHSVs and the functions of viral proteins encoded by these genes is provided in Table 1. A comprehensive list of deletion mutation oHSVs can be found in Table 2 and altered gene regulation or receptor retargeted oHSVs in Table 3.
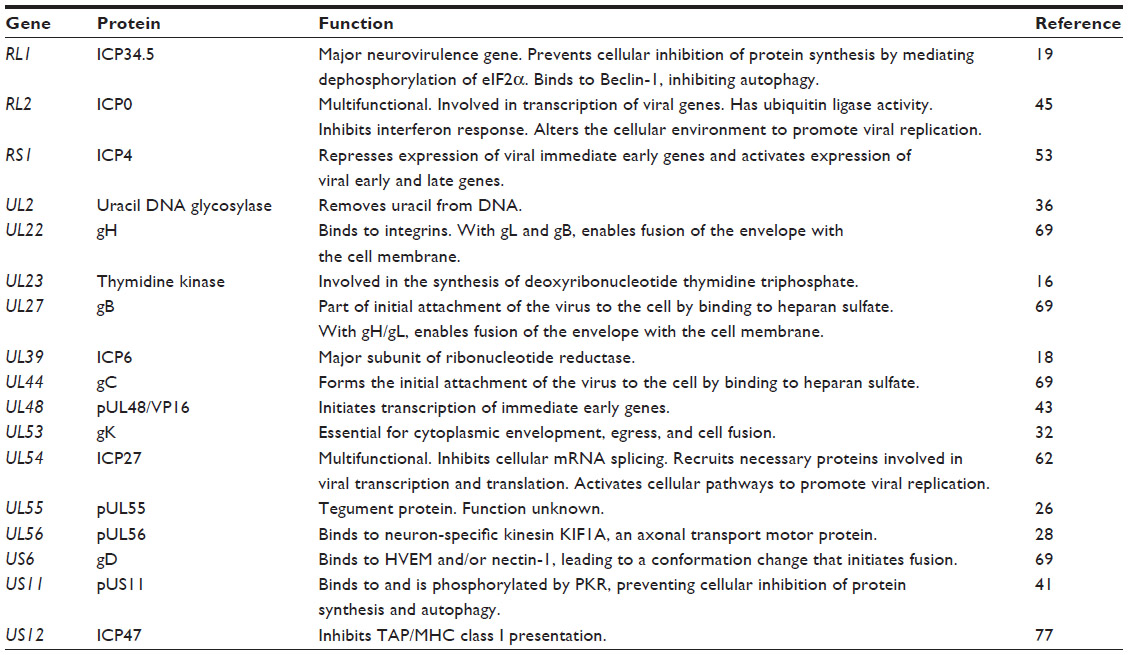 | Table 1 Summary of viral genes modified in oHSVs |
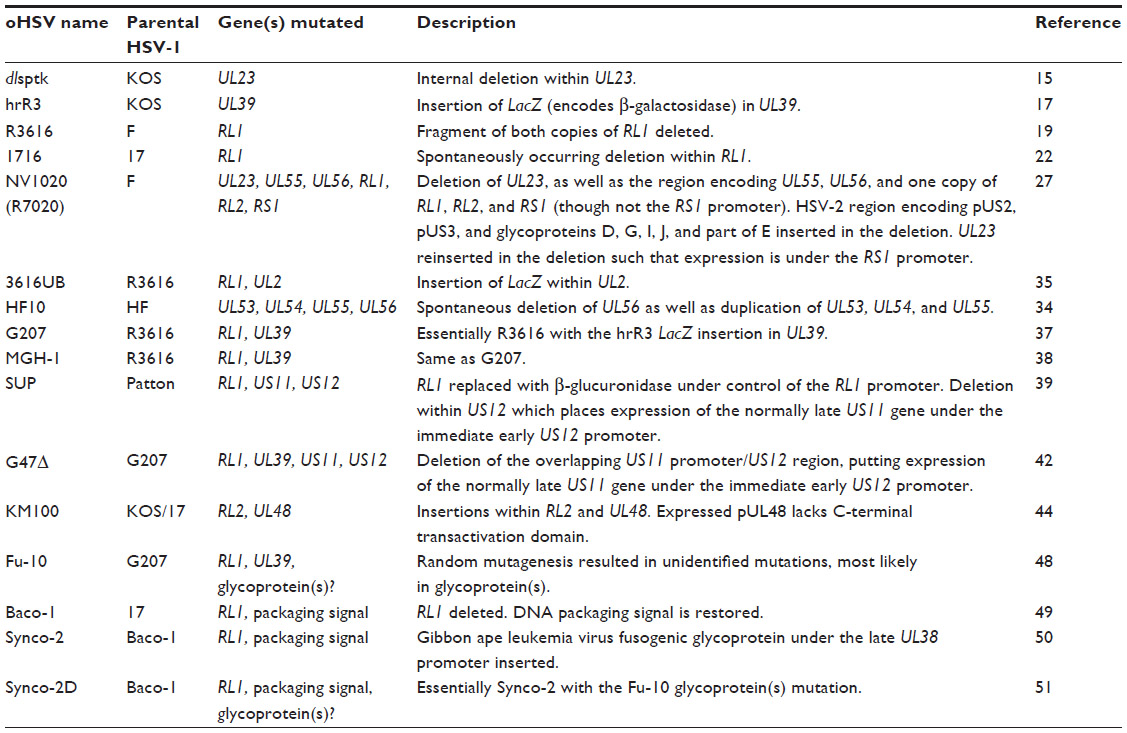 | Table 2 Summary of oHSVs incorporating viral gene mutations |
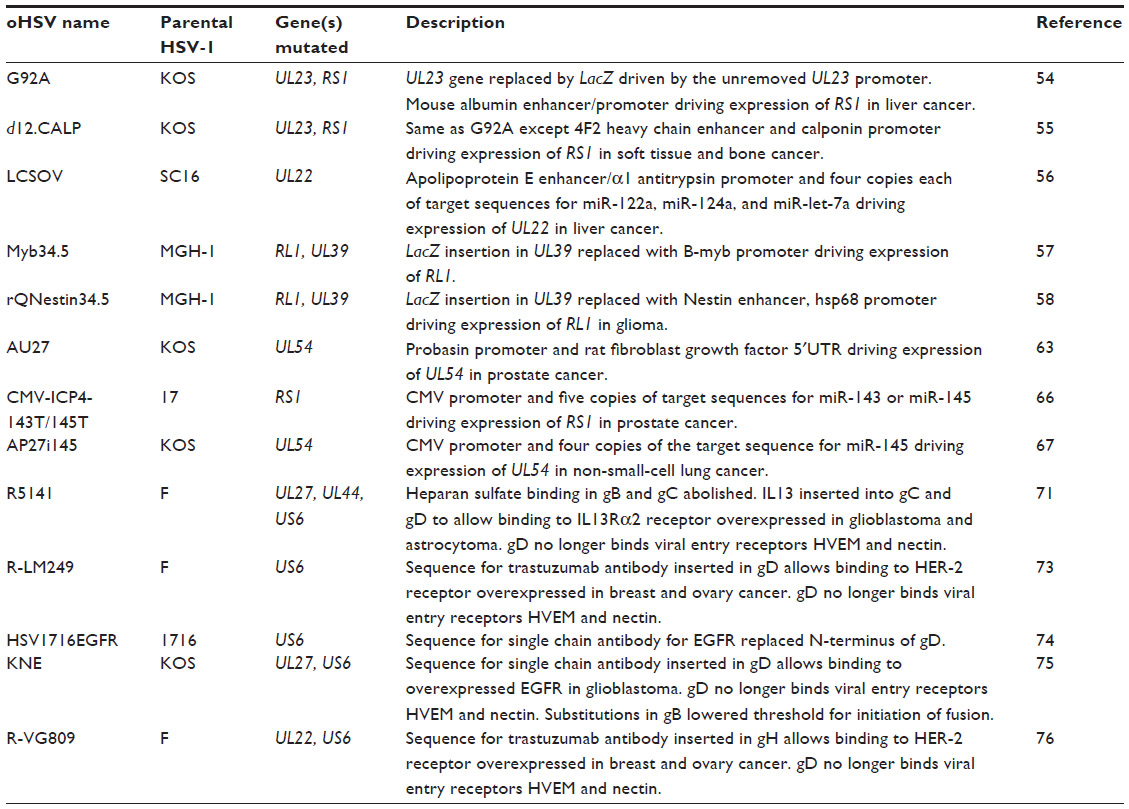 | Table 3 Summary of oHSVs incorporating gene regulated or receptor retargeting mutations |
Deletion mutants
HSV (types 1 and 2) infects nondividing cells such as neurons and therefore encodes viral homologues of various nucleotide metabolism and DNA synthesis enzymes.7 The dlsptk HSV-1 mutant contains a deletion within the unique long (UL)23 gene which encodes the viral homologue of thymidine kinase (TK).14–16 The hrR3 HSV mutant contains a LacZ (encodes β-galactosidase) insertion mutation of the HSV-1 large subunit of ribonucleotide reductase (RR), also designated infected cell protein (ICP) 6, encoded by gene UL39.17,18 Consequently, dlsptk and hrR3 HSV mutants only replicate in dividing cancer cells which overexpress TK and RR. Deletion of viral TK though does result in resistance of HSV dlsptk to current nucleoside analog inhibitor antivirals (eg, acyclovir), which are activated by the action of viral TK.14
The HSV-1 R3616 mutant contains deletions within both copies of the major neurovirulence determinant gene repeat long (RL)1 (encodes neurovirulence determinant ICP34.5).19 Host protein kinase R (PKR), in response to various stressors, including the presence of viral dsRNA, phosphorylates elongation initiation factor 2 (eIF2α), preventing the synthesis of proteins. One of the functions of ICP34.5 is to mediate dephosphorylation of eIF2α.20 Therefore, HSV-1 R3616 targets cancer cells that are characterized by uncontrollable protein synthesis.21 HSV-1 1716 mutant was also unable to express ICP34.5.22 It was generated by a recombination between 1714, which has the same inactivating deletion in both copies of RL1 (among other deletions), and wild type HSV-1 strain 17. While these four first generation oHSV mutants achieved mixed results in vivo,23–25 they highlighted the potential of oHSVs for the treatment of cancer.
Another HSV-1 mutant generated around the same time as hrR3, NV1020 (R7020), replaced five HSV-1 genes (UL55, encodes tegument protein (p)UL55;26 UL56, encodes envelope pUL56; and one copy each of repeat short (RS)1, encodes ICP4, RL1 and RL2, encodes ICP0) with the HSV-2 genomic region encoding several viral glycoproteins.27 The UL56 deletion, in addition to the deletion of RL1, most likely further reduces neurovirulence, as HSV-2 pUL56 associates with the neuron-specific kinesin KIF1A, a protein involved in the axonal transport of synaptic vesicle precursors.28 Although unsuccessful for its original purpose as a live attenuated vaccine for HSV-1 and -2,29 NV1020 showed potential as an oHSV in mice models of head and neck squamous cell carcinoma,30 epidermoid carcinoma, and prostate adenocarcinoma.31 The spontaneously generated oHSV HF10, which also includes a deletion of UL56 as well as duplication of UL53 (encodes glycoprotein gK),32 UL54 (encodes ICP27) and UL55, has had some success in animal models and human trials.33,34
