Back to Journals » Infection and Drug Resistance » Volume 12
Omadacycline: a novel aminomethylcycline
Authors Burgos RM, Rodvold KA
Received 26 March 2019
Accepted for publication 9 May 2019
Published 2 July 2019 Volume 2019:12 Pages 1895—1915
DOI https://doi.org/10.2147/IDR.S171352
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Joachim Wink
Rodrigo M Burgos,1 Keith A Rodvold1,2
1Department of Pharmacy Practice, University of Illinois at Chicago College of Pharmacy, Chicago, IL, USA; 2Department of Medicine, University of Illinois at Chicago College of Medicine, Chicago, IL, USA
Abstract: Tetracyclines have come a long way since they became available almost seven decades ago, with numerous enhancements allowing new agents to overcome bacterial mechanisms of resistance. However, these enhancements come with toxicities and pharmacokinetic disadvantages such as the gastrointestinal side-effects and poor oral bioavailability seen with the glycylcylcines. Omadacycline, a new and improved tetracycline, has demonstrated a broad spectrum of in vitro activity, has oral and intravenous formulations, improved safety compared to glycylcyclines, as well as clinical efficacy and safety for two types of infections: acute bacterial skin and skin structure infections and community-acquired bacterial pneumonia. This review will summarize salient points about its pharmacologic properties, available clinical efficacy, and safety data and omadacycline’s place in therapy.
Keywords: tetracyclines, community-acquired pneumonia, skin infections
Introduction
The tetracycline class of antibacterial agents was introduced for human use in the 1940s.1 Demeclocycline, tetracycline, oxytetracycline, minocycline, and doxycycline remain available in the US for systematic use. Synthetic processes have allowed for modifications within the four core rings of tetracyclines and the development of several new tetracycline analogs, including tigecycline as a glycylcycline, eravacycline as a fluorocycline, and omadacycline as an aminomethylcycline.2–4
Omadacycline (PTK0796, BAY 73–6944, MK-2764) is a first-in-class aminomethylcycline with broad-spectrum in vitro activity against Gram-positive aerobes, Gram-negative aerobes, anaerobes, and atypical bacteria.4,5 Omadacycline retains antibacterial activity against strains expressing the two most common tetracycline resistance mechanisms, bacterial ribosomal protection proteins, and tetracycline-specific efflux pumps. Both oral and intravenous (IV) formulations of omadacycline have been approved by the United States Food and Drug Administration (US FDA) for the treatment of acute bacterial skin and skin structure infections (ABSSSI) and community-acquired bacterial pneumonia (CABP). The recommended oral dosage of omadacycline in adults with ABSSSI is 450 mg orally once daily for Day 1 and 2 of therapy, followed by a maintenance dose of 300 mg orally once daily. The dosage of omadacycline in adults with CABP and ABSSSI is a loading dose of 200 mg by IV infusion over 60 minutes or 100 mg by IV infusion over 30 minutes twice on day 1, followed by a maintenance dose of 100 mg IV infusion over 30 minutes once daily or 300 mg orally once daily. The recommended treatment duration for omadacycline for both indications is 7–14 days.
This review provides an overview of the clinical microbiology, pharmacology, efficacy, and safety data of omadacycline. The current information will be provided as a foundation for understanding the clinical usefulness of the oral and IV dosage regimens of omadacycline for the treatment of ABSSSI and CABP in adults.
Chemistry
Omadacycline is available as monotosylate salt with molecular weight of 728.9, and the chemical name (4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-9-(2,2-dimethylpropylaminomethyl)-3,10,12,12a-tetrahydroxy-1,11-dioxo-1,4,4a,5,5a,6,11,12a- octahydrotetracene-2-carboxamide, 4-methylbenzenesulfonate.6 A chemical feature that distinguishes omadacycline from tigecycline and eravacycline is the aminomethyl substitution at the C9 position, which provides improved pharmacokinetic parameters, most notably oral bioavailability, lower dose-limiting nausea and vomiting, for which C9 glycylcyclines are infamous, and hindering of efflux-mediated resistance. Tigecycline and eravacycline, two C9-modified glycylcyclines, are also known to have improved potency related to the stability of ribosomal protection proteins and efflux pump resistance pathways.7,8
Mechanisms of action and resistance
Like all other tetracyclines, omadacycline is primarily a protein synthesis inhibitor, as confirmed by macromolecular synthesis experiments.9 Protein synthesis inhibition is thought to occur from binding to the 30S ribosomal subunit.9,10 Attachment to the 30S subunit blocks the acceptor site in the mRNA-ribosome complex, which prevents amino acid incorporation into the elongating peptide.3 Because this interaction is reversible, tetracyclines are generally thought to be bacteriostatic agents: However, omadacycline has demonstrated in vitro bactericidal activity against Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis.7 Additionally, modest inhibition of peptidoglycan synthesis may be a resulting effect of protein synthesis.9
Tetracycline resistance is thought to arise most commonly from organisms that encode for genes tet(K), tet(L), tet(A), and tet(B), which mediate efflux mechanisms, and from ribosomal protection genes tet(M), tet(O), and tet(S).11,12 Other mechanisms of tetracycline resistance include drug degradation and mutations in rRNA binding sites.3 The protein Tet(O) has been shown to protect the ribosome by promoting release of tetracycline from the 70S unit: Omadacycline demonstrated protein synthesis inhibition in both the presence and absence of Tet(O), however.9 Additionally, omadacycline demonstrated protein synthesis inhibition in Staphylococcus aureus harboring efflux genes tet(K) and tet(M) in whole-cell assays.9 Omadacycline could overcome ribosomal protection by higher affinity for the ribosome or by binding to the ribosome in a way that bypasses protection by proteins Tet(M) and Tet(O), however this is unclear at this point. Similarly, it is unknown how omadacycline overcomes efflux resistance. Intrinsic resistance to omadacycline in Pseudomonas aeruginosa and decreased susceptibility in Klebsiella pneumoniae have been described from overexpression of MexXY and AcrAB efflux pumps.13
Microbiology
A study by Pfaller et al14 assessed the in vitro activity of omadacycline in 69,246 isolates collected in 2010 and 2011 from around the world. A concentration of ≤2 µg/mL of omadacycline was able to inhibit growth in 99.9% of Staphylococcus aureus, 100% of methicillin susceptible Staphylococcus aureus (MSSA), 99.8% of methicillin resistant Staphylococcus aureus (MRSA), 100% of coagulase negative Staphylococcus, 100% of Enterococcus spp., including vancomycin susceptible and non-susceptible, and >99.9% of Enterococcus faecalis, including vancomycin susceptible and non-susceptible. A concentration of 0.5 µg/mL of omadacycline was able to inhibit 100% of Enterococcus faecium, including vancomycin susceptible and non-susceptible, 100% of other Enterococcus spp., 100% of Streptococcus pneumoniae, including penicillin susceptible or resistant, viridans group streptococci, and beta-hemolytic streptococci. Omadacycline also had activity against Enterobacteriaceae, notably Escherichia coli, Klebsiella aerogenes (formerly Enterobacter aerogenes), Klebsiella oxytoca, and Citrobacter spp.; also against Haemophilus influenzae, including beta-lactamase positive and negative, and Moraxella catarrhalis. The reported minimum inhibitory concentrations for 50% and 90% of isolates (MIC50/MIC90) for omadacycline in select organisms in this study can be found in Table 1. The reader is encouraged to consult this publication for susceptibilities of other organisms.
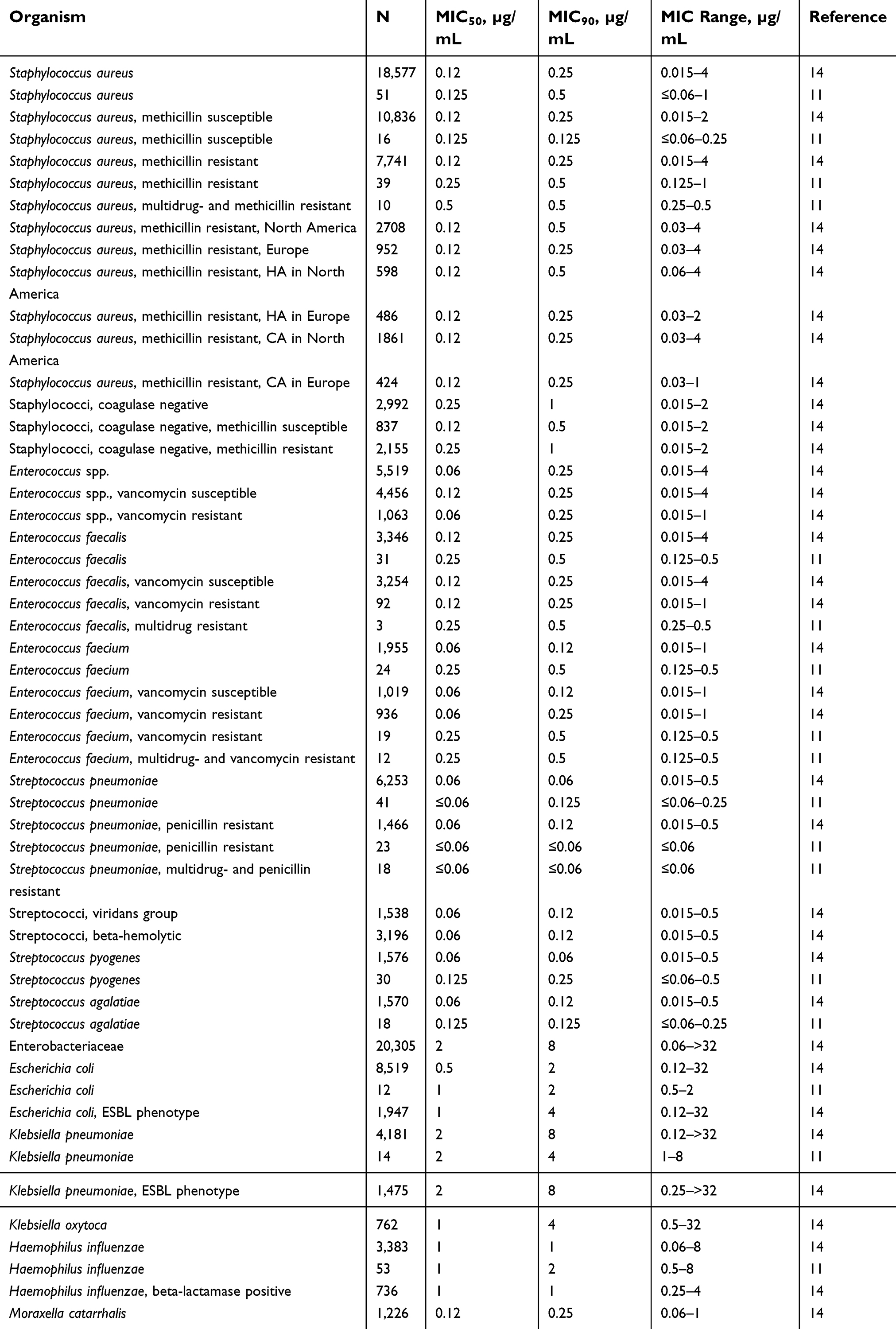 |
Table 1 In vitro antimicrobial activity of omadacycline against organisms in different studies |
Omadacycline has also demonstrated in vitro activity against gram-positive and gram-negative pathogens commonly responsible for ABSSSI and CABP from different centers across the US.11 When compared against tetracycline and doxycycline in susceptible organisms, omadacycline (but not tetracycline or doxycycline) retained similar MIC in the following organisms harboring tetracycline resistance genes: Staphylococcus aureus with tet(M) and tet(K); Enterococcus faecalis with tet(M), tet(L), both tet(M) and tet(L), and tet(S); Enterococcus faecium with tet(M), both tet(M) and tet(L), tet(K), and tet(O); Streptococcus pneumoniae with tet(M); beta-hemolytic streptococci (S. pyogenes and S. agalactiae) with tet(M) and tet(O); and Escherichia coli with tet(A). In this study, the MIC50/MIC90 (MIC range) for omadacycline was determined in a broad set of organisms, which are reported in Table 1. In vivo activity of omadacycline was also demonstrated in an intraperitoneal infection mouse model in this same study, were the 50% effective doses (ED50) were determined from a single-dose intravenous dose of omadacycline: For Streptococcus pneumoniae, 0.45–3.39 mg/kg; Staphylococcus aureus, including strains with tet(M) and tet(K) genes, 0.30–1.74 mg/kg; and Escherichia coli, 2.02 mg/kg.11
Pfaller et al15 evaluated the in vitro activity of omadacycline in MSSA and hospital- and community-acquired (HA-, CA-) MRSA. These were isolates from infections in 2014 in North America and Europe, which were obtained from a global surveillance program and compared to 2010 isolates from the SENTRY surveillance program. The authors reported no significant change in the MIC50/MIC90 values for omadacycline North American and European MSSA between 2010 (0.12/0.25 µg/mL) and 2014 (0.12/0.12 µg/mL). For MRSA in North America and Europe, the omadacycline MIC90 remained largely the same across time and place as well: 0.5 and 0.25 µg/mL in 2010, respectively, and 0.12 µg/mL for both in 2014. For HA-MRSA, omadacycline MIC90 in North America was 0.5 µg/mL in both 2010 and 2014; and in Europe it also remained stable at 0.25 µg/mL in 2010 and 0.12 µg/mL in 2014. Similarly, for CA-MRSA in North American and Europe, omadacycline MIC90 remained stable at 0.25 µg/mL in 2010 and 0.12 µg/mL in 2014.