The next generation oHSV combined gene mutations to reduce the chances of reversion to a virulent strain. The R3616 mutant (deleted RL1) was utilized to generate 3616UB, which also has an insertion of the LacZ gene into the UL2 gene which encodes uracil DNA deglycosylase.35,36 G207 (also known as MGH-1) combined the R3616 RL1 deletion and the hrR3 LacZ inactivating insertion in UL39.37,38 In RL1 deletion mutants, PKR inhibits the expression of late viral genes including unique short (US)11. Passaging of an RL1 deletion mutant in non-permissive cells leads to the natural generation of a mutant (known as SUP) with enhanced replication. SUP contains an additional deletion within US12 (encodes ICP47), thus losing expression of ICP47, but placing US11 under the immediate early US12 promoter.39,40 pUS11 is phosphorylated by PKR, which thus prevents phosphorylation of eIF2a.41 Hence, earlier expression of pUS11 allows it to inhibit PKR before PKR has the chance to inhibit pUS11 expression. This US12 deletion was also utilized in G207 to produce G47Δ.42
The KM100 mutant has insertions in UL48 (encodes the transactivator tegument protein pUL48 [VP16])43 and RL2 genes.44 KM100 no longer expresses the multifunctional protein ICP045 and while pUL48 is expressed it lacks the C-terminal transactivation domain. The resulting loss of expression of immediate early viral genes means KM100 only replicates well in cancer cells. Furthermore, KM100 activates antitumor immunity through interferon pathways normally suppressed by ICP0.46,47
The deletion of genes, whilst governing which cells oHSV can replicate in, also tends to attenuate the mutant virus. To boost virulence while maintaining selectivity for cancer cells, oHSVs with the capability to fuse cells have been generated. Fu-10 was created by inducing random mutations in G207 and selecting for mutations in the viral glycoproteins that enable syncytia formation.48 The lack of fusion in normal cells is due to the reduced replication of the HSV-1 genome, which strongly inhibits late gene expression (eg, glycoproteins). Synco-2, derived from Baco-1,49 utilized an RL1 deletion genome to insert a modified gibbon ape leukemia virus glycoprotein (constitutively fusogenic), under the control of a late HSV UL38 promoter.50 As both fusogenic mechanisms require different cellular receptors to enable syncytia formation, Synco-2D, also derived from Baco-1,49 employed both methods to ensure resistance to one would not inhibit cellular fusion.51 All three fusogenic oHSVs (Fu-10, Synco-2, and Synco-2D) demonstrated greater cytotoxicity in cancer cells than the HSV-1 parental strain.48,50,52
Gene regulated mutants
While deletion mutants have been proven to greatly limit virus replication to cancer cells, the attenuation caused by deleting one or more genes limits viral effectiveness. In an effort to retain the effectiveness of wild type HSV, oHSVs have been engineered to limit transcription and/or translation of an essential viral gene by replacing the viral promoters with tissue- or cancer-specific promoters.
G92A was targeted toward liver cancers by expressing the essential viral gene transcription regulator RS1 (encodes ICP4)53 from the exclusively liver expressed albumin promoter.54 Similar to G92A, d12.CALP has RS1 under the calponin promoter, which is aberrantly expressed in a variety of human soft tissue and bone tumors.55 However, due to the complicated nature of gene transcription regulation by RS1 encoded ICP4,53 both viruses have delayed and slow replication. Another liver targeted oHSV, LCSOV, was generated by placing UL22 (encodes viral glycoprotein gH) under the apolipoprotein E promoter.56
In an effort to boost replication of G207 in cancer cells, two mutants, Myb34.5 and rQNestin34.5, were generated by reinsertion of a copy of RL1 into the UL39/LacZ region, with expression of RL1 encoded ICP34.5 controlled by either the B-myb promoter or a nestin enhancer/heat shock protein 68 promoter cassette, respectively.57,58 B-myb is a transcriptional regulator which appears to be involved in cellular proliferation and differentiation and is downregulated in quiescent cells.59 Nestin is an intermediate filament protein whose expression is mostly switched off in adults, but expression is upregulated in malignant gliomas.58
Additional stringency in viral gene expression has been achieved by exploiting the overexpression of eukaryotic initiation factor 4E (eIF4E) in most cancers. eIF4E is the 5′cap binding and rate limiting protein of the eIF4F complex that is required to initiate capped translation of messenger RNAs (mRNAs). The 5′ untranslated region (UTR) of genes associated with malignant progression and metastasis is often rich in GC residues, forming hairpin structures that inhibit binding to eIF4E. While expression of these genes is normally regulated by expression of eIF4E,60 its constitutive overexpression in cancer allows unabated expression of these genes.61 The rat fibroblast growth factor gene has a GC rich 5′UTR that was inserted behind a prostate-specific probasin promoter, which replaced the promoter of the essential HSV-1 gene UL5462 to form the mutant AU27. Although UL54 was transcribed in both cancerous and noncancerous prostate cells, ICP27 protein expression was strongly limited to cancerous cells.63
An alternative viral gene translation control strategy takes advantage of the downregulation of certain miRNAs in cancers when compared to noncancerous tissue.64,65 miRNAs are short noncoding RNAs that regulate gene expression by binding to the 3′UTR of complementing mRNAs, repressing their transcription. The aforementioned LCSOV also contained four copies of target sequences for three different miRNAs (miR-122a, miR-124a, and miR-let-7a) in the 3′UTR of UL22 encoding gH to inhibit its translation in nonhepatocellular carcinoma cells.56
Multiple copies of target sequences for miR-143 and miR-145 were separately inserted into the 3′UTR of RS1 to generate CMV-ICP4-143T and CMV-ICP4-145T.66 AP27i145 was generated by inserting four copies of miR-145 target sequences into the 3′UTR of UL54.67 All three of these viruses were created by the amplicon helper virus system, whereby the HSV genome has been separated into two genomes.68 The helper genome comprises the complete HSV genome minus the gene that is to be controlled. The amplicon genome contains the missing gene (ie, RS1 or UL54) under control of a promoter from cytomegalovirus with the miRNAs target sequence in the 3′UTR. It also includes a viral origin of replication and DNA packaging signal so that the amplicon genome is synthesized and packaged when the helper genome is present.
Receptor retargeted mutants
The entry of HSV-1 into cells and the role of viral glycoproteins is well understood.69 Numerous attempts have been made to maintain wild type HSV effectiveness while limiting cell specificity by altering the receptors that HSV binds to enter cells. In the first instance, the region encoding binding to heparan sulfate in HSV-1 glycoproteins gC and gB was deleted and the sequence encoding interleukin (IL)-13 inserted into gC.70 The IL-13 sequence also replaced the N-terminal region of HSV-1 glycoprotein gD, which binds to herpesvirus entry mediator (HVEM). IL-13 was used to target the oHSV toward the IL-13Rα2 receptor that is overexpressed on malignant gliomas and high-grade astrocytomas.70 An additional single amino acid mutation in gD was also required to abolish binding to nectin-1, another entry receptor, with the final mutant known as R5141. However, the retargeting of R5141 caused an attenuation of the virus when compared to the parental virus.71
While creating another retargeting mutant, with urokinase plasminogen inserted into gD instead of IL-13, it was discovered that an 159 amino acid region in gD was dispensable for its function.72 Using this knowledge, R-LM249 was created by replacing this gD dispensable region with the sequence for the single chain antibody trastuzumab, which targets human epidermal growth factor receptor 2 (HER-2). HER-2 is overexpressed in approximately a quarter of breast and ovary carcinomas. Although R-LM249 displayed no binding to the normal HVEM or nectin-1 receptors, it also had a reduced replicative ability.73
In demonstrating that HSV tropism could be altered by replacing the 274 amino acid N-terminus of gD with a single chain antibody, HSV1716EGFR was created by replacing this region with a single chain antibody that targets human epidermal growth factor receptor.74 Another mutant, KNE, also incorporated the epidermal growth factor receptor antibody to replace the HVEM binding region of gD to target glioblastoma. KNE also required a single amino acid substitution in gD to ablate binding to nectin-1; however, as it did not effectively infect cells, it also needed a pair of entry enhancing mutations in the fusion glycoprotein gB.75
The most successful receptor retargeted mutant involves insertion of the trastuzumab antibody (targets HER-2) sequence into the N-terminal region of viral glycoprotein gH. This mutant, known as R-VG809, also contained deletions in gD to prevent binding to HVEM or nectin-1. R-VG809 was the first receptor retargeted mutant to show replication equal to the parental virus, at least in HER-2 positive ovarian cancer cells.76
Enhancing oHSVs
With the abundance of varied oHSVs that have been created, focus has shifted, to an extent, onto improving these viruses. The augmentation of these viruses utilizes two strategies, incorporating host genes into the virus and/or combining the oHSV with other treatments. The broad areas targeted by these strategies include: host immunity; the tumor microenvironment; and cancer cell replication and function.
Immune enhancement
Oncolytic virotherapies are occasionally viewed as immunotherapies due to the enhanced antitumor immune response that may be seen in patients after oncolytic vector treatment. Such a response could be due to numerous factors including greater release and thus detection of cancer antigens, an increase in infiltration of antitumor immune cells, and disruption of the immune toleragenic environment characteristic of many tumors. For this reason, significant effort has been made to further enhance the immune response during treatment with oncolytic viruses including oHSV.12,13
ICP47 (encoded by US12) normally inhibits major histocompatibility complex class I antigen presentation by binding to transporter associated with antigen presentation, blocking transport of antigenic peptides in the endoplasmic reticulum.77 Thus, deletion of US12 not only enhances viral replication, but also potentially allows a greater immune response.42 oHSV has also been used for expression of antitumor antigens. Following the success of dendritic cells loaded with mouse prostatic acid phosphatase (PAP) in eliciting an immune response to prostate cancer in a human clinical trial,78 the oHSV bPΔ6-hPAP was utilized to induce an antitumor immune response in mice bearing mouse prostate cancer via expression of human PAP.79 The combination of these two approaches remains an enticing prospect.