The in vitro activity of omadacycline against anaerobic organisms was also assessed in a study by Stapert et al.16 The isolates and their MIC50/MIC90 (range) values were: Bacteroides fragilis, 0.25/4 µg/mL (0.25–16 µg/mL); Bacteroides thetaiotaomicron, 1/4 µg/mL (0.12–16 µg/mL); Bacteroides vulgatus, 0.12/1 µg/mL (0.06–2 µg/mL); Bacteroides ovatus, 0.5/8 µg/mL (0.06–>16 µg/mL); Prevotella spp., 0.5/2 µg/mL (0.12–8 µg/mL); Prevotella asaccharolytica, 0.25/0.5 µg/mL (0.06–2 µg/mL); Clostridium difficile, 0.25/0.5 µg/mL (0.25–8 µg/mL); Clostridium perfringens, 4/6 µg/mL (0.12–16 µg/mL); and Peptostreptococcus spp., 0.12/1 µg/mL (0.06–2 µg/mL). These MIC were similar to those observed for tigecycline in this study. A different study by Steenbergen et al17 also determined that omadacycline has in vitro activity against Bacillus anthracis and Yersenia pestis, which reported MIC50/MIC90 of 0.03/0.06 µg/mL (range=≤0.03–0.006 µg/mL), and 1/1 µg/mL (range=0.12–2 µg/mL), respectively. The in vitro activity of omadacycline against Mycoplasma hominis, Mycoplasma pneumoniae, and Ureaplasma spp. was also reported by Waites et al,18 who determined MIC50/MIC90 (range) values as follow: 0.032/0.063 mg/mL (0.016–0.125 µg/mL), 0.125/0.25 µg/mL (0.125–0.25 µg/mL), and 1/2 µg/mL (0.25–2 µg/mL), respectively.
The in vitro extracellular activity of omadacycline against Legionella pneumophila in isolates from 1995 to 2014 remained unchanged: the MIC90 for serogroup 1 (n=90), the most commonly isolated L. pneumophila strain in respiratory tract infections and also the most resistant to erythromycin, remained at 0.25 µg/mL, which was lower than the MIC90 for doxycycline (1 µg/mL), erythromycin (1 µg/mL), and azithromycin (0.5 µg/mL).19 Against serogroups 2–6 (n=10), omadacycline had an MIC90 of 1 µg/mL for isolates from the same dates. The in vitro intracellular activity of omadacycline was evaluated in erythromycin-resistant (n=3) and susceptible (n=2) serogroup 1 strains as the minimal extracellular concentration inhibiting intracellular multiplication ≥50% (MIEC) in human monocytes.20 An MIEC of 0.06 µg/mL, an MIC of 0.25 µg/mL, and a ratio MIEC/MIC of 0.24 µg/mL were observed against 4/5 strains after 3 and 5 days of drug exposure. The omadacycline MIEC/MIC ratio was consistently lower than the ratio for doxycycline, moxifloxacin, and azithromycin. A different study by Kohlhoff et al21 assessed the in vitro activity of omadacycline against Chlamydia pneumoniae in cell cultures, with MIC as the lowest concentration at which no chlamydial antigen inclusions were observed, and the minimum bactericidal concentration (MBC) as the concentration at which no inclusions were passed onto new cells. Against Chlamydia pneumoniae, the omadacycline MIC90 was 0.25 µg/mL and the MBC90 was 0.5 µg/mL, which were comparable to those for azithromycin and doxycycline, and lower than those for moxifloxacin and levofloxacin.
An in vitro study by Goldstein et al22 assessed the MIC50 and MIC90 of omadacycline in isolates (n) from cat and dog bites in humans, which included the following: the aerobes Bergeyella zoohelcum (11), Neisseria weaveri (11), Neisseria zoodegmatis (11), Pasteurella canis (10), Pasteurella multocida subsp. multocida (11), Pasteurella multocida subsp. septica (10), Staphylococcus pseudintermedius (9); and the anaerobes Bacteroides pyogenes (10), Eikenella corrodens (10), Fusobacterium sp. (10), Porphyromonas sp. (12), Prevotella heparinolytica (10), and Prevotella sp. (10). For all aerobic and most anaerobic organisms, omadacycline displayed MIC50 and MIC90≤0.5 µg/mL; but for Eikenella corrodens, susceptibility to omadacycline and to the entire tetracycline class was diminished (omadacycline MIC50/MIC90 were 8/16 µg/mL, range=4–16 µg/mL). Overall, tetracycline and minocycline had activity against aerobes and most anaerobes, but activity against Prevotella heparinolytica and Prevotella spp. was reduced.
Pharmacokinetics
The pharmacokinetics of omadacycline have mainly been established in healthy adult subjects and subjects from special populations (eg, patients with renal or hepatic impairment, the elderly).6,23–30 No clinically significant differences in the pharmacokinetics of omadacycline were observed based on age, gender, race, weight, renal impairment or end-stage renal disease (ESRD), and hepatic impairment.6,26–28 A population pharmacokinetic model for describing oral and intravenous administration of omadacycline has been developed using pooled data from phase 1 studies in healthy subjects, one phase 1b study in patients with uncomplicated urinary tract infections, two phase 3 studies in patients with ABSSSIs, and a phase 3 study in patients with CABP.31,32 Pharmacokinetic parameters of omadacycline following single and multiple oral and intravenous doses in healthy adult subjects are summarized in Table 2.
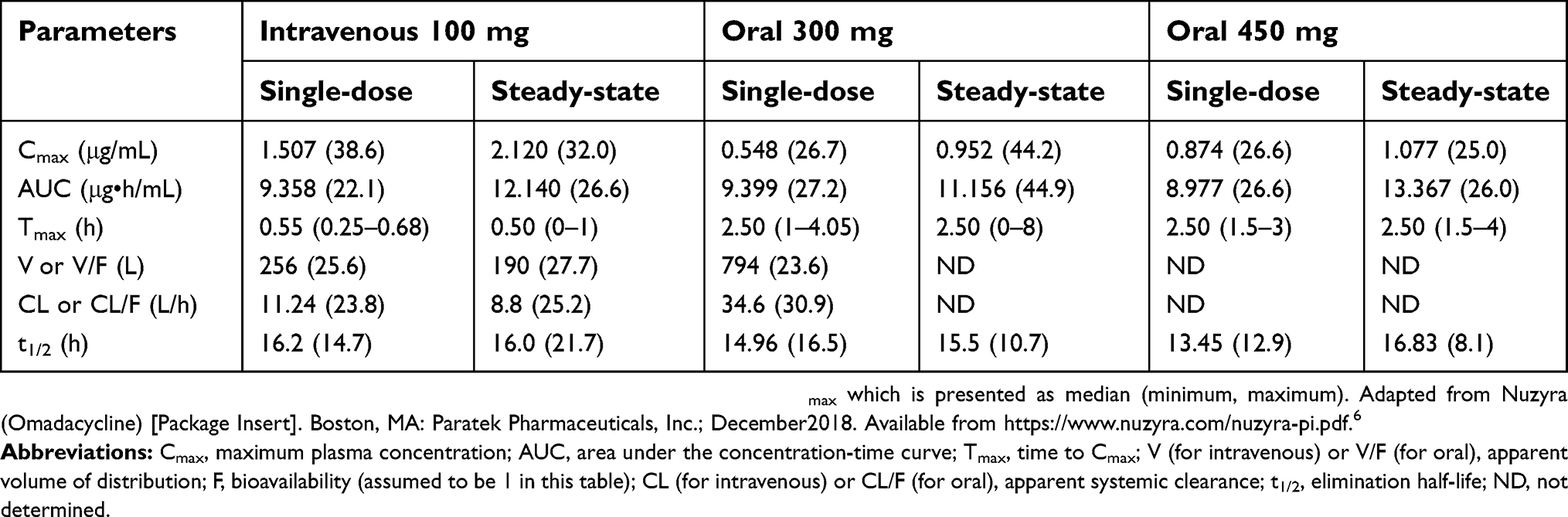 |
Table 2 Pharmacokinetic parameters of omadacycline in healthy adult subjects |
The exposure to omadacycline is similar between a 300-mg oral dose and a 100-mg intravenous dose of omadacycline in healthy fasted subjects (Figure 1).23 The ingestion of a high-fat non-dairy meal (800–1,000 calories; 50% calories from fat) 4 hours before administration of a single 300-mg oral dose of omadacycline did not substantially alter the rate (maximum plasma concentration, Cmax) and extent of absorption (area under the plasma concentration-time curve, AUC).29 Ingestion of a standard high-fat meal with and without dairy 2 hours before omadacycline administration, respectively, decreased Cmax by 42% and 40% and AUC by 63% and 59% compared to fasting conditions (Figure 2). The Cmax and AUC were not substantially altered following ingestion of either a light non-fat (300–350 calories; ≤5% calories from fat) or a standard low-fat (800–1,000 calories; 30% calories from fat) meal 2 hours after omadacycline administration.6,29,33
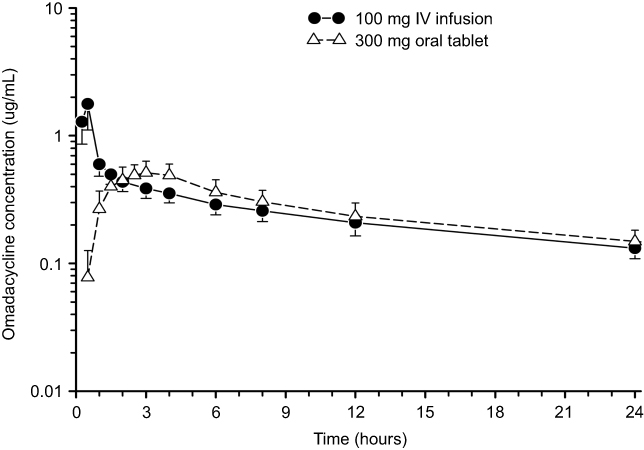 |
Figure 1 Mean±SD plasma concentration-time curve of omadacycline after administration of 300 mg oral dose (open triangles) and 100 mg intravenous dose (closed circles) in healthy subjects.Note: Data from Sun et al.23 |
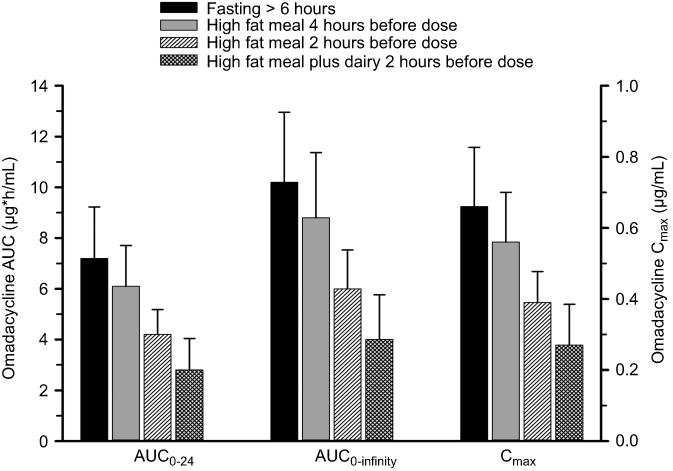 |
Figure 2 Mean±SD area under the plasma concentration-time curve (AUC) and maximum plasma concentration (Cmax) of omadacycline following oral administration of a 300 mg oral dose under fasting condition (solid black bar), when a standard high-fat non-dairy meal was ingested 4 hours pre-dose (solid gray bar), when a standard high-fat non-dairy meal was ingested 2 hours pre-dose (crossed line bar), and when a standard high-fat meal including dairy was ingested 2 hours pre-dose (confetti bar).Note: Data from Tzanis et al.29 |
The volume of distribution of omadacycline at steady-state following IV administration of omadacycline in healthy subjects ranges from 168–288 L.25,26,30,31 Plasma protein binding of omadacycline is approximately 21%, and is not concentration-dependent.34 The mean (±SD) concentrations over time for unbound plasma, epithelial lining fluid (ELF), and alveolar macrophages (AM) following intravenous administration of multiple doses of omadacycline, eravacycline, and tigecycline to healthy subjects are shown in Figure 3.29,35 The steady-state 24 hour AUC (AUC0–24) of omadacycline in ELF was approximately 1.5-fold higher than the total plasma AUC0–24, and the AUC0–24 in AM was 26-fold higher than total plasma AUC0–24. Although the pattern and time course of unbound plasma, ELF, and AM concentrations were similar among the three drugs, the magnitude of omadacycline concentrations was greater in all matrices compared to eravacycline and tigecycline.