Numerous immune stimulating genes have been inserted into various oHSVs including IL-12,80–83 IL-15,84 IL-18,81 tumor necrosis factor alpha,85 granulocyte macrophage-colony stimulating factor,80,86 CD80 (B7.1),81 and fms-like tyrosine kinase 3 ligand.87 These genes function to attract, activate, proliferate, differentiate, and maturate immune cells such as natural killer, cytotoxic CD8+ T, helper CD4+ T, dendritic, macrophage and B cells, as well as their progenitors. The local production of these proteins also proves an advantage where systemic injection proves to be quite toxic.85
These cytokine producing oHSVs have proven to be more efficacious and just as safe as their non-cytokine producing parents in mice models through various measures including overall survival rate,80,82,83,87 tumor reduction (especially in contralateral non-treated tumors),80,81,83,86 immune cell infiltration,80,82 and successful rejection of tumors when rechallenged.80,86 Hopefully, the success shown by T-VEC, a granulocyte macrophage-colony stimulating factor-producing oHSV with a deletion of RL1 and US12,86 in clinical trials will lead to more trials with other immune stimulating oHSVs. In Phase III clinical trials in late stage melanoma, T-VEC produced an objective response rate of 26%, a durable response rate of 16%, and a complete response rate of 11%.88
Tumor microenvironment
The composition of the microenvironment in tumors remains a barrier to effective treatment of cancer. Most tumors are characterized by a large interstitial area, high levels of collagen, and lack of or a poor lymphatic system, leading to high interstitial pressure.89 Despite the overexpression of angiogenic factors, the vascular system of these tumors is leaky, dilated, and haphazardly connected with highly varied and abnormal structures. The poor blood flow through these tumors minimizes the amount of drug which reaches the cancer cells, while the higher content of extracellular matrix inhibits the passage of larger molecules.90
A natural side effect of HSV infection is the induction of angiogenesis and hyperpermeability in infected cells. It has been shown that HSV infected cells suppress synthesis of extracellular matrix proteins fibronectin and collagen, as well as the antiangiogenesis factor thrombospondin.91 HSV infection also leads to increased expression of angiogenic vascular endothelial growth factor and extracellular matrix degrading matrix metalloproteinase-9.92,93 Combined with the general leakiness of tumor vessels, this increase in angiogenesis leads to rapid infiltration of antiviral immune cells that clear the virus. Since angiogenesis is a requirement for tumor growth, an argument can be made that suppressing angiogenesis benefits oHSVs directly.
A few of the oHSVs generated have shown antiangiogenic effects on their own. The 1716 and hrR3 mutants have demonstrated reduction in microvessel density by infecting and lysing proliferating endothelial cells lining the blood vessels.94,95 Unlike wild type or the parental G207 mutant, G47Δ does not exhibit downregulation of thrombospondin.96
Several oHSVs that express antiangiogenic factors such as thrombospondin-1,97 endostatin,95,98 angiostatin,98 and vaculostatin99 have also been constructed. These genes have a diverse role in inhibiting angiogenesis including regulating proangiogenesis elements, inhibiting endothelial cell migration, attachment and proliferation as well as inducing apoptosis. These antiangiogenic viruses generated mixed results, with significant antiangiogenesis and delay in disease progression predominantly seen in central nervous system tumors.98
Disruption of the extracellular matrix has been shown to be a great benefit to the spread of oHSV. Tumors overexpressing metalloproteinase-1, -8, or -9 proteins, which vary in the extracellular matrix fibers they degrade, demonstrate wider oHSV infection of the tumor.100,101 Furthermore, coinjection of oHSV with bacterial collagenase enables the virus to diffuse further throughout the tumor.102
Cancer cell replication and function
Chemotherapeutic agents are cytotoxic chemicals used to treat cancer. These agents act by enhancing stress conditions (eg, double stranded breaks in DNA, high levels of unfolded proteins) and promote cell apoptosis. Chemotherapeutic agents are nonspecific however, and rapidly proliferating, normal cells in the bone marrow, digestive tract, and hair follicles can undergo cell death in response to these cytotoxic agents. Another limitation of chemotherapy is that it often requires a functional apoptotic pathway, and cancer cells with mutations in this pathway are often resistant to chemotherapy-induced cell death.
oHSVs can reduce the limitations of other drug treatments by acting synergistically when combined. Such synergy is generally due to two possible factors, enhanced replication of the oHSV and/or enhanced induction of apoptosis. The order and timing of each treatment, the type of oHSV mutant utilized, and the cancer type being treated are all important considerations. Discussed below are some examples of combination treatments with others of note also in the literature.47,103
Etoposide, temozolomide, and doxorubicin are DNA damaging agents that lead to double stranded breaks and apoptosis if the DNA damage is not repaired. Combining these drugs with oHSVs leads to greater levels of apoptosis due to oHSV’s natural ability to downregulate the DNA damage repair pathway. Furthermore, cell lines that showed resistance to these drugs were still sensitive to oHSV treatment.104–106
The taxanes, docetaxel and paclitaxel, are microtubule stabilizing agents, preventing microtubule breakdown during mitosis, a necessary process for cell cycle progression. HSV-1 inhibits cell cycle progression at the G1 phase. Concomitant treatment of oHSV with taxanes induces greater levels of apoptosis through increased cell cycle arrest.107
Certain mutant oHSVs combine better with some treatments. Dipyridamole and dilazep are used for inhibiting clot formation and vasodilation. One of their mechanisms of action is to inhibit equilibrative nucleoside transporter 1, reducing the intracellular concentration of adenosine. Such an action increases the activity of cellular RR, which boosts replication of oHSV mutants lacking UL39 (encodes viral RR).108
Radiation therapy induces DNA damage and apoptosis. Its combinational effect with oHSV also varies with the mutant utilized and the cell line treated. Radiation treatment increases RR activity and upregulates growth arrest and DNA damage-inducible protein (GADD34), a cellular protein with homology to ICP34.5, improving replication of UL39 and/or RL1 mutants.109–111 However, in the absence of increased viral production, enhanced induction of apoptosis is the cause for synergy.112 oHSVs that produce IL-12 or tumor necrosis factor have an even greater effect when combined with radiation therapy.85,113
Bortezomib, a proteasome inhibitor, leads to a buildup of unfolded proteins, an increase in endoplasmic reticulum stress, and apoptosis. One of the cellular responses to proteasome inhibition is upregulation of molecular chaperones, including Hsp90, which is used by HSV-1 DNA polymerase to translocate to the nucleus. Cells pretreated with bortezomib lead to an increase in oHSV replication.114
The discovery that cancers often have aberrant histone acetylation patterns has led to a significant interest in the use of histone deacetylase inhibitors for treatment. Pretreatment with histone deacetylase inhibitors increases replication of oHSV115 with inhibition of interferon stimulated genes of the innate antiviral immunity and activation of NF-κB shown to be involved.116,117 However, the order of treatment (and thus increased oHSV replication) appears to have no effect on synergistic cell killing, with an increase in cell cycle arrest and decrease in angiogenesis implicated in this synergism.118
Alternatively, the specificity of effective replication in cancer cells of oHSVs means they can be used for activating or enhancing the local effect of a combination treatment. Examples of such a mechanism include production of noradrenaline transporter119 or sodium iodide symporter120 to enhance uptake of radioactive Iodine-131 labeled molecules for radiation therapy and expression of nitroreductase,121 rat CYP2B1,122 and yeast cytosine deaminase123 for bioactivation of CB1954, cyclophosphamide, and 5-fluorocytosine, respectively.
oHSV delivery
The route of delivery of oHSV to tumors is another area that is being addressed. Currently, oHSVs are either directly injected into the tumor or given intravenously. While direct tumoral injection ensures all the virus is delivered to the tumor, in the majority of cases, it is limited to the needle track into the tumor and the virus rarely spreads beyond the injected tumor. Intravenous injection gives the virus an opportunity to infect all tumors, which is especially important in the case of metastasis. However, considering that most oHSVs are nonspecific for the cell they infect, less virus generally infects cancer cells compared to normal cells.124 Intravenous virus must also face the host innate immune system, which can neutralize virions before they reach the target cells.125
A few approaches to improve delivery have already been discussed. Retargeting of the receptor oHSVs use for binding to and entering cells ensures the virus has a greater predisposition for entering cancer cells. Coinjection of oHSV with collagenase degrades part of the extracellular matrix in the tumor, allowing the virion to spread further away from the site of injection.