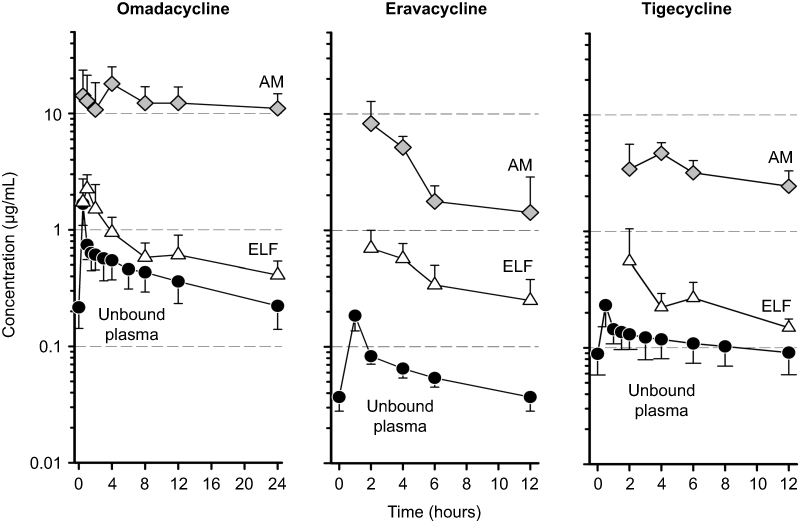 |
Figure 3 Mean±SD unbound plasma, epithelial lining fluid (ELF), and alveolar macrophages (AM) concentration-time curve of omadacycline, eravacycline, and tigecycline after multiple intravenous doses of 100 mg, 1 mg/kg, and 50 mg, respectively. Note that the duration of time represents the dosing interval of 24 hours for omadacycline and 12 hours for eravacycline and tigecycline.Note: Data from these studies30,35 |
A phase 1 study assessed the pharmacokinetics of omadacycline in eight ESRD vs eight matched healthy subjects.26 The following parameters were observed after an intravenous dose of omadacycline 100 mg before and after hemodialysis (HD) in ESRD subjects and in healthy subjects (pre-HD/post-HD/healthy subjects): AUC0–∞=10.20/10.30/9.76 µg•h/mL, Cmax =2.33/1.88/1.92 µg/mL, Tmax=0.59/0.58/0.58 h, V=194/214/204 L, CL=10.1/10.1/10.6 L/h, and t1/2=18.9/18.6/17.1 h. Based on these parameters, and tolerability and safety across ESRD and healthy subjects, omadacycline does not require dose adjustment in renal impairment or on HD days.
In vitro studies using human liver microsomes and hepatocytes demonstrated that omadacycline is not metabolized.6,36,37 In-vitro studies in human liver microsomes indicate that omadacycline does not inhibit nor induce metabolism mediated by CYP isoenzymes 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5, or UGT1A1. Omadacycline is not an inhibitor of P-glycoprotein (P-gp) and organic anion transporting polypeptide (OATP) 1B1 and OATP1B3. Omadacycline is not a substrate or inhibitor of the major organic anion transporters (OAT-1 and 3), breast cancer resistance protein (BCRP), or multidrug resistance-associated protein 2 (MRP2). Omadacycline was not an OATP1B1 or OATP1B3 substrate at supra-therapeutic concentrations (5–13 fold higher than clinically relevant concentrations).
Omadacycline is a substrate of P-gp. Administration of oral verapamil (P-gp inhibitor) 2 hours before a single 300 mg oral dose of omadacycline increased the AUC and Cmax of omadacycline by approximately 25% and 9%, respectively.33
In healthy male volunteers receiving 300-mg oral [14C] omadacycline, 77.5% to 84.0% of the dose was recovered in the feces, approximately 14.4% (range=10.8%–17.4%) in the urine, with 95.5% of the administered radioactive dose recovered after 7 days.37 Following intravenous administration of a 100-mg dose of omadacycline, 27% of the dose was recovered as unchanged omadacycline in the urine.26 The systemic and renal clearance of omadacycline in healthy subjects following intravenous administration ranged from 8.8–11.8 L/h and 2.4–3.3 L/h, respectively.26,27,30,31 The elimination t1/2 is approximately 16–17 hours (range=11–25 hours).23–26,30,31
Pharmacodynamics
A limited number of pharmacokinetic-pharmacodynamic studies for older tetracyclines and tigecycline have identified correlations between 24 hour unbound area under the concentration-time curve to minimum inhibitory concentration (fAUC0-24/MIC) and efficacy.38–40 Exposure-response relationships for omadacycline have also been shown to correlate with fAUC0–24/MIC ratios.41–44 Table 3 displays the magnitude of the plasma fAUC0–24/MIC ratio for omadacycline associated with net bacterial stasis as well as 1- and 2-log10 colony forming unit (CFU) reductions from baseline for Streptococcus pneumoniae and Staphylococcus aureus in the neutropenic murine thigh and lung infection models, respectively.41,42
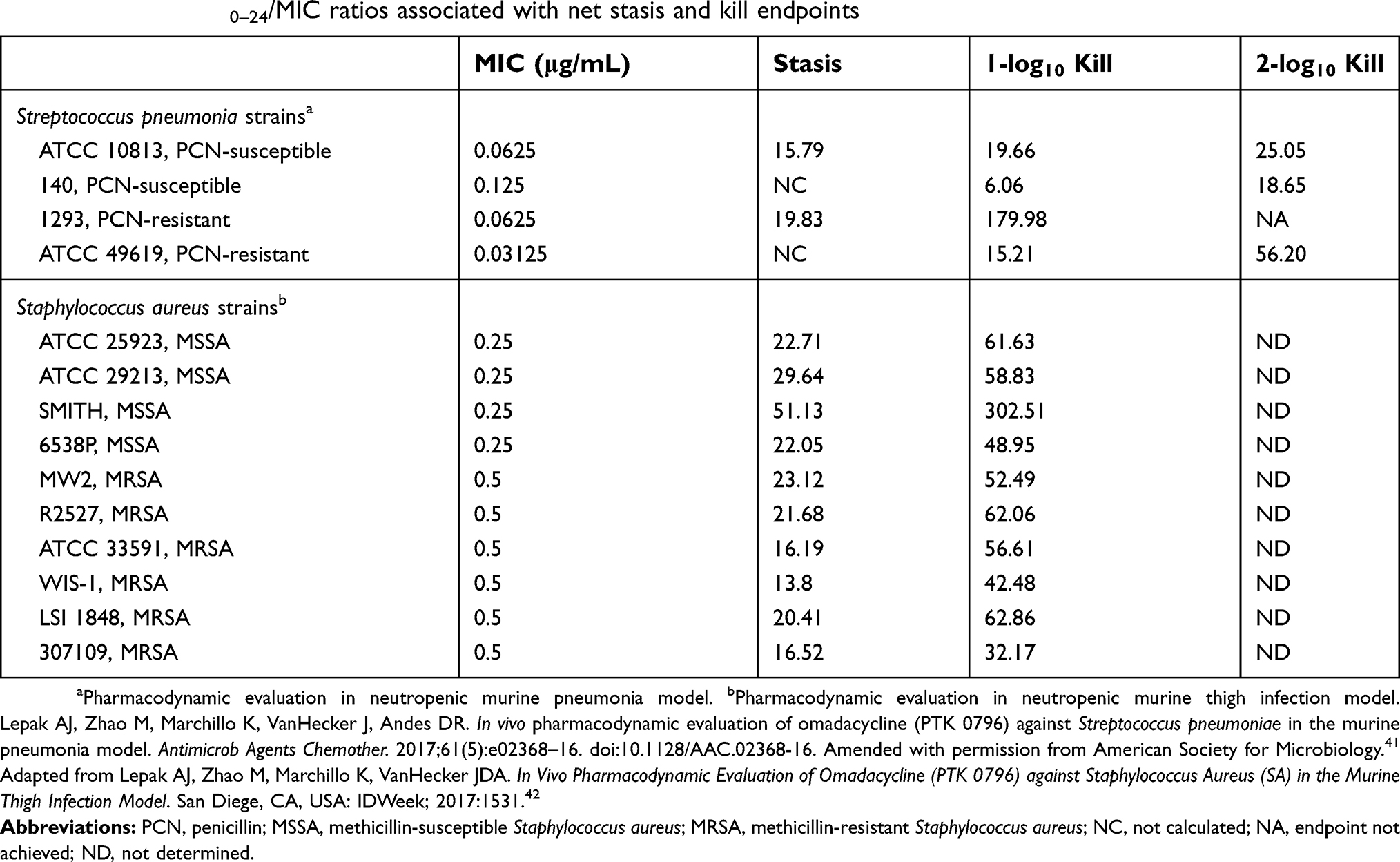 |
Table 3 Plasma AUC0–24/MIC ratios associated with net stasis and kill endpoints |
Omadacycline was slightly more potent than tigecycline in both neutropenic and non-neutropenic, immunocompetent murine thigh infection model. The presence of neutrophils enhanced the in vivo efficacy of omadacycline by over 6-fold and by approximately 2-fold against single strains of Streptococcus pneumoniae and Klebsiella pneumoniae, respectively.43 The total AUC0–24/MIC ratios for epithelial lining fluid (ELF) associated with bacterial stasis (14.18–17.80), 1-log10 kill (6.00–200.64), and 2-log10 kill (17.26–47.27) were similar to corresponding plasma fAUC0–24/MIC values (Table 3), since the penetration of omadacycline into ELF approached 100% (range=72−102%) in mice. Lung surfactant does not have an effect on the in vitro activity of omadacycline.45 Omadacycline has consistently demonstrated similar to or greater potency than minocycline, doxycycline, linezolid, and vancomycin for a wide range of mouse infections models and bacterial pathogens.7
The post-antibiotic effect (PAE) of omadacycline has ranged from 2.2–3.3 hours for Streptococcus pneumoniae and Staphylococcus aureus.46 The PAE of omadacycline for these pathogens, as well as for Escherichia coli (1.4 hours), were generally similar to those reported with tigecycline. A slightly longer PAE was observed with tigecycline (3.8–4.4 hours) compared to omadacycline (2.0–2.1 hours) for Enterococcus faecalis and Enterococcus faecium.
Dose justification for intravenous, intravenous-to-oral, and oral dosing regimens of omadacycline for treatment of ABSSSI and CABP were based on population pharmacokinetics of healthy subjects and patients, simulated probabilities of pharmacokinetic-pharmacodynamic target attainment by MIC values, and assessment of the relationships between efficacy and AUC0-24/MIC ratio.47 The probability of clinical success at the early clinical response visit increased in patients with ABSSSI as fAUC0–24/MIC ratio values increased. A successful early clinical response visit occurred in 20 of 25 patients (80%) when the plasma fAUC0–24/MIC ratio was <12.5 compared to a success rate of 96–100 patients (96%) when the ratio was ≥12.5. For patients with Staphylococcus aureus with a MIC value of 0.5 µg/mL, the predicted percent probabilities of a successful efficacy ranged from 87.2% to 95.6% for intravenous-to-oral dosing and oral only dosing. Simulated AUC/MIC ratios in ELF were similar or greater than non-clinical pharmacokinetic-pharmacodynamic target values associated with efficacy for Streptococcus pneumoniae and Haemophilus spp. in 11 omadacycline treated patients with CABP in the phase 3 clinical trial.
Clinical efficacy
Skin and skin structure infections
Noel et al48 investigated the efficacy of omadacycline vs linezolid for the treatment of complicated skin and skin structure infections (CSSSI) in hospitalized patients in a randomized, controlled, evaluator-blinded phase 2, non-inferiority trial in the US between 2007 and 2008. Subjects were randomized to receive intravenous treatment for CSSSI with either omadacycline 100 mg every 24 hours or linezolid 600 mg every 12 hours with an option to add aztreonam 2 g every 12 hours in cases of investigator-suspected or documented gram-negative pathogens. Based on the investigator’s judgment at hospital discharge, subjects could transition to oral therapy (two omadacycline 100 mg tablets daily, or one linezolid 600 mg tablet twice daily), at which point only the investigators remained blinded to study medication. Subjects were included if ≥18 years of age with one of the following: a wound infection, a major abscess, an infected ulcer in a lower extremity, or cellulitis. Subjects were excluded if they had infections potentially involving bone or if these were resolved with surgery alone. If the lower limit of the 95% confidence interval (95% CI) of the treatment difference was higher than −20%, omadacycline was considered non-inferior to linezolid.
There were 118 subjects randomized to omadacycline and 116 to linezolid with the following disposition: 94.1% (111/118) vs 93.1% (108/116) subjects in the safety (primary endpoint) population and intention-to-treat (ITT) populations who received ≥1 dose of study medication; 75.7% (84/111) vs 72.2% (78/108) in the ITT population who had a baseline pathogen (the modified ITT population, MITT); 90.1% (100/111) vs 81.5% (88/108) clinically evaluable (CE), which were subjects in the ITT population who had a defined infection, received ≥5 days of treatment, had all defined clinical evaluations, and didn’t receive antibiotics outside the study; 77% (77/100) vs 71.5% (63/88) were microbiologically evaluable (ME), which included subjects in the CE population with a baseline pathogen.48 The higher proportions of subjects in the omadacycline arm cascading from the MITT to the ME population were due to a higher number of subjects in the linezolid group who completed treatment, but were lost to follow-up and didn’t have a test-of-cure visit.
In this phase 2 clinical trial, efficacy (as the rate of successful clinical response) was a secondary endpoint, and was higher for the omadacycline arm in all four populations: 88.3% (98/111) vs 75.9% (82/108) in the ITT [difference of 12.4 percentage points (95% CI=1.9–22.9)]; 89.3% (75/84) vs 75.6% (59/78) in the MITT [difference of 13.6 percentage points (95% CI=1.4–25.9)]; 98.0% (98/100) vs 93.2% (82/88) in the CE [difference of 4.8 percentage points (95% CI=−1.7–11.3)]; and 97.4% (75/77) vs 93.7% (59/63) in the ME population [difference of 3.8 percentage points (95% CI=−4.0–11.5), respectively.48 For subjects in the CE population who had no prior antibiotics, clinical response was similar between groups: 96.3% (53/55) for omadacycline vs 95.2% (40/42) for linezolid. For subjects in the ME population, clinical responses by organism were as follow for omadacycline vs linezolid: 97.2% (70/72) vs 92.7% (51/55) in all infections with Staphylococcus aureus; 97.7% (43/44) vs 93.8% (30/32) in infections with methicillin-resistant S. aureus; 100% (3/3) vs 100% (7/7) in infections with gram-positive organisms other than S. aureus; and 100% (2/2) vs 100% (1/1) in infections with gram-negative organisms, respectively.
O’Riordan et al49 conducted a phase 3, double-blind, double-dummy, randomized clinical trial that assessed the non-inferiority of omadacycline compared to linezolid for the treatment of acute bacterial skin and skin structure infections (ABSSSI) in hospitalized subjects from Europe, the Americas, and South Africa between 2015 and 2016. This was known as the OASIS-1 study (Omadacycline in Acute Skin and Skin Structure Infections Study). Subjects randomly received a 7–14-day treatment course of either omadacycline 100 mg intravenously every 12 hours for two doses, then every 24 hours, with an optional switch to oral 300 mg every 24 hours ≥3 days later or linezolid 600 mg intravenously every 12 hours, with an optional switch to oral 600 mg every 12 hours ≥3 days later. Subjects included were ≥18 years of age, with a skin infection, which included wound infections, cellulitis, or erysipelas, and major abscesses (restricted to ≤30% of randomized subjects). Skin infections had to show a contiguous surface area of ≥75 cm2, defined edema, erythema or induration, and inflammatory response. Subjects who used ≥1 dose of potentially effective systemic or topical antibiotic treatment within 72 hours prior to the first dose of study medication, were expected to require long-term treatment for chronic skin infections, or had significant liver or renal insufficiency or immunocompromise, were disallowed into the study.
The primary efficacy endpoint was early clinical response in the modified intention-to-treat population (MITT), which comprised randomized subjects without only gram-negative organisms at baseline.49 This primary endpoint was assessed 48–72 hours after treatment initiation with study medication, and defined as survival with a decrease of ≥20% in lesion size. Secondary efficacy endpoints included: Investigator-assessed clinical response (survival with resolution or improvement in signs and symptoms without need for further treatment) at the end-of-treatment (EOT) and at the post-treatment evaluation (PTE) visit (7–14 days after the last dose of study medication) in both the MITT and the clinical per-protocol (cPP) populations; and microbiologic response at EOT and PTE visits in the microbiologic MITT and per-protocol (mMITT and mPP) populations. Diagnostic assays such as Gram stain, culture, and blood samples were collected for testing as appropriate. Safety data, including adverse events (AEs), clinical laboratories, vital signs, and electrocardiography were also obtained. For both primary and secondary endpoints, non-inferiority was met if the lower limit of the 95% confidence interval for the difference between groups was higher than −10 percentage points.
There were 655 subjects randomized (ITT population), 329 to receive omadacycline and 326 to receive linezolid: 645 received ≥1 dose of study medication (safety population), and 627 subjects in the MITT population (316 omadacycline, 311 linezolid).49 Baseline demographics and clinical characteristics were similar between treatment arms, including the median lesion area in the MITT group (299.5 cm2 omadacycline, 315 cm2 linezolid) and mean duration of treatment (4.4 days intravenous with 5.5 days oral omadacycline vs 4.4 intravenous with 5.4 oral linezolid). Based on the baseline pathogen associated with ABSSSI in the mMITT population, infections were gram-positive monomicrobial in 71.9%, gram-positive polymicrobial in 12.7%, and mixed gram-positive and gram-negative in 15.4% of subjects. These included the following organisms for the omadacycline vs linezolid arms: Staphylococcus aureus in 68.4% vs 66.5%, methicillin-resistant S. aureus (MRSA) in 30.3% vs 22.0%, Streptococcus anginosis group in 20.6% vs 16.3%, gram-positive anaerobes in 7.0% vs 6.6%, gram-negative aerobes in 12.3% vs 10.1%, and gram-negative anaerobes in 7.5% vs 5.7% of subjects. The proportions of subjects with a switch from intravenous to oral therapy (88.5% vs 87.9%) and ≥80% adherence (99.1% vs 98.8%) were similar between treatment omadacycline and linezolid, respectively.
In this study of subjects with ABSSSI, omadacycline demonstrated non-inferiority to linezolid with respect to the primary endpoint of early clinical response: 84.8% vs 85.5%, respectively, a difference of −0.7 percentage points (95% CI=−6.3–4.9).49 The efficacy of omadacycline was also non-inferior or similar to linezolid in secondary endpoints across different populations, including early and later clinical responses in the MITT, cPP, different skin infection types, and mMITT. Based on ABSSSI pathogens isolated at baseline, early and later clinical responses were also similar between treatments. Clinical response rates at the PTE visits were also similar for subjects with monomicrobial gram-positive (87.8% vs 84.8%), polymicrobial gram-positive (74.2% vs 81.5%), and polymicrobial mixed infections (80.5% vs 75.9%) for omadacycline vs linezolid, respectively. In subjects with bacteremia, the investigator-assessed clinical responses at PTE were 82% (9/11) vs 100% (9/9) for omadacycline vs linezolid, respectively. Given the small sample of subjects with bacteremia, it is unwise to draw interpretations about performance of either drug in subjects with bacteremia. Primary and select secondary endpoints from this study are summarized in Table 4.
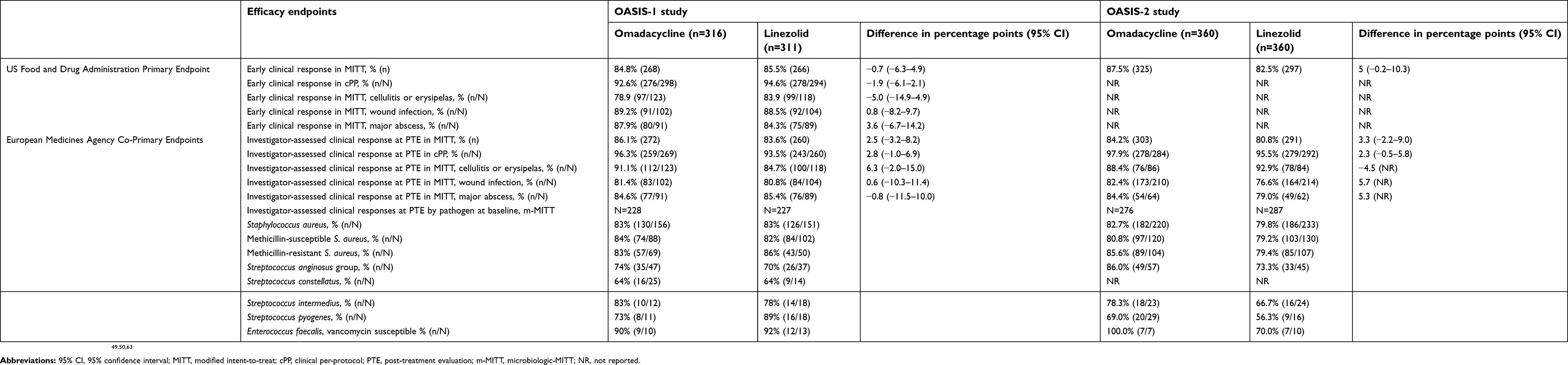 |
Table 4 Primary and select secondary endpoints in the OASIS-1 study, a phase 3 clinical trial comparing non-inferiority of omadacycline vs linezolid for acute bacterial skin and skin-structure infections |
A second phase 3, randomized, double-blind, non-inferiority clinical trial in multiple centers across the US evaluated the efficacy and safety of oral-only treatment of ABSSSI with omadacycline compared to linezolid.50,63 In this trial, known as the OASIS-2 study, 735 subjects were randomized to receive a 7–14-day treatment with either omadacycline 450 mg orally daily for 2 days, then 300 mg orally daily or linezolid 600 mg orally twice daily. Similar to OASIS-1, subjects ≥18 years of age with qualifying ABSSSI were included, and enrollment was limited for subjects with major abscesses (≤30%), but some (≤25%) subjects were allowed to participate if they had received a short-acting antibiotic within the previous 72 hours to study medication. Also similar to OASIS-1, the primary endpoint was early clinical response in the MITT population based on survival with lesion size reduction (≥20%), which was evaluated 48–72 hours after the initial dose of study medication. Secondary endpoints included the investigator-assessed clinical success at the EOT and PTE visits in different populations. Results from the OASIS-2 study are only available in abstract form, and have not been published in a peer-reviewed manner as of this writing.