The method for delivery of the oHSV can be enhanced or reduced by pretreatment with antiangiogenesis molecules. When oHSV was administered by direct injection, prior injection of cyclic RGD peptide, an antiangiogenic agent, reduced tumor vascular permeability and infiltration of leukocytes.126 Alternatively, systemic administration of oHSV after pretreatment with vascular endothelial growth factor antibody bevacuzimab reduced the amount of virus taken up by the tumor. However, the opposite treatment regime (oHSV prior to bevacuzimab) led to increased survival of mice with Ewing’s sarcoma.127
Targeting of tumors in the brain when oHSV is administered intravenously carries the added difficulty of having to cross the blood–brain barrier. In order to overcome this, it has been shown that blood–brain barrier disruption through a hypertonic solution of mannitol enhances the amount of virus that reaches the tumor.124 Alternatively, mesenchymal stem cells loaded with oHSV have been used to target glioblastoma. These cells are easily isolated from patients, routinely propagated in culture, and have been shown to home to tumors.128 However, these cells are also known to be immunosuppressive.129 Whether these cells would help (by enhancing the virus effect) or hinder (by inhibiting the antitumor immune response) treatment of the tumor was not determined as immunocompromised mice were used.
Another cell population that has been tested for delivery of oHSV is tumor antigen-specific lymphocytes. These cells were generated in a mouse host from exposure to inactivated cancer cells. After being harvested, they were infected with the oHSV before being reinjected into new mice bearing the same cancer. Cancer bearing mice injected with oHSV loaded lymphocytes survived longer than those treated with either single agent.130
Conclusion
Significant progress has been made in adapting HSV for the treatment of cancer. From the creation of numerous mutants to enhancing the efficacy of these mutants and the ability to deliver them to tumors, we have come a long way from early oncolytic virotherapy attempts utilizing wild type viruses. This progress is highlighted by several oHSVs that are or have been in Phase I and II clinical trials. These oHSVs have demonstrated excellent safety, with no deaths attributed to oHSVs to date, no encephalitis reported in clinical trials for brain tumors,131–134 and side effects predominantly limited to flu-like symptoms in nonbrain cancers.135,136 However, the majority of clinical trials have also shown limited long-term efficacy, as evident by lack of progression to Phase III trials, indicating there is still work to be done. The exception to this is T-VEC, which recently completed Phase III clinical trials in melanoma and just gained FDA approval.9,10,88
Given the safety of oHSVs observed in clinical trials to date, a detailed discussion of the testing of oHSVs in animal models for toxicity has not been included in this review. Though, it should be acknowledged that for oHSVs two predominantly used animal models for toxicity studies include owl monkeys and immunocompromised mice (mainly BALB/c) as both are extra sensitive to HSV encephalitis.22,29,35,37,42,86
As with much cancer research, the greatest limitation has been the models available. The immune response to both the virus and tumor seems to be a critical determinant to the effectiveness of oncolytic virotherapy, and hence models must incorporate this interplay. Immunocompromised mice are a poor model while syngeneic mice-cancer systems, with their intact immune system, offer a better view of how these factors interact and can be manipulated to enhance the antitumor effect. However, with such vast differences between preclinical and clinical results being evident, even these mice models still appear inadequate.
Zebrafish are a potential alternative model that is yet to be tested in oHSV virotherapy. Zebrafish have the ability to generate spontaneous tumors that are histopathologically and genetically similar to human tumors, or can be implanted with transgenic cells or xenotransplants.137 Additionally, due to a delay in developing an adaptive immune system, zebrafish can be used for metastatic and angiogenesis models of human cancers.138 Studies have revealed that zebrafish can be used as a model for HSV-1 infection, with similar infection and recognition mechanisms as in mice and humans.139,140
Better patient outcomes appear to be characterized by breaking the immune tolerance that is preventing clearance of the tumor by the host immune system itself. The virus could potentially achieve this through destruction of the immunosuppressive cells within the tumor and/or by inducing a more effective immune response.11 Stimulation of an antitumor immune response is epitomized by the aforementioned immune stimulating T-VEC, which demonstrated in melanoma patients not only an increase in melanoma-specific T cells, but also a decrease in regulatory and suppressor T cells, an effect which resulted in 15% of measurable visceral (non-injected tumors) shrinking by more than 50% in the Phase III trials.88,141
However, it is perhaps pertinent to also focus on patients in whom oncolytic virotherapy fails. What factors are limiting the response? Rapid clearance of the oHSV by the innate immune system may not allow adequate time for the virus to illicit an effective antitumor immune response, or patients may be incapable of mounting an effective antitumor response. In these cases, suppression of the innate immune system, even temporarily, may allow a more potent adaptive immune response to be mounted, or possibly allow the virus to eradicate the tumor itself. The enhanced replication and spread of oHSV when combined with immunosuppressive cyclophosphamide or rapamycin would indicate the potential feasibility for such a strategy.49,142
Oncolytic virotherapy is a complex treatment, whose success depends on the intricate interactions between the tumor and its microenvironment, as well as the virus and host antiviral and antitumor immune responses. HSV has proven to be a worthy virus for oncolytic virotherapy as it is amenable to genetic alterations, synergizes well with many current cancer treatments, is reasonably safe for patients, and has demonstrated some effectiveness. Future studies need to concentrate on improving outcomes through a combination of oHSV with targeted chemotherapies.143
Acknowledgment
This work was supported by an Australian postgraduate award (to NS).
Disclosure
The authors report no conflicts of interest in this work.
References
Dock G. The influence of complicating diseases upon leukaemia. Am J Med Sci. 1904;127:563–592. | |
Hoster HA, Zanes RP Jr, Von Haam E. Studies in Hodgkin’s syndrome; the association of viral hepatitis and Hodgkin’s disease; a preliminary report. Cancer Res. 1949;9(8):473–480. | |
Taylor AW. Effects of glandular fever infection in acute leukaemia. Br Med J. 1953;1(4810):589–593. | |
Huebner RJ, Rowe WP, Schatten WE, Smith RR, Thomas LB. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer. 1956;9(6):1211–1218. | |
Pack GT. Note on the experimental use of rabies vaccine for melanomatosis. AMA Arch Derm Syphilol. 1950;62(5):694–695. | |
Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15(4):651–659. | |
Roizman B, Knipe DM, Whitley RJ. Herpes simplex viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2013: 1824–1897. | |
Andtbacka RHI, Collichio FA, Amatruda T, et al. OPTiM: a randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol. 2013;31(Suppl; abstr LBA9008). | |
Johnson DB, Puzanov I, Kelley MC. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015;7(6):1–9. | |
Killock D. Skin cancer: T-VEC oncolytic viral therapy shows promise in melanoma. Nat Rev Clin Oncol. 2015;12(8):438. | |
Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe. 2014;15(3):260–265. | |
Coffin RS. From virotherapy to oncolytic immunotherapy: where are we now? Curr Opin Virol. 2015;13:93–100. | |
de Gruijl TD, Janssen AB, van Beusechem VW. Arming oncolytic viruses to leverage antitumor immunity. Expert Opin Biol Ther. 2015; 15(7):959–971. | |
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–856. | |
Coen DM, Kosz-Vnenchak M, Jacobson JG, et al. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A. 1989;86(12):4736–4740. | |
Wagner MJ, Sharp JA, Summers WC. Nucleotide sequence of the thymidine kinase gene of herpes simplex virus type 1. Proc Natl Acad Sci U S A. 1981;78(3):1441–1445. | |
Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol. 1988;62(1):196–205. | |
Preston VG, Palfreyman JW, Dutia BM. Identification of a herpes simplex virus type 1 polypeptide which is a component of the virus-induced ribonucleotide reductase. J Gen Virol. 1984;65 (Pt 9):1457–1466. | |
Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250(4985):1262–1266. | |
He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1997;94(3):843–848. | |
Chou J, Roizman B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci U S A. 1992;89(8):3266–3270. | |
MacLean AR, ul-Fareed M, Robertson L, Harland J, Brown SM. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J Gen Virol. 1991;72 (Pt 3):631–639. | |
Markert JM, Malick A, Coen DM, Martuza RL. Reduction and elimination of encephalitis in an experimental glioma therapy model with attenuated herpes simplex mutants that retain susceptibility to acyclovir. Neurosurgery. 1993;32(4):597–603. | |
Mineta T, Rabkin SD, Martuza RL. Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res. 1994;54(15):3963–3966. | |
Randazzo BP, Kesari S, Gesser RM, et al. Treatment of experimental intracranial murine melanoma with a neuroattenuated herpes simplex virus 1 mutant. Virology. 1995;211(1):94–101. | |
Loret S, Guay G, Lippe R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol. 2008; 82(17):8605–8618. | |
Meignier B, Longnecker R, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: construction and evaluation in rodents. J Infect Dis. 1988;158(3):602–614. | |
Koshizuka T, Kawaguchi Y, Nishiyama Y. Herpes simplex virus type 2 membrane protein UL56 associates with the kinesin motor protein KIF1A. J Gen Virol. 2005;86 (Pt 3):527–533. | |
Meignier B, Martin B, Whitley RJ, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020. II. Studies in immunocompetent and immunosuppressed owl monkeys (Aotus trivirgatus). J Infect Dis. 1990;162(2):313–321. | |
Wong RJ, Kim SH, Joe JK, Shah JP, Johnson PA, Fong Y. Effective treatment of head and neck squamous cell carcinoma by an oncolytic herpes simplex virus. J Am Coll Surg. 2001;193(1):12–21. | |
Advani SJ, Chung SM, Yan SY, et al. Replication-competent, nonneuroinvasive genetically engineered herpes virus is highly effective in the treatment of therapy-resistant experimental human tumors. Cancer Res. 1999;59(9):2055–2058. | |
David AT, Baghian A, Foster TP, Chouljenko VN, Kousoulas KG. The herpes simplex virus type 1 (HSV-1) glycoprotein K(gK) is essential for viral corneal spread and neuroinvasiveness. Curr Eye Res. 2008; 33(5):455–467. | |
Nakao A, Kasuya H, Sahin TT, et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011;18(3):167–175. | |
Takakuwa H, Goshima F, Nozawa N, et al. Oncolytic viral therapy using a spontaneously generated herpes simplex virus type 1 variant for disseminated peritoneal tumor in immunocompetent mice. Arch Virol. 2003;148(4):813–825. | |
Pyles RB, Warnick RE, Chalk CL, Szanti BE, Parysek LM. A novel multiply-mutated HSV-1 strain for the treatment of human brain tumors. Hum Gene Ther. 1997;8(5):533–544. | |
Mullaney J, Moss HW, McGeoch DJ. Gene UL2 of herpes simplex virus type 1 encodes a uracil-DNA glycosylase. J Gen Virol. 1989; 70 (Pt 2):449–454. | |
Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1(9):938–943. | |
Kramm CM, Chase M, Herrlinger U, et al. Therapeutic efficiency and safety of a second-generation replication-conditional HSV1 vector for brain tumor gene therapy. Hum Gene Ther. 1997;8(17):2057–2068. | |
Mohr I, Gluzman Y. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J. 1996;15(17):4759–4766. | |
Taneja S, MacGregor J, Markus S, Ha S, Mohr I. Enhanced antitumor efficacy of a herpes simplex virus mutant isolated by genetic selection in cancer cells. Proc Natl Acad Sci U S A. 2001;98(15):8804–8808. | |
Lussignol M, Queval C, Bernet-Camard MF, et al. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol. 2013;87(2):859–871. | |
Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A. 2001;98(11):6396–6401. | |
Wysocka J, Herr W. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci. 2003; 28(6):294–304. | |
Mossman KL, Smiley JR. Truncation of the C-terminal acidic transcriptional activation domain of herpes simplex virus VP16 renders expression of the immediate-early genes almost entirely dependent on ICP0. J Virol. 1999;73(12):9726–9733. | |
Smith MC, Boutell C, Davido DJ. HSV-1 ICP0: paving the way for viral replication. Future Virol. 2011;6(4):421–429. | |
Hummel JL, Safroneeva E, Mossman KL. The role of ICP0-Null HSV-1 and interferon signaling defects in the effective treatment of breast adenocarcinoma. Mol Ther. 2005;12(6):1101–1110. | |
Workenhe ST, Pol JG, Lichty BD, Cummings DT, Mossman KL. Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. Cancer Immunol Res. 2013;1(5):309–319. | |
Fu X, Zhang X. Potent systemic antitumor activity from an oncolytic herpes simplex virus of syncytial phenotype. Cancer Res. 2002; 62(8):2306–2312. | |
Fu X, Tao L, Rivera A, Zhang X. Rapamycin enhances the activity of oncolytic herpes simplex virus against tumor cells that are resistant to virus replication. Int J Cancer. 2011;129(6):1503–1510. | |
Fu X, Tao L, Jin A, Vile R, Brenner MK, Zhang X. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus potentiates the viral antitumor effect. Mol Ther. 2003;7(6):748–754. | |