In the OASIS-2 study, there were 360 subjects who received omadacycline and 360 subjects who received linezolid in the MITT population, with a mean duration of treatment of 8.2 days and 8.0 days, respectively.50 Baseline characteristics for omadacycline vs linezolid were relatively similar: A majority of males in each arm (65.8% vs 59.9%); with a mean age of 42.8 vs 44.5 years; a mean lesion area of 422 cm2 (range=75–2,601 cm2) vs 396 cm2 (range=75–2,243 cm2); and similar proportions in the types of ABSSSI, which included 58.3% vs 59.4% wound infections, 23.9% vs 23.3% cellulitis and erysipelas, and 17.8% vs 17.2% major abscesses, respectively. The proportions of subjects that met the primary endpoint were 87.5% vs 82.5% for omadacycline vs linezolid and a treatment difference of 5 percentage points (95% CI=−0.2–10.3) established non-inferiority. Secondary endpoints of investigator-assessed clinical success at PTE were 84.2% vs 80.8% in the MITT population (3.3 percentage points difference; 95% CI=-2.2–9.0) and 97.9% vs 95.5% in the CE (cPP) population (2.3 percentage points difference; 95% CI=−0.5–5.8), both for omadacycline vs linezolid, respectively. A summary of primary and secondary endpoints for the OASIS-2 study are presented in Table 4.
Community-acquired bacterial pneumonia
Stets et al51 conducted a phase 3, double-blind, double-dummy, randomized clinical trial that assessed the non-inferiority of omadacycline compared to moxifloxacin for the treatment of community-acquired bacterial pneumonia (CABP) in hospitalized subjects from Europe, the Americas, Africa, and Asia between 2015 and 2017. This was known as the OPTIC study (Omadacycline for Pneumonia Treatment in the Community). The 7–14-day treatment consisted of either omadacycline 100 mg intravenously every 12 hours for two doses, then every 24 hours, with an optional switch to oral 300 mg every 24 hours ≥3 days later, or moxifloxacin 400 mg intravenously every 24 hours, with an optional switch to oral 400 mg every 24 hours ≥3 days later. Subjects included were as follow: ≥18 years of age; with ≥3 symptoms of cough, purulent sputum, dyspnea, or pleuritic chest pain; with ≥2 abnormal vital signs; ≥1 clinical sign or laboratory associated with CABP; radiologic confirmation of pneumonia; classified as Pneumonia Severity Index (PSI) risk class II (in ≤15% of randomized subjects), III or IV; had not received ≥1 doses of potentially effective antibiotic treatment 72 hours prior to the first dose of study medication (except for one dose of a short-acting antibiotic in ≤25% of subjects); and did not have hospital-acquired pneumonia or empyema, significant liver or renal insufficiency, or immunocompromise.
The primary efficacy endpoint was early clinical response in the intent-to-treat (ITT) population, defined as survival with improvement of ≥1 levels compared to baseline in ≥2 symptoms of CABP and no worsening of ≥1 levels in other symptoms of CABP, without rescue antibiotic therapy.51 This primary efficacy endpoint was assessed 72–120 hours after the first dose of study medication, based on the investigator’s assessment of symptoms of CABP on a 4-point scale (absent, mild, moderate, severe). Secondary efficacy endpoints included: Investigator-assessed clinical response (survival with resolution or improvement in signs and symptoms without need for further treatment) at the end-of-treatment (EOT) and at the post-treatment evaluation (PTE) visit (5–10 days after the last dose of study medication) in both the ITT and the clinical per-protocol (cPP) populations; and microbiologic response at EOT and PTE visits in the microbiologic and per-protocol ITT (mITT and mPP) populations. Diagnostic assays such as sputum or other specimens from the lower respiratory tract, blood, and urine were collected for staining, culture, or antigen/serologic testing as appropriate. Safety data, including adverse events (AEs), clinical laboratories, vital signs, and electrocardiography were also obtained. For both primary and secondary endpoints, non-inferiority was met if the lower limit of the 95% confidence interval for the difference between groups was higher than −10 percentage points.
There were 774 subjects randomized (ITT population), 386 to receive omadacycline, and 388 to receive moxifloxacin: 770 received ≥1 dose of study medication (safety population), most of which (98.8%) were hospitalized during initiation of the study.51 Baseline demographics and clinical characteristics were similar between treatment arms, including the proportion of subjects ≥65 years old (41.9%) and PSI risk class III or IV (85.4%). A baseline pathogen responsible associated with CABP was isolated in 49.9% in the ITT population, and included: Mycoplasma pneumoniae (33%), Streptococcus pneumoniae (20%), Legionella pneumophila (19%), Chlamydia pneumoniae (15%), and Haemophilus influenzae (12%). The proportions of subjects with a switch from intravenous to oral therapy (77.2% vs 75.8%) and adherence (99.2% vs 99.5%) were similar between treatment omadacycline and moxifloxacin, respectively.
In this study of subjects with CABP, omadacycline demonstrated non-inferiority to moxifloxacin with respect to the primary endpoint of early clinical response: 81.1% vs 82.7%, respectively, a difference of −1.6 percentage points (95% CI=−7.1–3.8).51 The efficacy of omadacycline was also non-inferior or similar to moxifloxacin in secondary endpoints across different populations including early and later clinical responses in the ITT, cPP, different PSI risk classes, and mITT. Based on CABP pathogens isolated at baseline, early and later clinical responses were similar between treatments. In subjects with bacteremia, the investigator-assessed clinical responses at PTE were 73% (11/15) vs 83% (15/18) for omadacycline and moxifloxacin, respectively. Given the small sample of subjects with bacteremia and missing values reported by the investigators, it is unwise to draw interpretations about performance of either drug in subjects with bacteremia. Primary and select secondary endpoints from this study are summarized in Table 5.
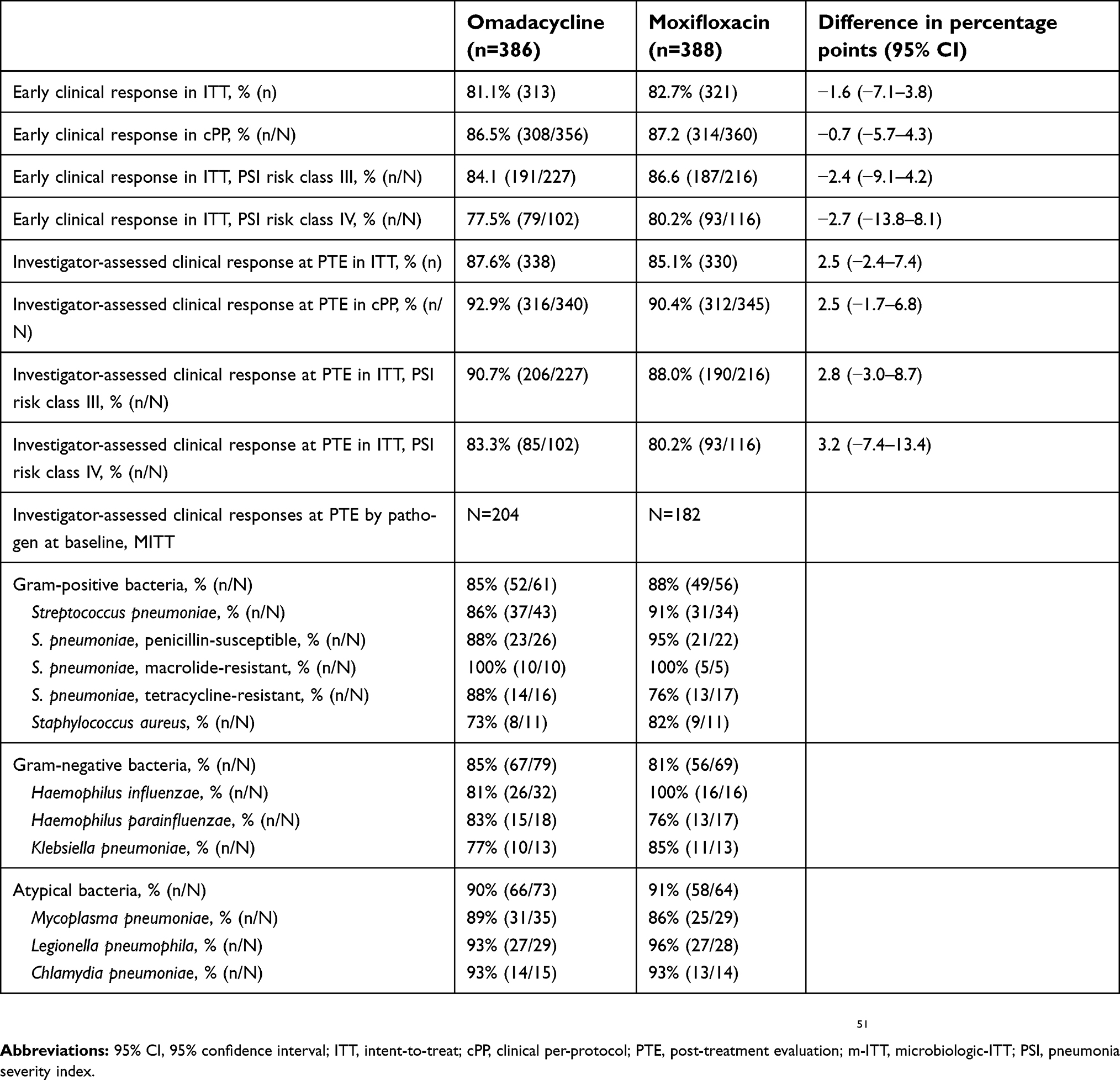 |
Table 5 Primary and select secondary endpoints in the OPTIC study, a phase 3 clinical trial comparing non-inferiority of omadacycline vs moxifloxacin for community-acquired bacterial pneumonia |
At this time, the Clinical and Laboratory Standards Institute (CLSI) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) do not offer susceptibility test interpretive criteria or “breakpoints” for omadacycline, but upcoming updates should provide these criteria in the future.52,53 However, based on the studies presented above, the US FDA has identified omadacycline breakpoints for a limited number of microorganisms causative of ABSSSI and CABP in minimum inhibitory concentrations and disk diffusion methods (Table 6).54
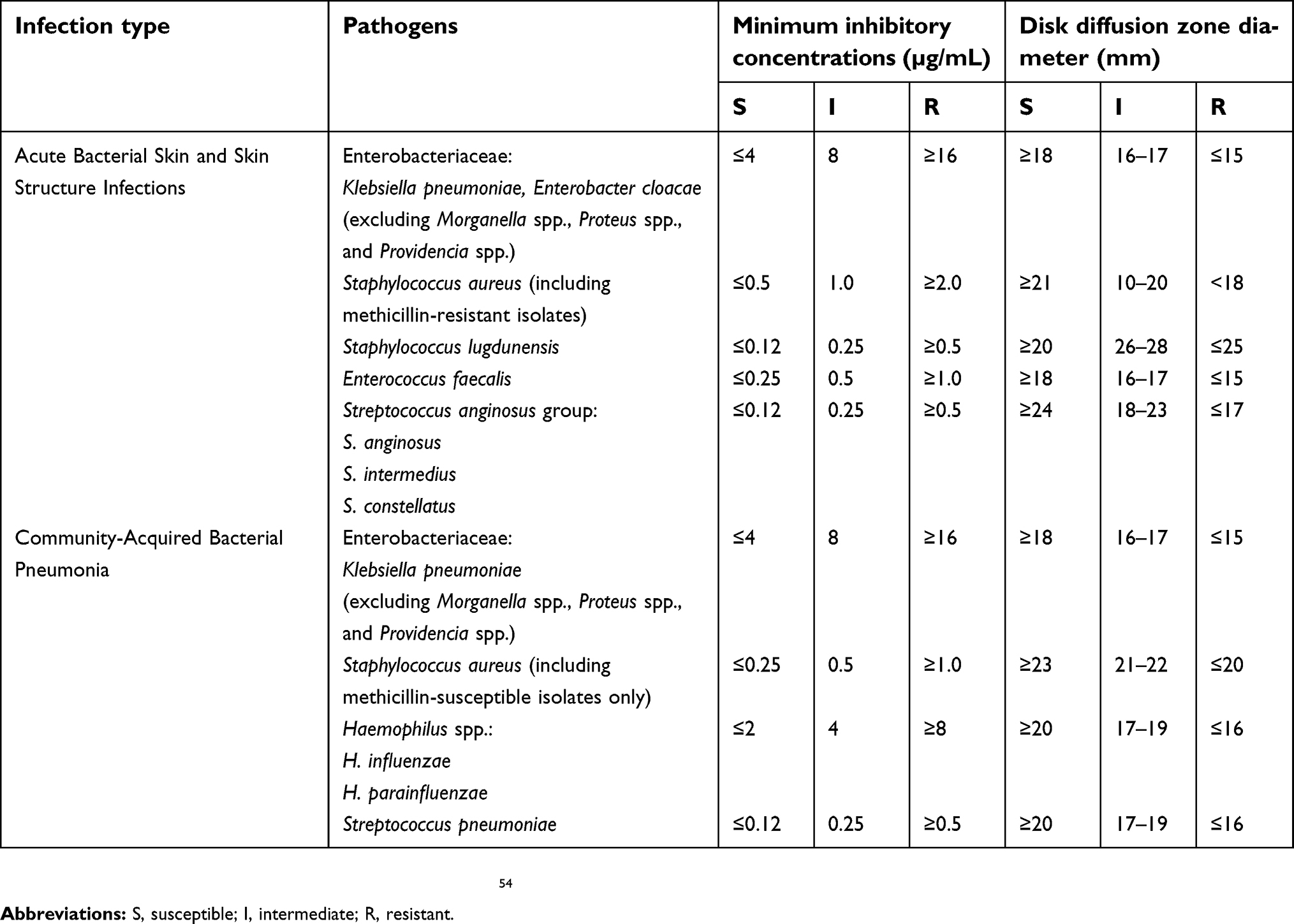 |
Table 6 United States food and drug administration identified breakpoints for omadacycline |
Clinical safety
A phase 2 clinical trial assessed safety of omadacycline compared to linezolid as a primary endpoint.48 The mean±standard deviation duration of exposure to omadacycline was 10.0±3.91 days and it was 9.6±4.36 days to linezolid, and 4.3-days was the mean duration of intravenous exposure in each treatment group. One or more treatment-emergent adverse events (TEAE) occurred in 41.6% (46/111) of subjects receiving omadacycline, and in 50.9% (55/108) of those receiving linezolid. Adverse events (AE) considered related to either treatment were 21.6% (24/111) in the omadacycline and 30.6% (33/108) in the linezolid groups. There was one serious AE (SAE) in the omadacycline and two in the linezolid groups. Discontinuations due to AE occurred in one subject treated with omadacycline (AE deemed unrelated to the study medication) and in two subjects receiving linezolid (heartburn and pruritic rash, both possibly related to the study medication). The most common AEs in both groups were gastrointestinal in nature. Safety endpoints, including lab abnormalities from this phase 2 study, are summarized in Table 7.