Nakamori M, Fu X, Meng F, et al. Effective therapy of metastatic ovarian cancer with an oncolytic herpes simplex virus incorporating two membrane fusion mechanisms. Clin Cancer Res. 2003;9(7):2727–2733. | |
Nakamori M, Fu X, Rousseau R, Chen SY, Zhang X. Destruction of nonimmunogenic mammary tumor cells by a fusogenic oncolytic herpes simplex virus induces potent antitumor immunity. Mol Ther. 2004;9(5):658–665. | |
Roizman B, Gu H, Mandel G. The first 30 minutes in the life of a virus: unREST in the nucleus. Cell Cycle. 2005;4(8):1019–1021. | |
Miyatake S, Iyer A, Martuza RL, Rabkin SD. Transcriptional targeting of herpes simplex virus for cell-specific replication. J Virol. 1997; 71(7):5124–5132. | |
Yamamura H, Hashio M, Noguchi M, et al. Identification of the transcriptional regulatory sequences of human calponin promoter and their use in targeting a conditionally replicating herpes vector to malignant human soft tissue and bone tumors. Cancer Res. 2001; 61(10):3969–3977. | |
Fu X, Rivera A, Tao L, De Geest B, Zhang X. Construction of an oncolytic herpes simplex virus that precisely targets hepatocellular carcinoma cells. Mol Ther. 2012;20(2):339–346. | |
Chung RY, Saeki Y, Chiocca EA. B-myb promoter retargeting of herpes simplex virus gamma34.5 gene-mediated virulence toward tumor and cycling cells. J Virol. 1999;73(9):7556–7564. | |
Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65(7):2832–2839. | |
Sala A, Kundu M, Casella I, et al. Activation of human B-MYB by cyclins. Proc Natl Acad Sci U S A. 1997;94(2):532–536. | |
Montero H, Garcia-Roman R, Mora SI. eIF4E as a control target for viruses. Viruses. 2015;7(2):739–750. | |
Graff JR, Zimmer SG. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metastasis. 2003;20(3):265–273. | |
Sandri-Goldin RM. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol. 2011;6(11):1261–1277. | |
Lee CY, Bu LX, DeBenedetti A, Williams BJ, Rennie PS, Jia WW. Transcriptional and translational dual-regulated oncolytic herpes simplex virus type 1 for targeting prostate tumors. Mol Ther. 2010;18(5):929–935. | |
Naidu S, Magee P, Garofalo M. MiRNA-based therapeutic intervention of cancer. J Hematol Oncol. 2015;8:68. | |
Ruiz AJ, Russell SJ. MicroRNAs and oncolytic viruses. Curr Opin Virol. 2015;13:40–48. | |
Lee CY, Rennie PS, Jia WW. MicroRNA regulation of oncolytic herpes simplex virus-1 for selective killing of prostate cancer cells. Clin Cancer Res. 2009;15(16):5126–5135. | |
Li JM, Kao KC, Li LF, et al. MicroRNA-145 regulates oncolytic herpes simplex virus-1 for selective killing of human non-small cell lung cancer cells. Virol J. 2013;10:241. | |
Fraefel C, Marconi P, Epstein AL. Herpes simplex virus type 1 (HSV-1)- derived amplicon vectors for gene transfer and gene therapy. Methods Mol Biol. 2015;1254:295–316. | |
Salameh S, Sheth U, Shukla D. Early events in herpes simplex virus lifecycle with implications for an infection of lifetime. Open Virol J. 2012;6:1–6. | |
Zhou G, Ye GJ, Debinski W, Roizman B. Engineered herpes simplex virus 1 is dependent on IL13Ralpha 2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc Natl Acad Sci U S A. 2002;99(23):15124–15129. | |
Zhou G, Roizman B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc Natl Acad Sci U S A. 2006;103(14):5508–5513. | |
Zhou G, Roizman B. Separation of receptor-binding and profusogenic domains of glycoprotein D of herpes simplex virus 1 into distinct interacting proteins. Proc Natl Acad Sci U S A. 2007;104(10):4142–4146. | |
Menotti L, Nicoletti G, Gatta V, et al. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc Natl Acad Sci U S A. 2009;106(22):9039–9044. | |
Conner J, Braidwood L, Brown SM. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein D fusion protein. Gene Ther. 2008;15(24):1579–1592. | |
Uchida H, Marzulli M, Nakano K, et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther. 2013;21(3):561–569. | |
Gatta V, Petrovic B, Campadelli-Fiume G. The engineering of a novel ligand in gH confers to HSV an expanded tropism independent of gD activation by its receptors. PLoS Pathog. 2015;11(5):e1004907. | |
Hill A, Jugovic P, York I, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375(6530):411–415. | |
Fong L, Brockstedt D, Benike C, et al. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol. 2001; 167(12):7150–7156. | |
Castelo-Branco P, Passer BJ, Buhrman JS, et al. Oncolytic herpes simplex virus armed with xenogeneic homologue of prostatic acid phosphatase enhances antitumor efficacy in prostate cancer. Gene Ther. 2010;17(6):805–810. | |
Wong RJ, Patel SG, Kim S, et al. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum Gene Ther. 2001;12(3):253–265. | |
Ino Y, Saeki Y, Fukuhara H, Todo T. Triple combination of oncolytic herpes simplex virus-1 vectors armed with interleukin-12, interleukin-18, or soluble B7-1 results in enhanced antitumor efficacy. Clin Cancer Res. 2006;12(2):643–652. | |
Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A. 2000;97(5):2208–2213. | |
Antoszczyk S, Spyra M, Mautner VF, et al. Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro Oncol. 2014;16(8):1057–1066. | |
Gaston DC, Odom CI, Li L, et al. Production of bioactive soluble interleukin-15 in complex with interleukin-15 receptor alpha from a conditionally-replicating oncolytic HSV-1. PLoS One. 2013;8(11):e81768. | |
Han ZQ, Assenberg M, Liu BL, et al. Development of a second-generation oncolytic Herpes simplex virus expressing TNFalpha for cancer therapy. J Gene Med. 2007;9(2):99–106. | |
Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. | |
Barnard Z, Wakimoto H, Zaupa C, et al. Expression of FMS-like tyrosine kinase 3 ligand by oncolytic herpes simplex virus type I prolongs survival in mice bearing established syngeneic intracranial malignant glioma. Neurosurgery. 2012;71(3):741–748. | |
Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. | |
Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987;47(12):3039–3051. | |
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. | |
Kefalides NA, Ziaie Z. Herpes simplex virus suppression of human endothelial matrix protein synthesis is independent of viral protein synthesis. Lab Invest. 1986;55(3):328–336. | |
Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J Virol. 2001; 75(20):9828–9835. | |
Lee S, Zheng M, Kim B, Rouse BT. Role of matrix metalloproteinase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J Clin Invest. 2002;110(8):1105–1111. | |
Benencia F, Courreges MC, Conejo-Garcia JR, et al. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum Gene Ther. 2005;16(6):765–778. | |
Goodwin JM, Schmitt AD, McGinn CM, et al. Angiogenesis inhibition using an oncolytic herpes simplex virus expressing endostatin in a murine lung cancer model. Cancer Invest. 2012;30(3):243–250. | |
Aghi M, Rabkin SD, Martuza RL. Angiogenic response caused by oncolytic herpes simplex virus-induced reduced thrombospondin expression can be prevented by specific viral mutations or by administering a thrombospondin-derived peptide. Cancer Res. 2007;67(2):440–444. | |
Tsuji T, Nakamori M, Iwahashi M, et al. An armed oncolytic herpes simplex virus expressing thrombospondin-1 has an enhanced in vivo antitumor effect against human gastric cancer. Int J Cancer. 2013; 132(2):485–494. | |
Zhang G, Jin G, Nie X, et al. Enhanced antitumor efficacy of an oncolytic herpes simplex virus expressing an endostatin-angiostatin fusion gene in human glioblastoma stem cell xenografts. PLoS One. 2014;9(4):e95872. | |
Hardcastle J, Kurozumi K, Dmitrieva N, et al. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol Ther. 2010;18(2):285–294. | |
Mok W, Boucher Y, Jain RK. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007;67(22):10664–10668. | |
Hong CS, Fellows W, Niranjan A, et al. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010;17(10):1200–1205. | |
McKee TD, Grandi P, Mok W, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66(5):2509–2513. | |
Kanai R, Wakimoto H, Martuza RL, Rabkin SD. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res. 2011;17(11):3686–3696. | |
Cheema TA, Kanai R, Kim GW, et al. Enhanced antitumor efficacy of low-dose Etoposide with oncolytic herpes simplex virus in human glioblastoma stem cell xenografts. Clin Cancer Res. 2011; 17(23):7383–7393. | |
Kanai R, Rabkin SD, Yip S, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104(1):42–55. | |
Zhuang X, Zhang W, Chen Y, et al. Doxorubicin-enriched, ALDH(br) mouse breast cancer stem cells are treatable to oncolytic herpes simplex virus type 1. BMC Cancer. 2012;12:549. | |
Passer BJ, Castelo-Branco P, Buhrman JS, Varghese S, Rabkin SD, Martuza RL. Oncolytic herpes simplex virus vectors and taxanes synergize to promote killing of prostate cancer cells. Cancer Gene Ther. 2009;16(7):551–560. | |
Passer BJ, Cheema T, Zhou B, et al. Identification of the ENT1 antagonists dipyridamole and dilazep as amplifiers of oncolytic herpes simplex virus-1 replication. Cancer Res. 2010;70(10):3890–3895. | |
Stanziale SF, Petrowsky H, Joe JK, et al. Ionizing radiation potentiates the antitumor efficacy of oncolytic herpes simplex virus G207 by upregulating ribonucleotide reductase. Surgery. 2002;132(2):353–359. | |
Jarnagin WR, Zager JS, Hezel M, et al. Treatment of cholangiocarcinoma with oncolytic herpes simplex virus combined with external beam radiation therapy. Cancer Gene Ther. 2006;13(3):326–334. | |
Adusumilli PS, Chan MK, Hezel M, et al. Radiation-induced cellular DNA damage repair response enhances viral gene therapy efficacy in the treatment of malignant pleural mesothelioma. Ann Surg Oncol. 2007;14(1):258–269. | |
Dai MH, Zamarin D, Gao SP, et al. Synergistic action of oncolytic herpes simplex virus and radiotherapy in pancreatic cancer cell lines. Br J Surg. 2010;97(9):1385–1394. | |
Gillory LA, Megison ML, Stewart JE, et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of neuroblastoma. PLoS One. 2013;8(10):e77753. | |
Yoo JY, Hurwitz BS, Bolyard C, et al. Bortezomib-induced unfolded protein response increases oncolytic HSV-1 replication resulting in synergistic antitumor effects. Clin Cancer Res. 2014;20(14):3787–3798. | |
Cody JJ, Markert JM, Hurst DR. Histone deacetylase inhibitors improve the replication of oncolytic herpes simplex virus in breast cancer cells. PLoS One. 2014;9(3):e92919. | |
Otsuki A, Patel A, Kasai K, et al. Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol Ther. 2008;16(9):1546–1555. | |
Katsura T, Iwai S, Ota Y, Shimizu H, Ikuta K, Yura Y. The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther. 2009; 16(3):237–245. | |
Liu TC, Castelo-Branco P, Rabkin SD, Martuza RL. Trichostatin A and oncolytic HSV combination therapy shows enhanced antitumoral and antiangiogenic effects. Mol Ther. 2008;16(6):1041–1047. | |
Sorensen A, Mairs RJ, Braidwood L, et al. In vivo evaluation of a cancer therapy strategy combining HSV1716-mediated oncolysis with gene transfer and targeted radiotherapy. J Nucl Med. 2012;53(4):647–654. | |
Li H, Nakashima H, Decklever TD, Nace RA, Russell SJ. HSV-NIS, an oncolytic herpes simplex virus type 1 encoding human sodium iodide symporter for preclinical prostate cancer radiovirotherapy. Cancer Gene Ther. 2013;20(8):478–485. | |
Braidwood L, Dunn PD, Hardy S, Evans TR, Brown SM. Antitumor activity of a selectively replication competent herpes simplex virus (HSV) with enzyme prodrug therapy. Anticancer Res. 2009; 29(6):2159–2166. | |
Chase M, Chung RY, Chiocca EA. An oncolytic viral mutant that delivers the CYP2B1 transgene and augments cyclophosphamide chemotherapy. Nat Biotechnol. 1998;16(5):444–448. | |
Yamada S, Kuroda T, Fuchs BC, et al. Oncolytic herpes simplex virus expressing yeast cytosine deaminase: relationship between viral replication, transgene expression, prodrug bioactivation. Cancer Gene Ther. 2012;19(3):160–170. | |
Liu R, Martuza RL, Rabkin SD. Intracarotid delivery of oncolytic HSV vector G47Delta to metastatic breast cancer in the brain. Gene Ther. 2005;12(8):647–654. | |
Wakimoto H, Ikeda K, Abe T, et al. The complement response against an oncolytic virus is species-specific in its activation pathways. Mol Ther. 2002;5(3):275–282. | |
Kurozumi K, Hardcastle J, Thakur R, et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J Natl Cancer Inst. 2007;99(23):1768–1781. | |
Eshun FK, Currier MA, Gillespie RA, Fitzpatrick JL, Baird WH, Cripe TP. VEGF blockade decreases the tumor uptake of systemic oncolytic herpes virus but enhances therapeutic efficacy when given after virotherapy. Gene Ther. 2010;17(7):922–929. | |
Duebgen M, Martinez-Quintanilla J, Tamura K, et al. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J Natl Cancer Inst. 2014;106(6):dju090. | |
Krause MN, Sancho-Martinez I, Izpisua Belmonte JC. RE: stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J Natl Cancer Inst. 2015;107(1):368. | |
Kanzaki A, Kasuya H, Yamamura K, et al. Antitumor efficacy of oncolytic herpes simplex virus adsorbed onto antigen-specific lymphocytes. Cancer Gene Ther. 2012;19(4):292–298. | |
Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7(10):859–866. | |
Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867–874. | |
Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9(6):398–406. | |
Harrow S, Papanastassiou V, Harland J, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004; 11(22):1648–1658. | |
Geevarghese SK, Geller DA, de Haan HA, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther. 2010;21(9):1119–1128. | |
Senzer NN, Kaufman HL, Amatruda T, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27(34):5763–5771. | |
Stoletov K, Klemke R. Catch of the day: zebrafish as a human cancer model. Oncogene. 2008;27(33):4509–4520. | |
Teng Y, Xie X, Walker S, White DT, Mumm JS, Cowell JK. Evaluating human cancer cell metastasis in zebrafish. BMC Cancer. 2013;13:453. | |
Antoine TE, Jones KS, Dale RM, Shukla D, Tiwari V. Zebrafish: modeling for herpes simplex virus infections. Zebrafish. 2014; 11(1):17–25. | |
Ge R, Zhou Y, Peng R, et al. Conservation of the STING-mediated cytosolic DNA sensing pathway in zebrafish. J Virol. 2015;89(15):7696–7706. | |
Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010; 17(3):718–730. | |
Wakimoto H, Fulci G, Tyminski E, Chiocca EA. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide’s enhancement of viral oncolysis. Gene Ther. 2004;11(2):214–223. | |
Baudino TA. Targeted cancer therapy: The next generation of cancer treatment. Curr Drug Discov Technol. 2015;12(1):3–20. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.