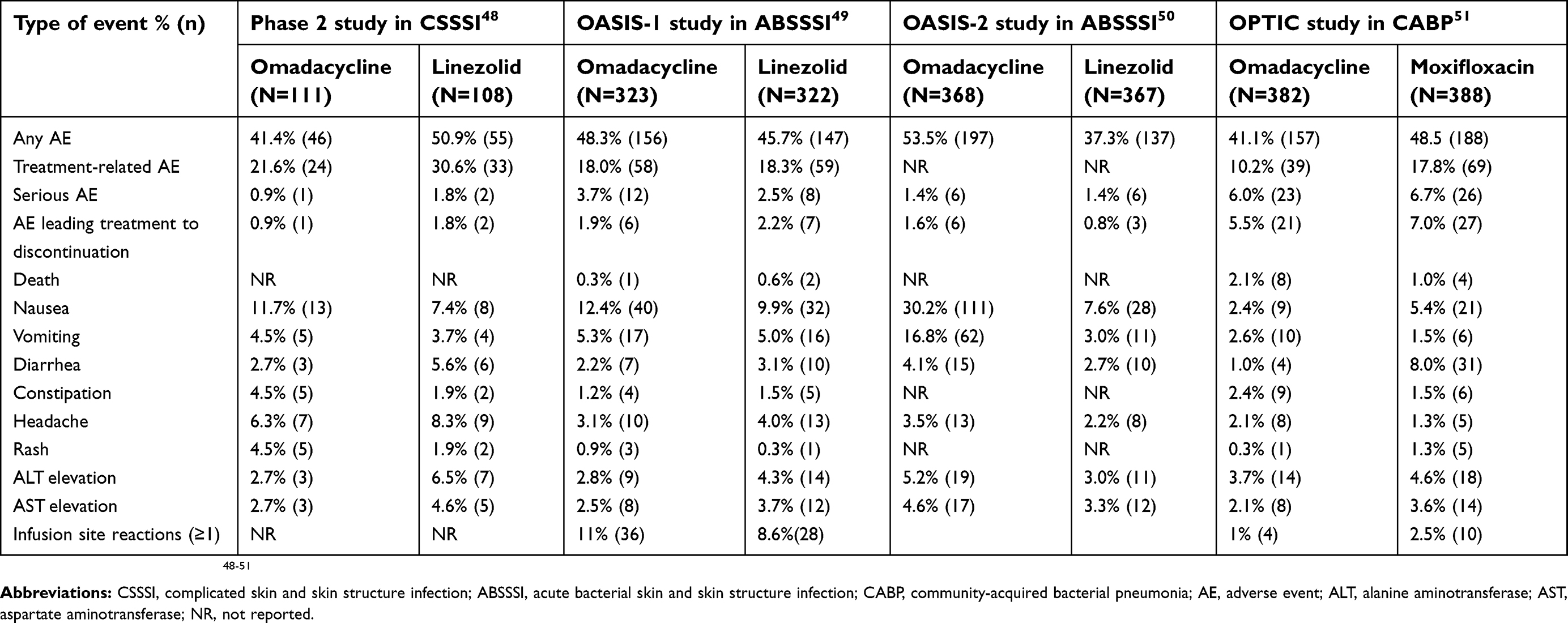 |
Table 7 Safety outcomes of omadacycline and comparators in phase 2 and 3 studies for the treatment of different infections |
In the OASIS-1 study, TEAE occurred in 48.3% (156/323) of subjects on omadacycline vs 45.7% (147/322) on linezolid.49 The AE deemed related to either treatment by blinded investigators occurred in 18.0% (58/323) vs 18.3% (59/322) of subjects on omadacycline vs linezolid, respectively. Serious AE occurred in 3.7% (12/323) of subjects on omadacycline and 2.5% (8/322) on linezolid, and AE leading to treatment discontinuation occurred in 1.9% (6/323) vs 2.2% (7/322) of subjects, respectively. There was one death reported in the omadacycline arm due to opiate overdose, and two in the linezolid arm considered unrelated to study medication (one cardiac arrest and one cardiac failure). Similar to the phase 2 study, the most common AE were gastrointestinal in nature (nausea and vomiting), all of which were mild-to-moderate and transient, except for severe vomiting in one subject on linezolid; gastrointestinal AE led to discontinuation of study medications in one subject in each group. There were no significant changes in vital signs, electrocardiogram, or laboratories reported in this phase 3 study. Safety endpoints from the OASIS-1 study are summarized in Table 7.
In the OASIS-2 study, the number of subjects who received ≥1 dose of study medication was 368 in the omadacycline and 367 in the linezolid arms: of these subjects, TEAE were reported in 53.5% vs 37.3%, respectively.50 Serious AE occurred in 1.4% of subjects in each arm of this population, but AE leading to discontinuation were reported in 1.6% vs 0.8% of subjects on omadacycline vs linezolid, respectively. Similar to previous studies, the most common AE were gastrointestinal in nature and mild or moderate: Nausea in 30.2% vs 7.6% and vomiting in 16.8% vs 3.0% receiving omadacycline vs linezolid, respectively. It is important to note that the overall higher rates of gastrointestinal AE in the omadacycline group were driven by the higher incidence of nausea and vomiting on Day 1 and 2 with oral treatment (25.5% and 12.5%, respectively), which consisted of higher loading daily doses of 450 mg; but these rates were lower on Day 3 through EOT (4.1% and 4.1%, respectively) when lower 300 mg daily doses were used. Other safety endpoints, which were comparable between treatment arms, are summarized in Table 7.
In the OPTIC study there were TEAE reported in 41.1% (151/382) and 48.5% (188/388) of subjects receiving omadacycline and moxifloxacin, but only 10.2% (39/382) and 17.8% (69/388) were deemed related to either treatment, respectively.51 There were SAE reported in 6% (23/382) and 6.7% (26/388) of subjects on omadacycline and moxifloxacin, and treatment discontinuations due to AE occurred in 5.5% (21/382) vs 7% (27/388) of subjects on these study drugs, respectively. Deaths were reported in eight subjects on omadacycline and four subjects on moxifloxacin, from progression of underlying pneumonia, respiratory compromise, hospital-acquired pneumonia, cardiac or vascular causes, and cancer, and all 12 subjects were >65 years of age. Consistent with other studies, gastrointestinal AE were most common. There were no significant changes in vital signs, electrocardiogram, or laboratories reported in this phase 3 study. Safety endpoints from the OPTIC study are summarized in Table 7.
Dosage and administration
The US FDA-approved dosage and duration of therapy of omadacycline for the treatments of adult patients with ABSSSI and CABP caused by susceptible microorganisms are listed in Table 8.1 A loading dose for both intravenous and oral routes of administration is recommended because the long elimination half-life of omadacycline allows for once daily dosing. No dose adjustments are required for patients with renal or hepatic impairment.
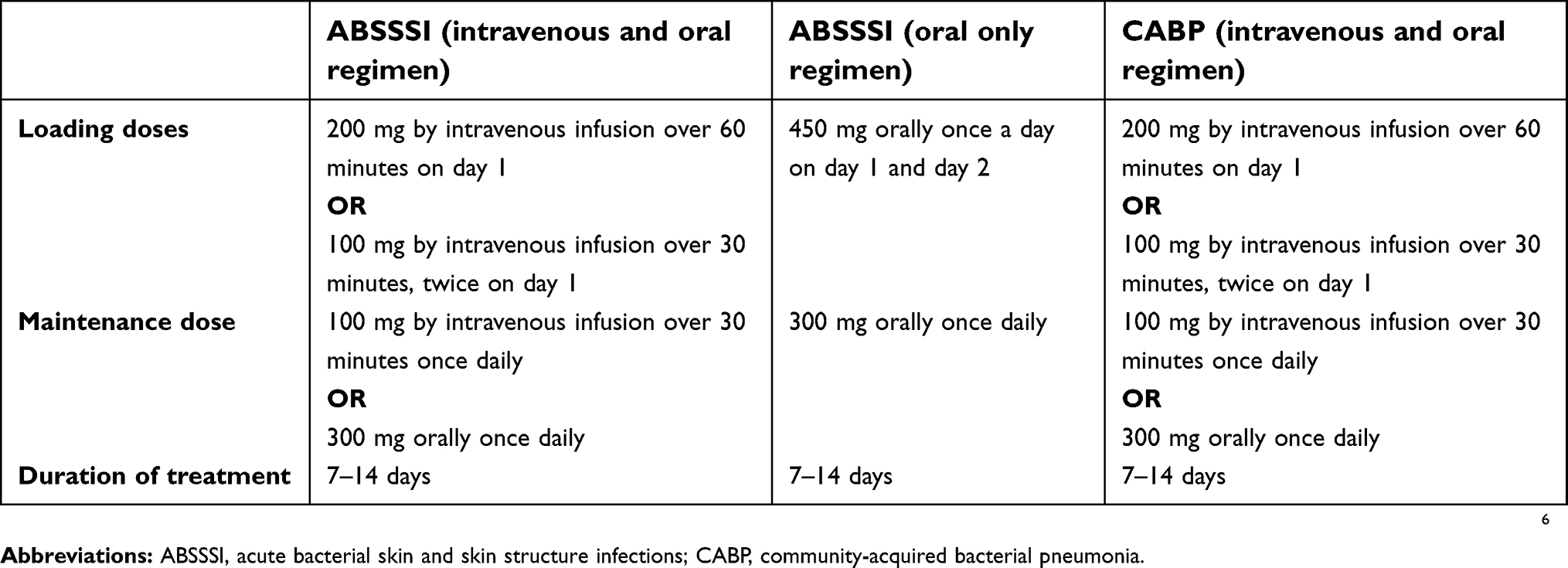 |
Table 8 Dosage of omadacycline for adult patients with ABSSSI and CABP |
Omadacycline for injection is available as sterile lyophilized powder in single-dose vials containing 100 mg of omadacycline (equivalent to 131 mg omadacycline tosylate).6 Each vial is reconstituted with sterile water and subsequently further diluted in 100 mL of normal saline or 5% dextrose. The intravenous dose of 200 mg should be administered as an infusion over 60 minutes whereas the 100 mg dose can be administered as a 30 minute intravenous infusion. Intravenous omadacycline should be administered through a dedicated intravenous line or through a Y-site. Omadacycline for injection should not be administered through the same intravenous line or with solutions containing multivalent cations (eg, calcium, magnesium). No further information is available about the compatibility of omadacycline with other intravenous solutions or drugs.
Omadacycline for oral administration is available as tablets containing 150 mg of omadacycline (equivalent to 196 mg omadacycline tosylate).6 The systematic exposure (eg, AUC) is similar between 300 mg oral dose and a 100 mg intravenous dose of omadacycline.23 Oral tablets of omadacycline should be administered in a fasting state (ie, no food or beverages (other than water) for at least 4 hours before and 2 hours after dosing).6,29 Similar to other tetracyclines, no dairy products, antacids, or multivitamins should be administered for 4 hours after oral dosing of omadacycline. This precaution is highlighted by the observed decrease in AUC and Cmax when administered with a meal that contained dairy.29 Other warnings and precautions include permanent tooth discoloration, enamel hypoplasia, bone growth inhibition in the fetus, and up to 8 years of age, hypersensitivity reactions, the potential for Clostridium difficile-associated diarrhea, photosensitivity, and other warnings associated with the tetracycline class of antibiotics, including crossing the placenta and excretion in human milk, which may pose a risk to the fetus and children.6
Place of omadacycline in therapy
Omadacycline has broad-spectrum in vitro activity, allowing monotherapy for commonly encountered community-acquired infections including ABSSSI and CABP. Pharmacokinetic and pharmacodynamic characteristics have established an initial loading dose followed by once daily maintenance dose regimens for both the intravenous and oral formulations of omadacycline. No adjustment in dose is required in patients with renal or liver impairment, and the potential of drug–drug interactions with cytochrome isoenzymes and transporters appears to be minimal. The bioequivalent oral doses of omadacycline allows empiric outpatient therapy in patients with elevated risk for polymicrobial infections caused by staphylococci (including methicillin-resistant isolates), streptococci (including penicillin- and macrolide-resistant isolates), and multiple Gram-negative organisms or have contraindications to first-line antimicrobial agents. Intravenous administration of omadacycline allows patients in the emergency room or admitted to the hospital to receive effective empiric antibiotic therapy as well as a shorter length of stay because of the potential for intravenous to oral sequential therapy with the same antibiotic. The incidence of adverse events has been low, and the common adverse reactions (≥2% of patients) are gastrointestinal, elevations in liver transaminases, and infusion site reactions with intravenous administration. While the use of linezolid as a comparator was endorsed by regulatory agencies in the US and Europe, it is possible that other strategies (eg, narrow spectrum beta-lactam, trimethoprim-sulfamethoxazole, or older tetracyclines) may be more appropriate based on local standard of care or local antimicrobial susceptibilities.
The role of omadacycline is limited by the data currently available from clinical trials.48–51 The challenge is to appropriately use omadacycline to optimize clinical outcomes, minimize adverse events, improve healthcare efficiency, and reduce overall healthcare costs among patients with ABSSSI and CABP. In both phase 2 and 3 clinical trials, omadacycline demonstrated efficacy and safety for the treatment of ABSSSI.48–50 Oral omadacycline may allow treatment as monotherapy in the ambulatory care setting as well as sequential therapy after intravenous omadacycline or switch therapy following other broad spectrum intravenous antibiotics. Current susceptibility patterns of microorganisms, patient allergies, low potential of serious adverse events (including nephrotoxicity), and drug–drug interactions, lack of significant risks for causing Clostridium difficile infection or QTc prolongation, oral and intravenous formulations, and cost will all be part of the decisions on whether to use omadacycline to treat ABSSSI. Omadacycline will need to establish its niche among the numerous antibiotic choices available to treat adult patients with ABSSSI.55,56
Omadacycline can be considered as an alternative therapy choice for adult patients with CABP when current first-line agents such as fluoroquinolones or the combination of a beta-lactam plus a macrolide are not treatment options because of hypersensitivity, adverse effect profile, or concerns about resistant pathogens.57 Low protein binding and high concentrations of omadacycline in the epithelial lining fluid and alveolar macrophages provide pharmacokinetic advantages for this antibiotic to be effective in the treatment of lower respiratory tract infections caused by susceptible microorganisms. The increasing resistance and safety concerns with fluoroquinolones makes omadacycline a reasonable alternative for intravenous and oral monotherapy for inpatient and ambulatory care settings.58 Despite the approval by regulatory agencies to use moxifloxacin as a comparator in the phase 3 study, local standards of care and local antimicrobial susceptibility should guide antimicrobial therapy. Because of insufficient clinical data to assess the observed mortality imbalance in the phase 3 clinical trial (OPTIC) comparing omadacycline to moxifloxacin, the US FDA has required a postmarketing commitment of Paratek Pharmaceuticals to conduct another active-controlled safety and efficacy study in adults with CABP.59 In addition, an active-controlled safety study in children between 8 and 17 years old with CABP will also be conducted as part of the pediatric assessment requirement of a new drug application to US FDA.9 These additional studies will further define the potential roles of omadacycline for the treatment of adult and pediatric patients with CABP.
Ongoing clinical trials with omadacycline include phase-2 studies for the treatment of uncomplicated urinary tract infections and acute pyelonephritis.60,61 Paratek Pharmaceuticals also has a cooperative research agreement with the US Army Medical Research Institute of Infectious Diseases to study omadacycline against biodefense pathogens, including Yersinia pestis (plague) and Bacillus anthracis (anthrax).17,62 Omadacycline has not be evaluated for the treatment of infections caused by multiple-drug-resistant Gram-negative infections, and clinical trials are required to define the role of omadacycline against pathogens such as carbapenam-resistant Enterobacteriaceae and Acinetobacter spp.55
When deciding how to approach antimicrobial therapy, cost is always a concern, particularly with new agents. A 3-year budget impact model to evaluate omadacycline use for the treatment of CABP in the US showed that, although an initial treatment acquisition cost would be incurred ($20,643), the impact from transitioning from intravenous to oral treatment on the 3-year cost decreased by reducing 1 day of hospital stay ($2,384), provided cost-savings by reducing 2 days of hospital stay ($15,875), and provided a substantial cumulative cost-saving by shifting inpatient care to the outpatient management ($112,843).64 Similar hypothetical models of patients with ABSSSI also showed significant cost-savings by reducing hospital stay and shifting inpatient to outpatient care or from avoiding inpatient admission for intravenous antibiotic administration.65,66 Hence, in theory, the upfront costs of implementing omadacycline use for CABP and ABSSSI could be offset by decreased hospital stay or avoidance in the US.
Conclusion
Three phase 3 clinical trials have established the novel aminomethylcycline, omadacycline, as an effective and safe antibiotic for the treatment of ABSSSI and CABP. The oral and intravenous formulations will allow monotherapy to be used in both the outpatient and inpatient settings. No dosage adjustment in renal or hepatic impairment, minimal chances of major drug–drug interactions, and a low incidence of adverse effects are positive features of this new tetracycline analog. While omadacycline retains antibacterial activity against bacterial strains expressing the two most common tetracycline resistance mechanisms (ie, bacterial ribosomal protection proteins and tetracycline-specific efflux pumps), further microbiological evidence and clinical efficacy against multidrug-resistant pathogens are still needed. Omadacycline will need to establish its place among the available antibiotic choices for the treatment of community-acquired infections.
Disclosure
Dr Rodvold is a consultant for Paratek Pharmaceuticals, Inc. and reports personal fees from Paratek Pharmaceuticals, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Nelson ML, Levy SB. The history of tetracyclines. Ann N Y Acad Sci. 2011;1241:17–32. doi:10.1111/j.1749-6632.2011.06354.x
2. Zhanel GG, Hornenuik K, Nichol K, et al. The glycylcyclines: a comparative review with the tetracyclines. Drugs. 2004;64(1):63–88. doi:10.2165/00003495-200464010-00005
3. Zhanel GG, Cheung D, Adam H, et al. Review of eravacycline, a novel fluorocycline antibacterial agent. Drugs. 2016;76(5):567–588. doi:10.1007/s40265-016-0625-9
4. Markham A, Keam SJ. Omadacycline: first global approval. Drugs. 2018;78(18):1931–1937. doi:10.1007/s40265-018-1015-2
5. Villano S, Steenbergen J, Loh E. Omadacycline: development of a novel aminomethylcycline antibiotic for treating drug-resistant bacterial infections. Future Microbiol. 2016;11:1421–1434. doi:10.2217/fmb-2016-0100
6. Nuzyra (Omadacycline) [Package Insert]. Boston, MA: Paratek Pharmaceuticals, Inc.; December 2018. Available from https://www.nuzyra.com/nuzyra-pi.pdf.
7. Tanaka KS, Steenbergen J, Villano S. Discovery, pharmacology, and clinical profile of omadacycline, a novel aminomethylcycline antibiotic. Bioorg Med Chem. 2016;24(24):6409–6419. doi:10.1016/j.bmc.2016.06.043
8. Honeyman L, Ismail M, Nelson ML, et al. Structure-activity relationship of the aminomethylcyclines and the discovery of omadacycline. Antimicrob Agents Chemother. 2015;59(11):7044–7053. doi:10.1128/AAC.01536-15
9. Draper MP, Weir S, Macone A, et al. Mechanism of action of the novel aminomethylcycline antibiotic omadacycline. Antimicrob Agents Chemother. 2014;58(3):1279–1283. doi:10.1128/AAC.01066-13
10. Grossman TH. Tetracycline antibiotics resistance. Cold Spring Harb Perspect Med. 2016;6(4):a025387. doi:10.1101/cshperspect.a025387
11. Macone AB, Caruso BK, Leahy RG, et al. In vitro and in vivo antibacterial activities of omadacycline, a novel aminomethylcycline. Antimicrob Agents Chemother. 2014;58(2):1127–1135. doi:10.1128/AAC.02045-12
12. Ng LK, Martin I, Alfa M, Mulvey M. Multiplex PCR for the detection of tetracycline resistant genes. Mol Cell Probes. 2001;15(4):209–215. doi:10.1006/mcpr.2000.0344
13. Ruzin A, Dzink-Fox J, Jones AK, Dean CR, Bradford PA Studies on the mechanism of resistance to PTK796 in pseudomonas aeruginosa and klebsiella pneumoniae.
14. Pfaller MA, Huband MD, Rhomberg PR, Flamm RK. Surveillance of omadacycline activity against clinical isolates from a global collection (North America, Europe, Latin America, Asia-Western Pacific), 2010–2011. Antimicrob Agents Chemother. 2017;61(5):e00018–17. doi:10.1128/AAC.00018-17
15. Pfaller MA, Rhomberg PR, Huband MD, Flamm RK. Activities of omadacycline and comparator agents against Staphylococcus aureus isolates from a surveillance program conducted in North America and Europe. Antimicrob Agents Chemother. 2017;61(3):e02411–16. doi:10.1128/AAC.02411-16
16. Stapert L, Wolfe C, Shinabarger D, et al. In vitro activities of omadacycline and comparators against anaerobic bacteria. Antimicrob Agents Chemother. 2018;62(4):e02551–18. doi:10.1128/AAC.02551-17
17. Steenbergen J, Tanaka SK, Miller LL, Halasohoris SA, Hershfield JR. In vitro and in vivo activity of omadacycline against two biothreat pathogens, bacillus anthracis and yersinia pestis. Antimicrob Agents Chemother. 2017;61(5):e02434–16. doi:10.1128/AAC.02434-16
18. Waites KB, Crabb DM, Liu Y, Duffy LB. In vitro activities of omadacycline (PTK 0796) and other antimicrobial agents against human mycoplasmas and ureaplasmas. Antimicrob Agents Chemother. 2016;60(12):7502–7504.
19. Dubois J, Dubois M, Martel J-F, Tanaka SK. In vitro activity of omadacycline against Legionella pneumophila.
20. Dubois J, Dubois M, Martel J-F, Tanaka SK. In vitro intracellular activity of omadacycline against Legionella pneumophila.
21. Kohlhoff SA, Huerta N, Hammerschlag MR. In vitro activity of omadacylines against Chlamydia pneumoniae. Antimicrob Agents Chemother. 2019;63(2):e01907–18.
22. Goldstein EJC, Citron DM, Tyrrell KL, Leoncio E, Merriam CV. Comparative in vitro activity of omadacycline against dog and cat bite wound isolates. Antimicrob Agents Chemother. 2018;62(4):e02551–17. doi:10.1128/AAC.02551-17
23. Sun H, Ting L, Machineni S, et al. Randomized, open-label study of the pharmacokinetics and safety of oral and intravenous administration of omadacycline to healthy subjects. Antimicrob Agents Chemother. 2016;60(12):7431–7435. doi:10.1128/AAC.01393-16
24. Bundrant LA, Tzanis E, Garrity-Ryan L, et al. Safety and pharmacokinetics of the aminomethylcycline antibiotic omadacycline administered to healthy subjects in oral multiple-dose regimens. Antimicrob Agents Chemother. 2018;62(2):e01487–17. doi:10.1128/AAC.01487-17
25. Tanaka SK, Villano S, Tzanis E. Single and multiple dose pharmacokinetics and tolerability of intravenous omadacycline in healthy volunteers.
26. Berg JK, Tzanis E, Garrity-Ryan L, et al. Pharmacokinetics and safety of omadacycline in subjects with impaired renal function. Antimicrob Agents Chemother. 2018;62(2):e02057–17. doi:10.1128/AAC.02057-17
27. Ting L, Kovacs SJ, Praestgaard J, et al. Pharmacokinetics of omadacycline (PTK0796) in subjects with hepatic impairment.
28. Tanaka SK, Tzanis E, Villano S. Effect of age and gender on the pharmacokinetics of oral and IV omadacycline, a new class of aminomethylcyclines.
29. Tzanis E, Manley A, Villano S, Tanaka SK, Bai S, Loh E. Effect of food on the bioavailability of omadacycline in healthy participants. J Clin Pharmacol. 2017;57(3):321–327. doi:10.1002/jcph.814
30. Gotfried MH, Horn K, Garrity-Ryan L, et al. Comparison of omadacycline and tigecycline pharmacokinetics in the plasma, epithelial lining fluid, and alveolar cells of healthy adult Subjects. Antimicrob Agents Chemother. 2017;61(9):e01135–17. doi:10.1128/AAC.01135-17
31. Overcash JS, Bhiwandi P, Tzanis E, et al. Pharmacokinetics and safety of omadacycline in patients with uncomplicated urinary tract infections.
32. Lakota EA, van Wart SA, Tzanis E, et al. Population Pharmacokinetic Analysis of Omadacycline Using Phase 1 and 3 Data. Atlanta, GA: ASM Microbe; 2016. Saturday 628.
33. Hunt T. Personal communication: a phase 1, open-label, 3-period, single-sequence study to evaluate the effect of verapamil extended release and a light meal on the pharmacokinetics, safety, and tolerability of omadacycline in healthy adult subjects. Protocol PTK0796-DDI-17106; 2018.
34. Villano S, Tzanis E, Tanaka SK. In Vitro Protein Binding with Omadacycline, a First in Class Aminomethylcycline Antibiotic. Boston, USA: American Society of Microbiology Microbe; 2016:518.
35. Connors KP, Housman ST, Pope JS, et al. Phase I, open-label, safety and pharmacokinetic study to assess bronchopulmonary disposition of intravenous eravacycline in healthy men and women. Antimicrob Agents Chemother. 2014;58(4):2113–2118. doi:10.1128/AAC.02036-13
36. Lin W, Flarakos J, Du Y, et al. Pharmacokinetics, distribution, metabolism, and excretion of omadacycline following a wingle intravenous or oral dose of 14C-omadacycline in rats. Antimicrob Agents Chemother. 2017;61(1):e01784–16. doi:10.1128/AAC.01784-16
37. Flarakos J, Du Y, Gu H, et al. Clinical disposition, metabolism and in vitro drug-drug interaction properties of omadacycline. Xenobiotica. 2017;47(8):682–696. doi:10.1080/00498254.2016.1213465
38. Andes D, Craig WA. Pharmacokinetics and pharmacodynamics of tetracyclines. In: Nightingale CH, Ambrose PG, Drusano GL, Murakawa T, editors. Pharmacodynamics in Theory and Clinical Practice.
39. Agwuh KN, MacGowan A. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J Antimicrob Chemother. 2006;58(2):256–265. doi:10.1093/jac/dkl224
40. MacGowan AP. Tigecycline pharmacokinetic/pharmacodynamic update. J Antimicrob Chemother. 2008;62(Suppl 1):i11–16. doi:10.1093/jac/dkn242
41. Lepak AJ, Zhao M, Marchillo K, VanHecker J, Andes DR. In vivo pharmacodynamic evaluation of omadacycline (PTK 0796) against Streptococcus pneumoniae in the murine pneumonia model. Antimicrob Agents Chemother. 2017;61(5):e02368–16. doi:10.1128/AAC.02368-16
42. Lepak AJ, Zhao M, Marchillo K, VanHecker J,DA. In Vivo Pharmacodynamic Evaluation of Omadacycline (PTK 0796) against Staphylococcus Aureus (SA) in the Murine Thigh Infection Model. San Diege, CA, USA: IDWeek; 2017:1531.
43. Craig WA, Andes D, Odinecs A In vivo pharmacodynamics of MK-2764/PTK 0796 against gram-positive and gram-negative bacteria in the thighs of neutropenic and normal mice.
44. Tessier PR, Fan HW, Tanaka SK, Nicolau DP. Pharmacokinetic/pharmacodynamics profile of MK-2764/PTK 0796 against S. pneumoniae in a murine pneumonia model.
45. Macone AB, Donatelli JE, Draper MP, Tanaka SK. In vitro activity of omadacycline (PTK796) in broth, broth plus lung surfactant or human serum.
46. Hinshaw R, Stapert L, Shinabarger D, Pillar C. Post-antibiotic effect of omadacycline against target pathogens.
47. Paratek Pharmaceuticals. Omadacycline p-toluenesulfonate tablets and injection for the treatment of acute bacterial skin and skin structure infections (ABSSSI) and community-acquired bacterial pneumonia (CAPD). Briefing Document for FDA Antimicrobial Drugs Advisory Committee; August 8, 2018. Available from: https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM615849.pdf. Accessed May 22, 2019.
48. Noel GJ, Draper MP, Hait H, et al. A randomized, evaluator-blind, phase 2 study comparing the safety and efficacy of omadacycline to those of linezolid for treatment of complicated skin and skin structure infections. Antimicrob Agents Chemother. 2012;56(11):5650–5656. doi:10.1128/AAC.06446-11
49. O’Riordan W, Green S, Overcash JS, et al. Omadacycline for acute bacterial skin and skin-structure infections. N Engl J Med. 2019;380:528–538. doi:10.1056/NEJMoa1800170
50. O’Riordan W, Cardenas C, Sirbu A, et al. A phase-3 randomized, double-blind, multi-centre study to compare the safety and efficacy of oral omadacycline to oral linezolid for treating adult subjects with ABSSSI (OASIS-2 study), presentation O0425.
51. Stets R, Popescu M, Gonong JR, et al. Omadacycline for community-acquired bacterial pneumonia. N Engl J Med. 2019;380:517–527. doi:10.1056/NEJMoa1800201
52. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing.
53. The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. Version 9.0. 2019. Available from: http://www.eucast.org. Accessed May 22, 2019.
54. U.S. Food and Drug Administration. Omadacycline injection and oral products: FDA identified breakpoints. Available from: https://www.fda.gov/drugs/development-resources/omadacycline-injection-and-oral-products. Accessed May 22, 2019.
55. Chambers HF. Omadacycline: the newest tetracycline. N Engl J Med. 2019;380(6):588–589. doi:10.1056/NEJMe1900188
56. Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the infectious diseases society of America. Clin Infect Dis. 2014;59(2):147–159. doi:10.1093/cid/ciu296
57. Wunderink RG, Waterer G. Advances in the causes and management of community acquired pneumonia in adults. BMJ. 2017;358:j2471. doi:10.1136/bmj.j2471
58. U.S. Food and Drug Administration. Fluoroquinolone antimicrobial drugs information. Available from: https://www.fda.gov/Drugs/DrugSafety/ucm346750.htm. Accessed May 22, 2019.
59. Center for Drug Evaluation and Research. Approval package for application number: 209816Orig1s000 and 209817Orig1s000. October 2, 2018. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/209816Orig1s000,209817Orig1s000Approv.pdf. Accessed May 22, 2019.
60. NIH U.S. National Library of Medicine. Oral omadacycline vs oral nitrofurantoin for the treatment of cystitis (NCT03425396). ClinicalTrails.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03425396?term=omadacycline&rank=3. Accessed May 22, 2019.
61. NIH U.S. National Library of Medicine. IV or IV/PO omadacycline vs IV/PO levofloxacin for the treatment of acute pyelonephritis (NCT03757234). ClinicalTrails.gov, https://clinicaltrials.gov/ct2/show/NCT03757234?term=omadacycline&rank=1. Accessed May 22, 2019.
62. Global Biodefence. DOD to study omadacycline as countermeasure for plague, anthrax. Oct 12, 2016. Available from: https://globalbiodefense.com/2016/10/12/dod-study-omadacycline-countermeasure-plague-anthrax/. Accessed May 22, 2019.
63. Bassetti M, Armstrong ES, Steenbergen JN, et al. Efficacy of oral omadacycline versus linezolid for treating adult subjects with ABSSSI: analysis by infection type and pathogen in the OASIS-2 study, poster P0274.
64. LaPensee K, Mistry R, Lodise T. Budget impact of omadacycline for the treatment of patients with community-acquired bacterial pneumonia in the United States from the hospital perspective. Am Health Drug Benefits. 2019;12(Supplement 1):S1–12.
65. LaPensee K, Mistry R, Lodise T. Budget impact model of omadacycline on replacing a proportion of existing treatment options among patients who present to the emergency department with acute cacterial skin and skin structure infections. Am Health Drug Benefits. 2019;12(Supplement 2):S13–24.
66. LaPensee K, Lodise T. Potential cost-savings with once-daily aminomethylcycline antibiotic versus vancomycin in hospitalized patients with acute bacterial skin and skin structure infections. Am Health Drug Benefits. 2018;11(9):449–459.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.